Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling


Abstract
1. Introduction
2. Results
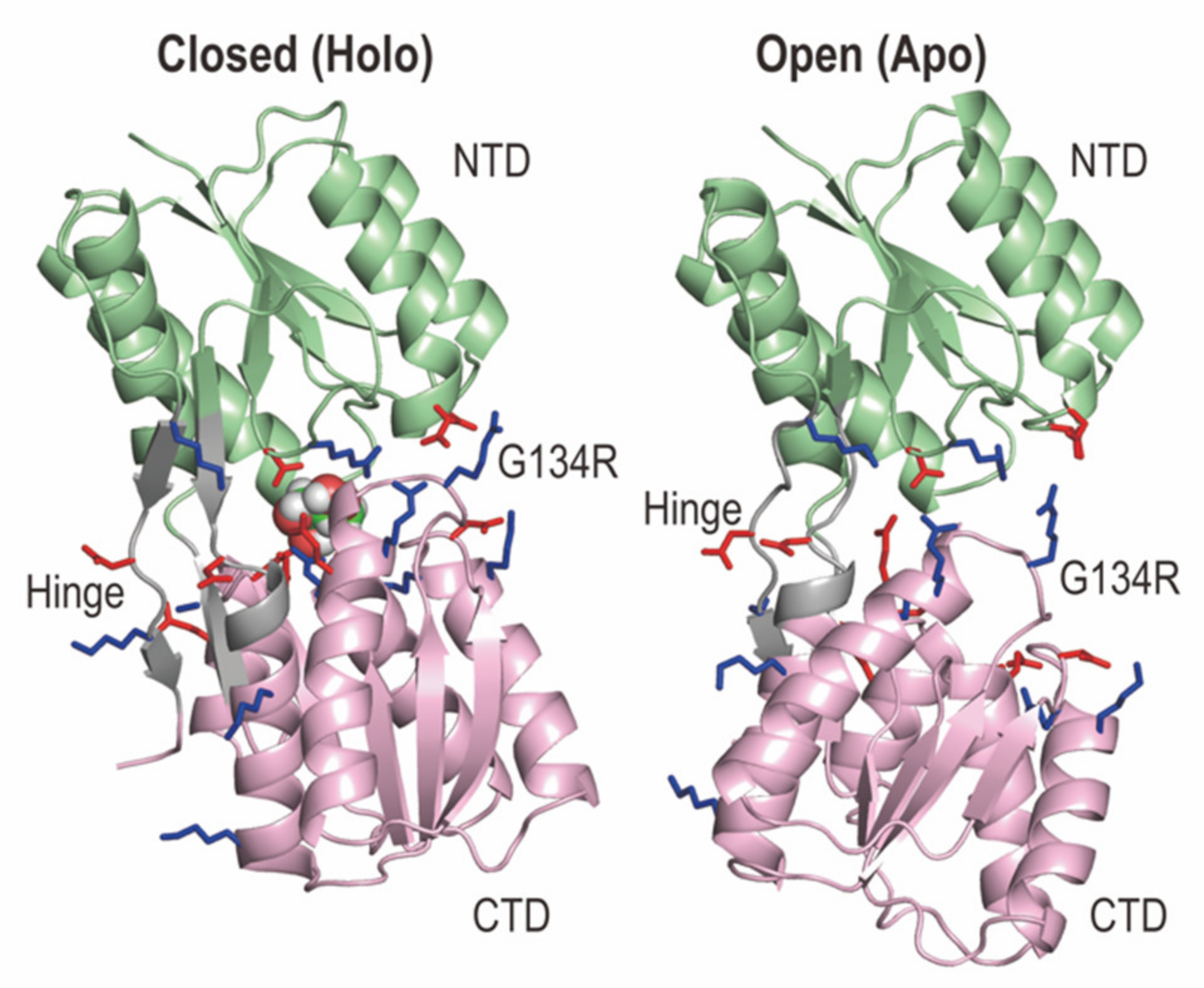
2.1. Structures of RBPG134R in the Apo and Holo States
2.2. gREST_SSCR Simulations of RBPG134R in the Apo and Holo States
2.2.1. How gREST_SSCR Works in RBPG134R Simulations
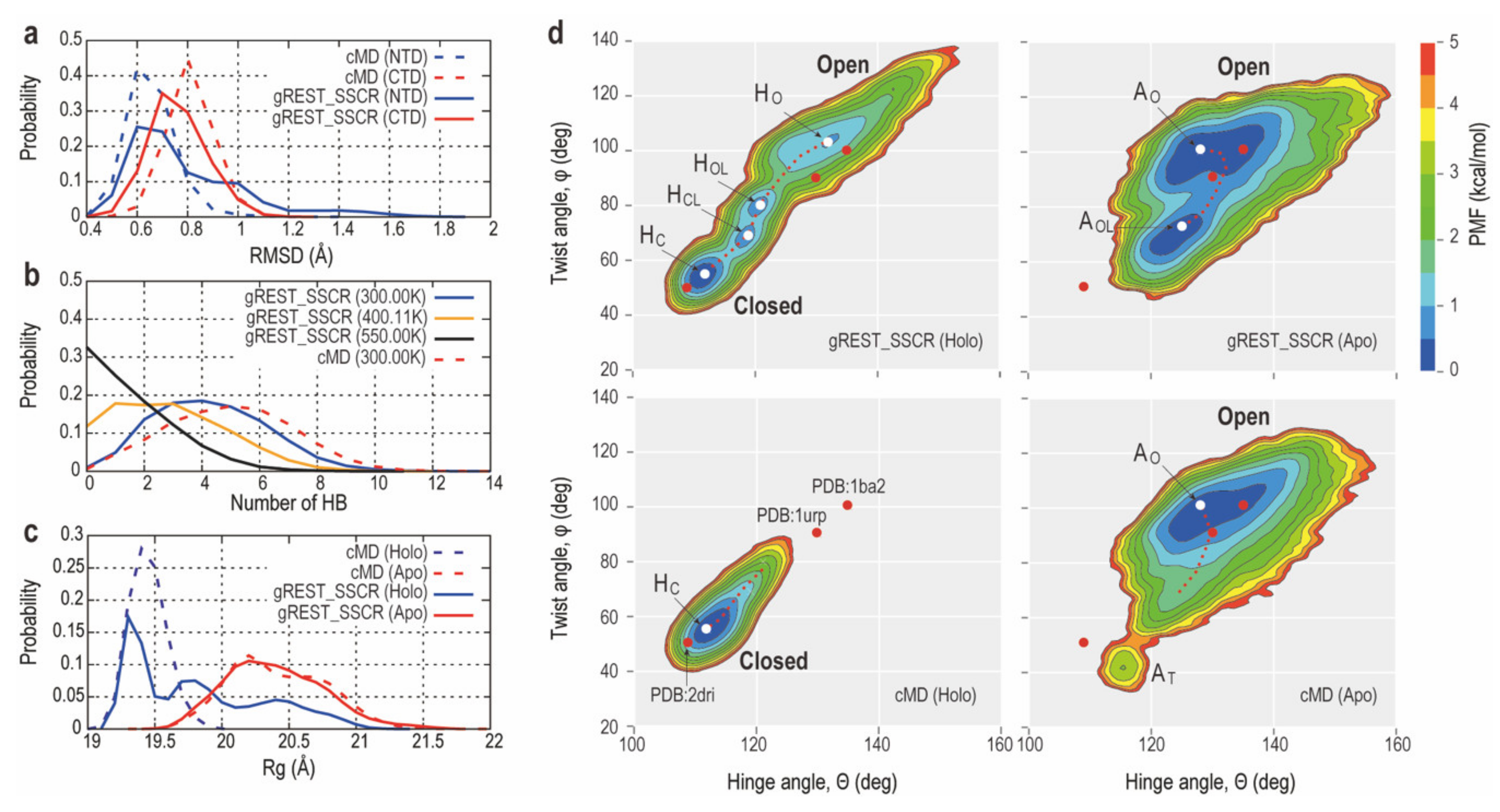
2.2.2. Comparison of Conformational Sampling Abilities between cMD and gREST_SSCR
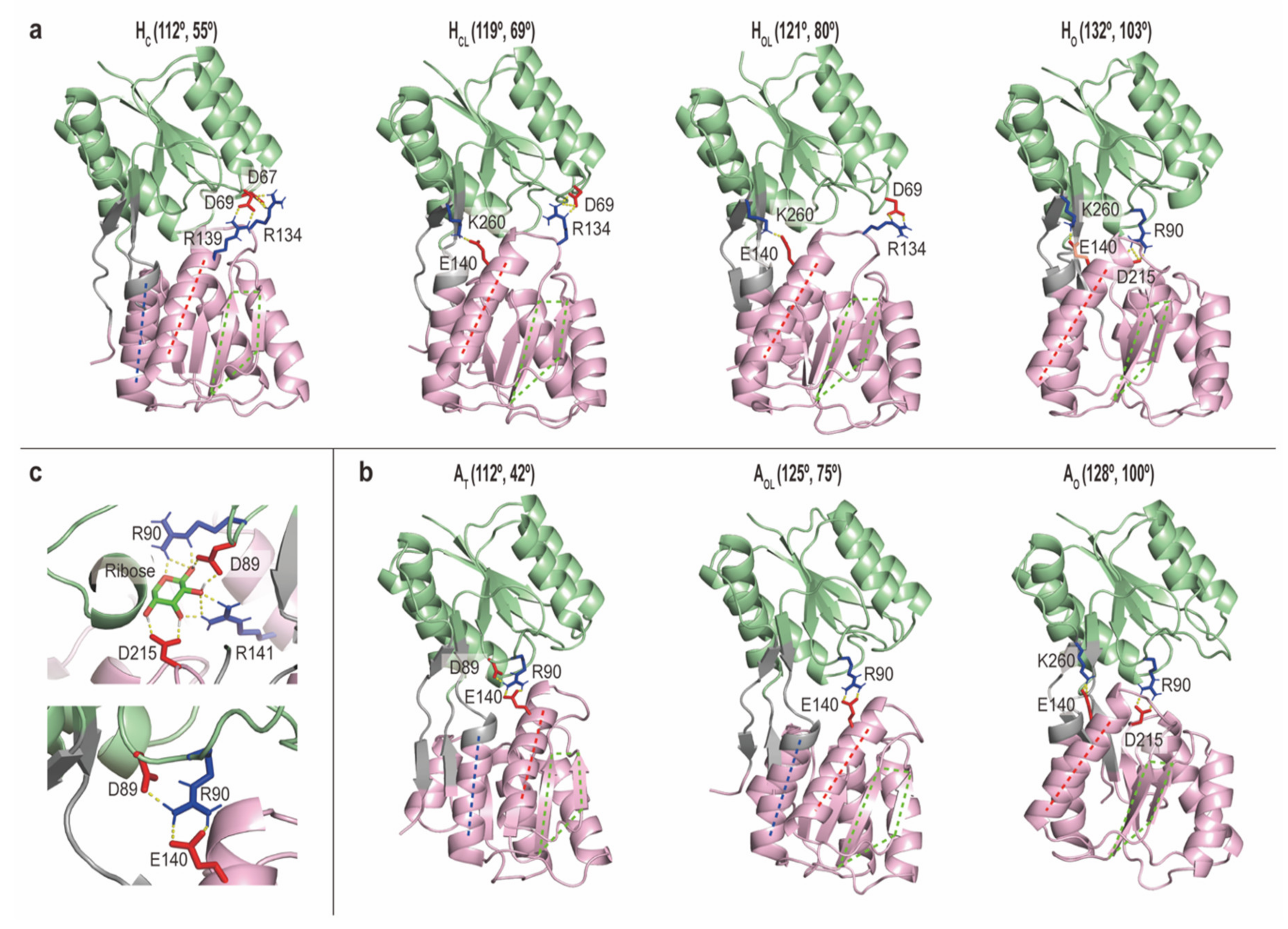
2.2.3. Intermediate Structures of RBPG134R Stabilized by the Inter-Domain Salt-Bridge Interactions
3. Discussion
3.1. How gREST_SSCR Can Enhance Conformational Sampling of Large-Scale Domain Motions of Proteins
3.2. Molecular Mechanisms Underlying Ligand-Induced Conformational Changes of RBP
3.3. General Applications of gREST and gREST_SSCR
4. Materials and Methods
4.1. Modeling of RBPG134R for MD Simulations
4.2. cMD Simulations
4.3. gREST_SSCR Simulations
4.4. Simulation Trajectory Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular dynamics |
| gREST | Generalized replica exchange with solute tempering |
| gREST_SSCR | gREST selected surface charged residues |
| RBP | Ribose binding protein |
| NTD | N-terminal domain |
| CTD | C-terminal domain |
| Rg | Radius of gyration |
| RMSD | Root mean square deviation |
References
- Gerstein, M.; Lesk, A.M.; Chothia, C. Structural Mechanisms for Domain Movements in Proteins. Biochemistry 1994, 33, 6739–6749. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.A.; Duderstadt, K.; Watkins, L.P.; Bhattacharyya, S.; Brokaw, J.; Chu, J.-W.; Yang, H. Illuminating the mechanistic roles of enzyme conformational dynamics. Proc. Natl. Acad. Sci. USA 2007, 104, 18055–18060. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.; Zuiderweg, E.R. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 2003, 13, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Bashton, M.; Kerrison, N.D.; Chothia, C.; Teichmann, S.A. Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol. 2004, 14, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, S.L.; Björkman, A. Conformational changes of ribose-binding protein and two related repressors are tailored to fit the functional need. J. Mol. Biol. 1999, 294, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Vishwanath, S.; De Brevern, A.G.; Srinivasan, N. Same but not alike: Structure, flexibility and energetics of domains in multi-domain proteins are influenced by the presence of other domains. PLoS Comput. Biol. 2018, 14, e1006008. [Google Scholar] [CrossRef] [PubMed]
- Michielssens, S.; De Groot, B.L.; Grubmüller, H. Binding Affinities Controlled by Shifting Conformational Equilibria: Opportunities and Limitations. Biophys. J. 2015, 108, 2585–2590. [Google Scholar] [CrossRef]
- Tiefenbrunn, T.; Forli, S.; Baksh, M.M.; Chang, M.W.; Happer, M.; Lin, Y.-C.; Perryman, A.L.; Rhee, J.-K.; Torbett, B.E.; Olson, A.J.; et al. Small Molecule Regulation of Protein Conformation by Binding in the Flap of HIV Protease. ACS Chem. Biol. 2013, 8, 1223–1231. [Google Scholar] [CrossRef]
- Millet, O.; Hudson, R.P.; Kay, L.E. The energetic cost of domain reorientation in maltose-binding protein as studied by NMR and fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 12700–12705. [Google Scholar] [CrossRef]
- Kooshapur, H.; Ma, J.; Tjandra, N.; Bermejo, G.A. Nmr analysis of apo glutamine-binding protein exposes challenges in the study of interdomain dynamics. Angew. Chem. Int. Ed. Engl. 2019, 58, 16899–16902. [Google Scholar] [CrossRef]
- Tang, C.; Schwieters, C.D.; Clore, G.M. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature 2007, 449, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Han, W.; Liu, C.; Zang, Y.; Li, J.; Wang, F.; Wang, Y.; Cong, Y. An ensemble of cryo-EM structures of TRiC reveal its conformational landscape and subunit specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 19513–19522. [Google Scholar] [CrossRef] [PubMed]
- Gershenson, A.; Gosavi, S.; Faccioli, P.; Wintrode, P.L. Successes and challenges in simulating the folding of large proteins. J. Biol. Chem. 2020, 295, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Orellana, L. Large-Scale Conformational Changes and Protein Function: Breaking the in silico Barrier. Front. Mol. Biosci. 2019, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Barz, B.; Loschwitz, J.; Strodel, B. Large-scale, dynamin-like motions of the human guanylate binding protein 1 revealed by multi-resolution simulations. PLoS Comput. Biol. 2019, 15, e1007193. [Google Scholar] [CrossRef]
- Sacquin-Mora, S. Motions and mechanics: Investigating conformational transitions in multi-domain proteins with coarse-grain simulations. Mol. Simul. 2013, 40, 229–236. [Google Scholar] [CrossRef]
- Roy, A.; Hua, D.P.; Post, C.B. Analysis of Multidomain Protein Dynamics. J. Chem. Theory Comput. 2016, 12, 274–280. [Google Scholar] [CrossRef]
- Wriggers, W.; Schulten, K. Protein domain movements: Detection of rigid domains and visualization of hinges in comparisons of atomic coordinates. Proteins 1997, 29, 1–14. [Google Scholar] [CrossRef]
- Hayward, S.; Kitao, A.; Berendsen, H.J.C. Model-free methods of analyzing domain motions in proteins from simulation: A comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins Struct. Funct. Bioinform. 1997, 27, 425–437. [Google Scholar] [CrossRef]
- Poornam, G.P.; Matsumoto, A.; Ishida, H.; Hayward, S. A method for the analysis of domain movements in large biomolecular complexes. Proteins Struct. Funct. Bioinform. 2009, 76, 201–212. [Google Scholar] [CrossRef]
- Veevers, R.; Hayward, S. Methodological improvements for the analysis of domain movements in large biomolecular complexes. Biophys. Phys. 2018, 16, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Lee, R.; Hayward, S. A comprehensive and non-redundant database of protein domain movements. Bioinformatics 2005, 21, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Cawley, G.C.; Hayward, S. Classification of Domain Movements in Proteins Using Dynamic Contact Graphs. PLoS ONE 2013, 8, e81224. [Google Scholar] [CrossRef] [PubMed]
- Koike, R.; Ota, M.; Kidera, A. Hierarchical description and extensive classification of protein structural changes by motion tree. J. Mol. Biol. 2014, 426, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Moritsugu, K.; Koike, R.; Yamada, K.; Kato, H.; Kidera, A. Motion Tree Delineates Hierarchical Structure of Protein Dynamics Observed in Molecular Dynamics Simulation. PLoS ONE 2015, 10, e0131583. [Google Scholar] [CrossRef] [PubMed]
- Shinobu, A.; Kobayashi, C.; Matsunaga, Y.; Sugita, Y. Building a Macro-mixing Dual-basin Go Model using the Multistate Bennett Acceptance Ratio. Biophys. J. 2019, 16, 310–321. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Sugita, Y.; Kitao, A.; Okamoto, Y. Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys. 2000, 113, 6042–6051. [Google Scholar] [CrossRef]
- Fukunishi, H.; Watanabe, O.; Takada, S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: Application to protein structure prediction. J. Chem. Phys. 2002, 116, 9058–9067. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; McCammon, J.A. Chapter six—Gaussian accelerated molecular dynamics: Theory, implementation, and applications. In Annual Reports in Computational Chemistry; Dixon, D.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 13, pp. 231–278. [Google Scholar]
- Camilloni, C.; Provasi, D.; Tiana, G.; Broglia, R.A. Exploring the protein G helix free-energy surface by solute tempering metadynamics. Proteins Struct. Funct. Bioinform. 2008, 71, 1647–1654. [Google Scholar] [CrossRef]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica exchange with solute scaling: A more efficient version of replica exchange with solute tempering (rest2). J. Phys. Chem. B 2011, 115, 9431–9438. [Google Scholar] [CrossRef]
- Moors, S.L.C.; Michielssens, S.; Ceulemans, A. Improved Replica Exchange Method for Native-State Protein Sampling. J. Chem. Theory Comput. 2011, 7, 231–237. [Google Scholar] [CrossRef]
- Terakawa, T.; Kameda, T.; Takada, S. On easy implementation of a variant of the replica exchange with solute tempering in GROMACS. J. Comput. Chem. 2011, 32, 1228–1234. [Google Scholar] [CrossRef]
- Kamiya, M.; Sugita, Y. Flexible selection of the solute region in replica exchange with solute tempering: Application to protein-folding simulations. J. Chem. Phys. 2018, 149, 072304. [Google Scholar] [CrossRef]
- Re, S.; Oshima, H.; Kasahara, K.; Kamiya, M.; Sugita, Y. Encounter complexes and hidden poses of kinase-inhibitor binding on the free-energy landscape. Proc. Natl. Acad. Sci. USA 2019, 116, 18404–18409. [Google Scholar] [CrossRef]
- Niitsu, A.; Re, S.; Oshima, H.; Kamiya, M.; Sugita, Y. De Novo Prediction of Binders and Nonbinders for T4 Lysozyme by gREST Simulations. J. Chem. Inf. Model. 2019, 59, 3879–3888. [Google Scholar] [CrossRef]
- Matsuoka, D.; Kamiya, M.; Sato, T.; Sugita, Y. Role of the N-Terminal Transmembrane Helix Contacts in the Activation of FGFR3. J. Comput. Chem. 2020, 41, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Tam, R.; Saier, M.H. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 1993, 57, 320–346. [Google Scholar] [CrossRef] [PubMed]
- Björkman, A.J.; Binnie, R.A.; Zhang, H.; Cole, L.B.; Hermodson, M.A.; Mowbray, S.L. Probing protein-protein interactions. The ribose-binding protein in bacterial transport and chemotaxis. J. Biol. Chem. 1994, 269, 30206–30211. [Google Scholar] [PubMed]
- Unione, L.; Ortega, G.; Mallagaray, A.; Corzana, F.; Pérez-Castells, J.; Canales, A.; Jimenez-Barbero, J.; Millet, O. Unraveling the Conformational Landscape of Ligand Binding to Glucose/Galactose-Binding Protein by Paramagnetic NMR and MD Simulations. ACS Chem. Biol. 2016, 11, 2149–2157. [Google Scholar] [CrossRef]
- Loeffler, H.H.; Kitao, A. Collective Dynamics of Periplasmic Glutamine Binding Protein upon Domain Closure. Biophys. J. 2009, 97, 2541–2549. [Google Scholar] [CrossRef]
- Björkman, A.; Mowbray, S.L. Multiple open forms of ribose-binding protein trace the path of its conformational change. J. Mol. Biol. 1998, 279, 651–664. [Google Scholar] [CrossRef]
- Cuneo, M.J.; Beese, L.S.; Hellinga, H.W. Ligand-induced conformational changes in a thermophilic ribose-binding protein. BMC Struct. Biol. 2008, 8, 50. [Google Scholar] [CrossRef]
- Ravindranathan, K.P.; Gallicchio, E.; Levy, R.M. Conformational Equilibria and Free Energy Profiles for the Allosteric Transition of the Ribose-binding Protein. J. Mol. Biol. 2005, 353, 196–210. [Google Scholar] [CrossRef]
- Borrok, M.J.; Kiessling, L.L.; Forest, K.T. Conformational changes of glucose/galactose-binding protein illuminated by open, unliganded, and ultra-high-resolution ligand-bound structures. Protein Sci. 2007, 16, 1032–1041. [Google Scholar] [CrossRef]
- Sun, Y.-J.; Rose, J.; Wang, B.-C.; Hsiao, C.-D. The structure of glutamine-binding protein complexed with glutamine at 1.94 Å resolution: Comparisons with other amino acid binding proteins. J. Mol. Biol. 1998, 278, 219–229. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, L.; Wu, S.; Liu, Z.; Gao, X.; Zhang, X.; Liu, M.; Liu, J.; Huang, X.; Wang, W. Conformational dynamics of apo-glnbp revealed by experimental and computational analysis. Angew. Chem. Int. Ed. Engl. 2016, 55, 13990–13994. [Google Scholar] [CrossRef] [PubMed]
- Bucher, D.; Grant, B.J.; McCammon, J.A. Induced Fit or Conformational Selection? The Role of the Semi-closed State in the Maltose Binding Protein. Biochemistry 2011, 50, 10530–10539. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. Charmm-gui input generator for namd, gromacs, amber, openmm, and charmm/openmm simulations using the charmm36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Mori, T.; Kobayashi, C.; Matsunaga, Y.; Yoda, T.; Feig, M.; Sugita, Y. GENESIS: A hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.; Jung, J.; Matsunaga, Y.; Mori, T.; Ando, T.; Tamura, K.; Kamiya, M.; Sugita, Y. GENESIS 1.1: A hybrid-parallel molecular dynamics simulator with enhanced sampling algorithms on multiple computational platforms. J. Comput. Chem. 2017, 38, 2193–2206. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, S.R.B.L.D.G.H.; MacKerell, J.A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W., 2nd; Mackerell, A.D., Jr. Charmm additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Berendsen, H.J.C. A Leap-frog Algorithm for Stochastic Dynamics. Mol. Simul. 2007, 1, 173–185. [Google Scholar] [CrossRef]
- Quigley, D.; Probert, M.I.J. Langevin dynamics in constant pressure extended systems. J. Chem. Phys. 2004, 120, 11432–11441. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kobayashi, C.; Sugita, Y. Kinetic energy definition in velocity Verlet integration for accurate pressure evaluation. J. Chem. Phys. 2018, 148, 164109. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 1.4; Schrödinger, LLC: New York, NY, USA, 2011.




{kind=link}
{kind=link}
{kind=link}
| Method (State) | gREST_SSCR (Holo) | gREST_SSCR (Apo) | cMD (Holo) | cMD (Apo) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Residue (domain) | Residue (domain) | HC | HCL | HOL | HO | AOL | AO | All | HC | All | AT |
| Asp67 * (NTD) | Arg134 * (CTD) | 82.8 ± 1.4 | 5.0 ± 0.6 | 0 | 0 | 0 | 0 | 47.7 ± 3.8 | 79.8 ± 1.1 | <0.1 | 0 |
| Asp69 * (NTD) | Arg134 * (CTD) | 19.4 ± 1.3 | 78.0 ± 2.4 | 30.2 ± 4.5 | <0.4 | <0.1 | <0.1 | 19.0 ± 1.8 | 16.0 ± 0.8 | <0.1 | 0 |
| Asp69 (NTD) | Arg139 (CTD) | 23.5 ± 1.0 | <0.5 | 0 | 0 | 0.1 | 0 | 18.0 ± 2.5 | 39.7 ± 2.4 | 0 | <0.1 |
| Arg90 (NTD) | Glu140 (CTD) | 0 | 0 | 0 | 0 | 86.8 ± 0.9 | <1.1 | 11.8 ± 3.0 | 0 | 1.8 ± 0.3 | 74.5 |
| Arg90 (NTD) | Asp215 (CTD) | 0 | 0 | <0.5 | 34.8 ± 4.2 | 0 | 37.2 ± 3.9 | 0 | 0 | 30.9 ± 2.3 | 0 |
| Glu140 (CTD) | Lys260 (Hinge) | 3.9 ± 0.3 | 44.5 ± 3.8 | 80.4 ± 1.2 | 79.2 ± 1.4 | <0.7 | 59.0 ± 3.5 | 6.7 ± 1.3 | 3.6 ± 0.1 | 62.4 ± 1.1 | 0 |
| Glu221 (CTD) | Lys266 (Hinge) | 24.0 ± 1.2 | 26.9 ± 1.1 | <0.2 | 0 | <0.6 | <0.3 | 19.4 ± 1.8 | 31.2 ± 0.8 | <0.3 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dokainish, H.M.; Sugita, Y. Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling. Int. J. Mol. Sci. 2021, 22, 270. https://doi.org/10.3390/ijms22010270
Dokainish HM, Sugita Y. Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling. International Journal of Molecular Sciences. 2021; 22(1):270. https://doi.org/10.3390/ijms22010270
Chicago/Turabian StyleDokainish, Hisham M., and Yuji Sugita. 2021. "Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling" International Journal of Molecular Sciences 22, no. 1: 270. https://doi.org/10.3390/ijms22010270
APA StyleDokainish, H. M., & Sugita, Y. (2021). Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling. International Journal of Molecular Sciences, 22(1), 270. https://doi.org/10.3390/ijms22010270