The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
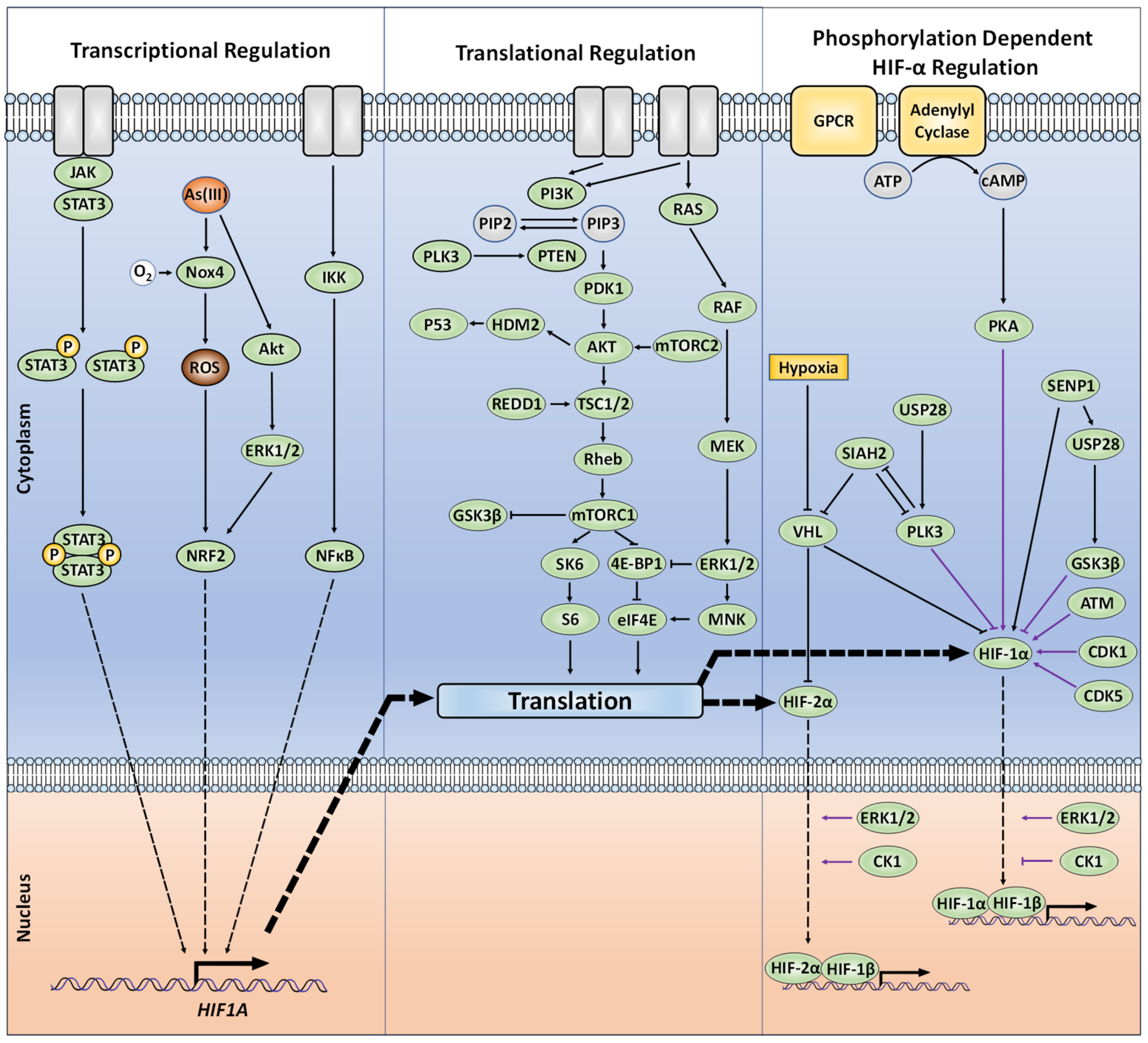
2. Canonical Regulation of HIF-α, the Role of Ubiquitination
3. Non-Canonical PTMs Regulating HIF-α Subunits
4. Phosphorylation
4.1. Phosphorylation by GSK-3β
4.2. Phosphorylation by PLK3
4.3. Phosphorylation by Cyclin-Dependent Kinases (CDKs)
4.4. Phosphorylation by Protein Kinase A (PKA)
4.5. Phosphorylation by pATM (Ataxia Telangiectasia Mutated protein)
4.6. HIF-1α vs HIF-2α Phosphorylation, Similarities, and Differences
4.7. Beyond Direct HIF Protein Phosphorylation; Indirect Kinase-Dependent HIF-α Regulation
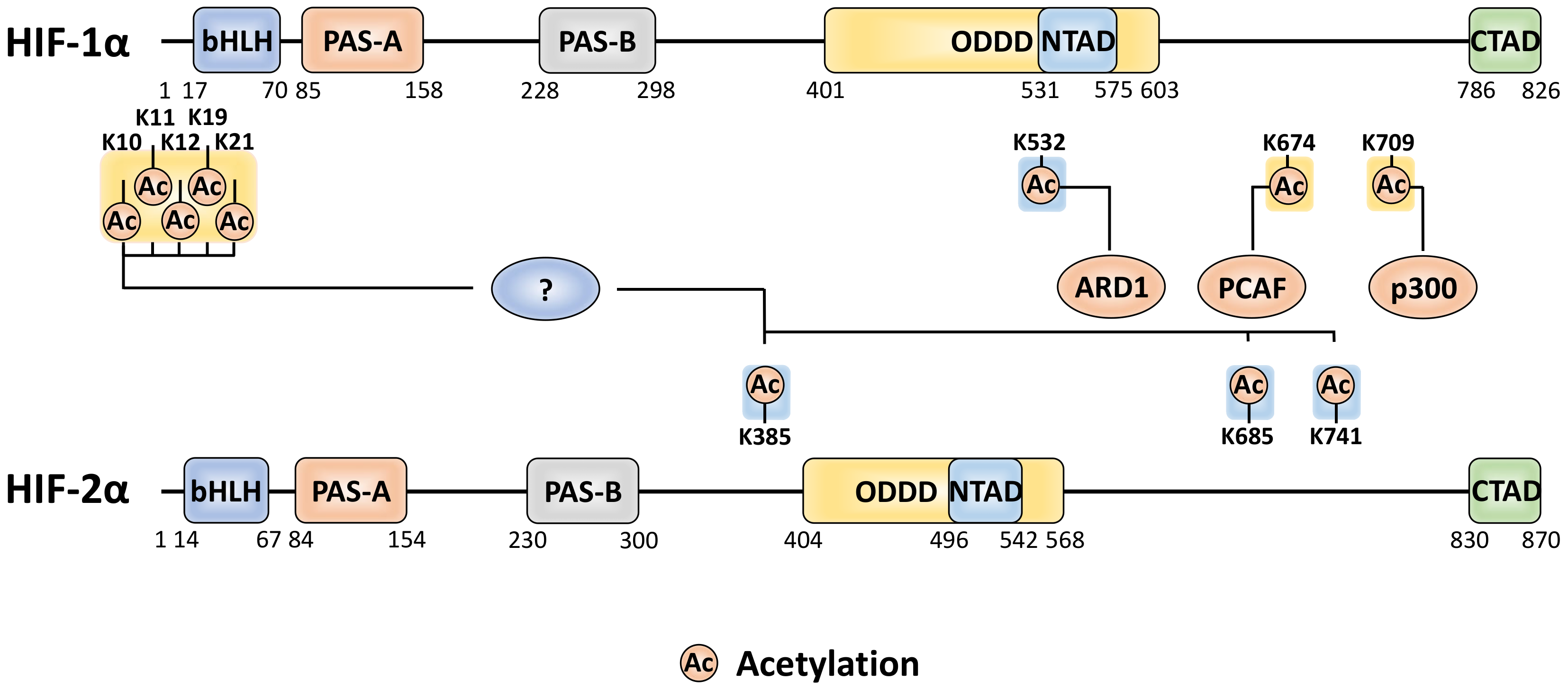
5. Acetylation
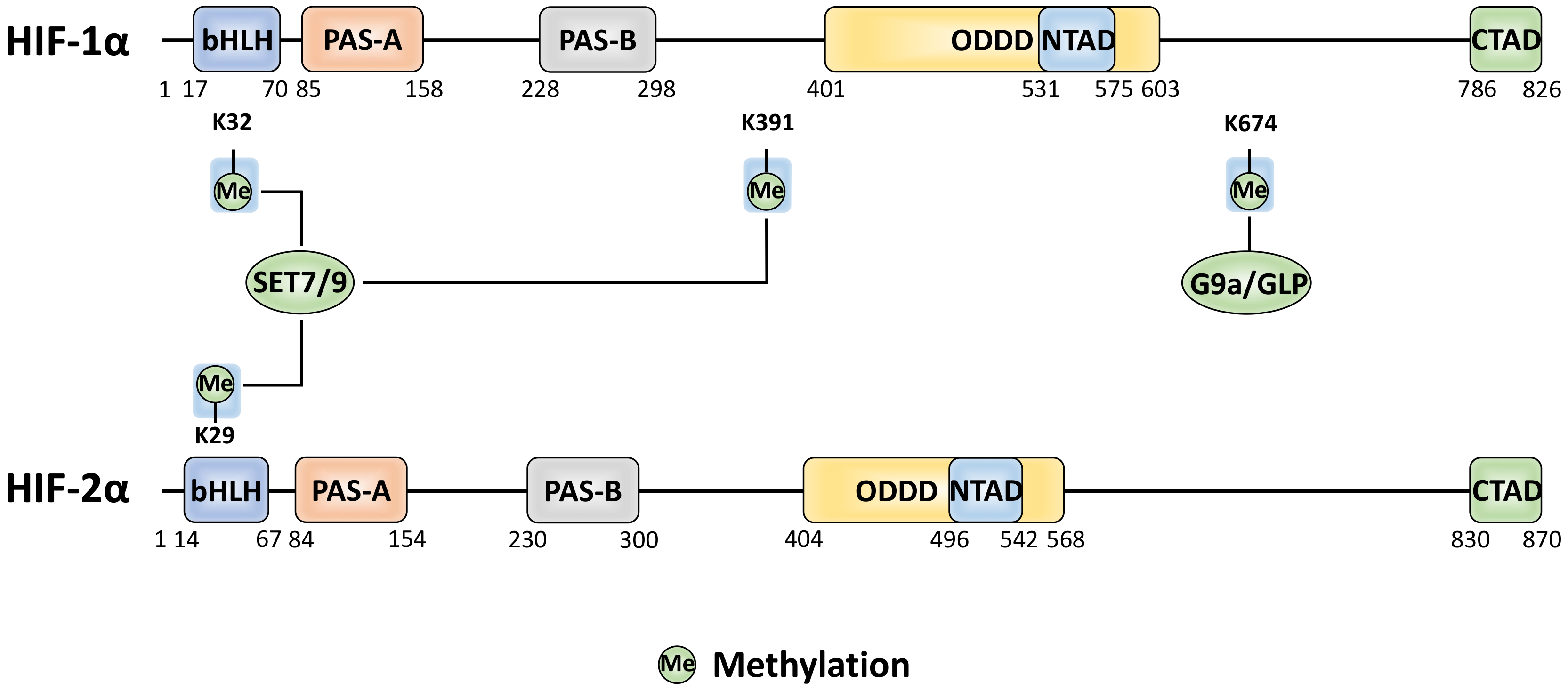
6. Methylation
7. SUMOylation
8. S-Nitrosylation
9. Uncharacterised PTMs
9.1. Glycosylation
9.2. S-Glutathionylation
9.3. Neddylation
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Taylor, S.E.; Bagnall, J.; Mason, D.; Levy, R.; Fernig, D.G.; See, V. Differential sub-nuclear distribution of hypoxia-inducible factors (HIF)-1 and -2 alpha impacts on their stability and mobility. Open Biol. 2016, 6. [Google Scholar] [CrossRef]
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA-binding specificities of the HIF-1alpha and HIF-2alpha transcription factors in chromatin. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Menrad, H.; Werno, C.; Schmid, T.; Copanaki, E.; Deller, T.; Dehne, N.; Brune, B. Roles of hypoxia-inducible factor-1alpha (HIF-1alpha) versus HIF-2alpha in the survival of hepatocellular tumor spheroids. Hepatology 2010, 51, 2183–2192. [Google Scholar] [CrossRef] [PubMed]
- Lofstedt, T.; Fredlund, E.; Holmquist-Mengelbier, L.; Pietras, A.; Ovenberger, M.; Poellinger, L.; Pahlman, S. Hypoxia inducible factor-2alpha in cancer. Cell Cycle 2007, 6, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3 alpha locus. J. Biol. Chem. 2002, 277, 32405–32408. [Google Scholar] [CrossRef]
- Pasanen, A.; Heikkila, M.; Rautavuoma, K.; Hirsila, M.; Kivirikko, K.I.; Myllyharju, J. Hypoxia-inducible factor (HIF)-3a is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. Int. J. Biochem. Cell Biol. 2010, 42, 1189–1200. [Google Scholar] [CrossRef]
- Heikkila, M.; Pasanen, A.; Kivirikko, K.I.; Myllyharju, J. Roles of the human hypoxia-inducible factor (HIF)-3 alpha variants in the hypoxia response. Cell. Mol. Life Sci. 2011, 68, 3885–3901. [Google Scholar] [CrossRef]
- Makino, Y.; Uenishi, R.; Okamoto, K.; Isoe, T.; Hosono, O.; Tanaka, H.; Kanopka, A.; Poellinger, L.; Haneda, M.; Morimoto, C. Transcriptional up-regulation of inhibitory PAS domain protein gene expression by hypoxia-inducible factor 1 (HIF-1)—A negative feedback regulatory circuit in HIF-1-mediated signaling in hypoxic cells. J. Biol. Chem. 2007, 282, 14073–14082. [Google Scholar] [CrossRef]
- Tolonen, J.P.; Heikkila, M.; Malinen, M.; Lee, H.M.; Palvimo, J.J.; Wei, G.H.; Myllyharju, J. A long hypoxia-inducible factor 3 isoform 2 is a transcription activator that regulates erythropoietin. Cell. Mol. Life Sci. 2020, 77, 3627–3642. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Cossart, P. Ubiquitin, SUMO, and NEDD8: Key Targets of Bacterial Pathogens. Trends Cell. Biol. 2018, 28, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A. Regulation of HIF-1alpha at the transcriptional level. Curr. Pharm. Des. 2009, 15, 3844–3852. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [PubMed]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef]
- Maynard, M.A.; Qi, H.; Chung, J.; Lee, E.H.L.; Kondo, Y.; Hara, S.; Conaway, R.C.; Conaway, J.W.; Ohh, M. Multiple splice variants of the human HIF-3 alpha locus are targets of the von Hippel-Lindau E3 uhiquitin ligase complex. J. Biol. Chem. 2003, 278, 11032–11040. [Google Scholar] [CrossRef]
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1 alpha in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef]
- Bagnall, J.; Leedale, J.; Taylor, S.E.; Spiller, D.G.; White, M.R.; Sharkey, K.J.; Bearon, R.N.; See, V. Tight control of hypoxia-inducible factor-alpha transient dynamics is essential for cell survival in hypoxia. J. Biol. Chem. 2014, 289, 5549–5564. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell. Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Paltoglou, S.; Roberts, B.J. HIF-1alpha and EPAS ubiquitination mediated by the VHL tumour suppressor involves flexibility in the ubiquitination mechanism, similar to other RING E3 ligases. Oncogene 2007, 26, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef]
- Stiehl, D.P.; Wirthner, R.; Koditz, J.; Spielmann, P.; Camenisch, G.; Wenger, R.H. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. J. Biol. Chem. 2006, 281, 23482–23491. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef]
- Rodriguez, J.; Haydinger, C.H.D.; Peet, D.J.; Nguyen, L.; von Kriegsheim, A. Asparagine hydroxylation is a reversible post-translational modification. Mol. Cell. Proteom. 2020. [Google Scholar] [CrossRef]
- Mennerich, D.; Kubaichuk, K.; Kietzmann, T. DUBs, hypoxia, and cancer. Trends Cancer 2019, 5, 632–653. [Google Scholar] [CrossRef]
- Schober, A.S.; Berra, E. DUBs, new members in the hypoxia signaling club. Front Oncol. 2016, 6, 53. [Google Scholar] [CrossRef]
- Semenza, G.L. A compendium of proteins that interact with HIF-1 alpha. Exp. Cell Res. 2017, 356, 128–135. [Google Scholar] [CrossRef]
- Hardman, G.; Perkins, S.; Brownridge, P.J.; Clarke, C.J.; Byrne, D.P.; Campbell, A.E.; Kalyuzhnyy, A.; Myall, A.; Eyers, P.A.; Jones, A.R.; et al. Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO J. 2019, 38, e100847. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R. Molecular-cloning and expression of glycogen-synthase kinase-3 factor-A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.; Ramakrishna, S.; Benjamin, W.B.; Woodgett, J.R. Identification of multifunctional atp-citrate lyase kinase as the alpha-isoform of glycogen-synthase kinase-3. Biochem. J. 1992, 288, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Force, T.; Woodgett, J.R. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J. Biol. Chem. 2009, 284, 9643–9647. [Google Scholar] [CrossRef]
- Flugel, D.; Gorlach, A.; Michiels, C.; Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol. Cell. Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef]
- Mottet, D.; Dumont, V.; Deccache, Y.; Demazy, C.; Ninane, N.; Raes, M.; Michiels, C. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J. Biol. Chem. 2003, 278, 31277–31285. [Google Scholar] [CrossRef]
- Cassavaugh, J.M.; Hale, S.A.; Wellman, T.L.; Howe, A.K.; Wong, C.; Lounsbury, K.M. Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J. Cell. Biochem. 2011, 112, 3882–3890. [Google Scholar] [CrossRef]
- Flugel, D.; Gorlach, A.; Kietzmann, T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood 2012, 119, 1292–1301. [Google Scholar] [CrossRef]
- Du, S.C.; Zhu, L.; Wang, Y.X.; Liu, J.; Zhang, D.; Chen, Y.L.; Peng, Q.; Liu, W.; Liu, B. SENP1-mediated deSUMOylation of USP28 regulated HIF-1alpha accumulation and activation during hypoxia response. Cancer Cell Int. 2019, 19, 4. [Google Scholar] [CrossRef]
- Xu, D.; Yao, Y.; Lu, L.; Costa, M.; Dai, W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1alpha. J. Biol. Chem. 2010, 285, 38944–38950. [Google Scholar] [CrossRef]
- Xu, D.; Yao, Y.; Jiang, X.; Lu, L.; Dai, W. Regulation of PTEN stability and activity by Plk3. J. Biol. Chem. 2010, 285, 39935–39942. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, K.S.; Sunderman, F.W., Jr.; Salnikow, K. Nickel carcinogenesis. Mutat. Res. 2003, 533, 67–97. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Park, S.; Zhang, X.; Dai, W.; Xu, D. Mutual regulation between Polo-like kinase 3 and SIAH2 E3 ubiquitin ligase defines a regulatory network that fine-tunes the cellular response to hypoxia and nickel. J. Biol. Chem. 2017, 292, 11431–11444. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Qi, J.; Ronai, Z. The ubiquitin ligase Siah2 and the hypoxia response. Mol. Cancer Res. 2009, 7, 443–451. [Google Scholar] [CrossRef]
- Qi, J.; Kim, H.; Scortegagna, M.; Ronai, Z.A. Regulators and effectors of Siah ubiquitin ligases. Cell Biochem. Biophys. 2013, 67, 15–24. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nature Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Kshitiz; Gilkes, D.M.; Rey, S.; Wong, C.C.; Luo, W.; Kim, D.H.; Dang, C.V.; Levchenko, A.; Semenza, G.L. A nontranscriptional role for HIF-1alpha as a direct inhibitor of DNA replication. Sci. Signal. 2013, 6, ra10. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz; Ahmed, I.; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1alpha to promote cell-cycle progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef]
- Warfel, N.A.; Dolloff, N.G.; Dicker, D.T.; Malysz, J.; El-Deiry, W.S. CDK1 stabilizes HIF-1alpha via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle 2013, 12, 3689–3701. [Google Scholar] [CrossRef]
- Li, Q.; Kluz, T.; Sun, H.; Costa, M. Mechanisms of c-myc degradation by nickel compounds and hypoxia. PLoS ONE 2009, 4, e8531. [Google Scholar] [CrossRef]
- Herzog, J.; Ehrlich, S.M.; Pfitzer, L.; Liebl, J.; Frohlich, T.; Arnold, G.J.; Mikulits, W.; Haider, C.; Vollmar, A.M.; Zahler, S. Cyclin-dependent kinase 5 stabilizes hypoxia-inducible factor-1alpha: A novel approach for inhibiting angiogenesis in hepatocellular carcinoma. Oncotarget 2016, 7, 27108–27121. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, S.; Feron, O.; Raes, M.; Michiels, C. Intermittent hypoxia changes HIF-1 alpha phosphorylation pattern in endothelial cells: Unravelling of a new PKA-dependent regulation of HIF-1 alpha. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Bullen, J.W.; Tchernyshyov, I.; Holewinski, R.J.; deVine, L.; Wu, F.; Venkatraman, V.; Kass, D.L.; Cole, R.N.; van Eyk, J.; Semenza, G.L. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci. Signal. 2016, 9, ra56. [Google Scholar] [CrossRef] [PubMed]
- Daly, L.A.; Brownridge, P.J.; See, V.; Eyers, C.E. Oxygen-dependent changes in HIF binding partners and post-translational modifications regulate stability and transcriptional activity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lucia, K.; Wu, Y.H.; Garcia, J.M.; Barlier, A.; Buchfelder, M.; Saeger, W.; Renner, U.; Stalla, G.K.; Theodoropoulou, M. Hypoxia and the hypoxia inducible factor 1 alpha activate protein kinase A by repressing RII beta subunit transcription. Oncogene 2020. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Wang, P.; Guan, D.; Zhang, X.P.; Liu, F.; Wang, W. Modeling the regulation of p53 activation by HIF-1 upon hypoxia. FEBS Lett. 2019, 593, 2596–2611. [Google Scholar] [CrossRef]
- Zhou, C.H.; Zhang, X.P.; Liu, F.; Wang, W. Modeling the interplay between the HIF-1 and p53 pathways in hypoxia. Sci. Rep. 2015, 5, 13834. [Google Scholar] [CrossRef]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1 alpha. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef]
- Ousset, M.; Bouquet, F.; Fallone, F.; Biard, D.; Dray, C.; Valet, P.; Salles, B.; Muller, C. Loss of ATM positively regulates the expression of hypoxia inducible factor 1 (HIF-1) through oxidative stress. Cell Cycle 2010, 9, 2814–2822. [Google Scholar] [CrossRef]
- Gradin, K.; Takasaki, C.; Fujii-Kuriyama, Y.; Sogawa, K. The transcriptional activation function of the HIF-like factor requires phosphorylation at a conserved threonine. J. Biol. Chem. 2002, 277, 23508–23514. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, D.E.; McNeill, L.A.; McDonough, M.A.; Aplin, R.T.; Hewitson, K.S.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem. J. 2004, 383, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Ahn, D.R.; Park, H.; Yang, E.G. Modulation of p300 binding by posttranslational modifications of the C-terminal activation domain of hypoxia-inducible factor-1alpha. FEBS Lett. 2007, 581, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Hubert, A.; Paris, S.; Piret, J.P.; Ninane, N.; Raes, M.; Michiels, C. Casein kinase 2 inhibition decreases hypoxia-inducible factor-1 activity under hypoxia through elevated p53 protein level. J. Cell Sci. 2006, 119, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Mottet, D.; Ruys, S.P.D.; Demazy, C.; Raes, M.; Michiels, C. Role for casein kinase 2 in the regulation of HIF-1 activity. Int. J. Cancer 2005, 117, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Chachami, G.; Paraskeva, E.; Simos, G. Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1 alpha by MAPK. J. Biol. Chem. 2008, 283, 27620–27627. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1 alpha. J. Biol. Chem. 2006, 281, 33095–33106. [Google Scholar] [CrossRef]
- Han, H.J.; Kwon, N.; Choi, M.A.; Jung, K.O.; Piao, J.Y.; Ngo, H.K.C.; Kim, S.J.; Kim, D.H.; Chung, J.K.; Cha, Y.N.; et al. Peptidyl prolyl isomerase PIN1 directly binds to and stabilizes hypoxia-inducible factor-1 alpha. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Jalouli, M.; Dery, M.A.C.; Lafleur, V.N.; Lamalice, L.; Zhou, X.Z.; Lu, K.P.; Richard, D.E. The prolyl isomerase Pin1 regulates hypoxia-inducible transcription factor (HIF) activity. Cell. Signal. 2014, 26, 1649–1656. [Google Scholar] [CrossRef]
- Driver, J.A.; Zhou, X.Z.; Lu, K.P. Pin1 dysregulation helps to explain the inverse association between cancer and Alzheimer’s disease. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 2069–2076. [Google Scholar] [CrossRef]
- Lonati, E.; Brambilla, A.; Milani, C.; Masserini, M.; Palestini, P.; Bulbarelli, A. Pin1, a new player in the fate of HIF-1 alpha degradation: An hypothetical mechanism inside vascular damage as Alzheimer’s disease risk factor. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Kalousi, A.; Mylonis, I.; Politou, A.S.; Chachami, G.; Paraskeva, E.; Simos, G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 2010, 123, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Gkotinakou, I.M.; Befani, C.; Simos, G.; Liakos, P. ERK1/2 phosphorylates HIF-2 alpha and regulates its activity by controlling its CRM1-dependent nuclear shuttling. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Pangou, E.; Befani, C.; Mylonis, I.; Samiotaki, M.; Panayotou, G.; Simos, G.; Liakos, P. HIF-2alpha phosphorylation by CK1delta promotes erythropoietin secretion in liver cancer cells under hypoxia. J. Cell. Sci. 2016, 129, 4213–4226. [Google Scholar] [CrossRef]
- To, K.K.; Sedelnikova, O.A.; Samons, M.; Bonner, W.M.; Huang, L.E. The phosphorylation status of PAS-B distinguishes HIF-1alpha from HIF-2alpha in NBS1 repression. EMBO J. 2006, 25, 4784–4794. [Google Scholar] [CrossRef] [PubMed]
- BelAiba, R.S.; Bonello, S.; Zahringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Gorlach, A. Hypoxia up-regulates hypoxia-inducible factor-1 alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappa B in pulmonary artery smooth muscle cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-kappaB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef]
- Obacz, J.; Pastorekova, S.; Vojtesek, B.; Hrstka, R. Cross-talk between HIF and p53 as mediators of molecular responses to physiological and genotoxic stresses. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef]
- Taylor, C.T. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J. Physiol. 2008, 586, 4055–4059. [Google Scholar] [CrossRef]
- Vollmer, S.; Kappler, V.; Kaczor, J.; Flugel, D.; Rolvering, C.; Kato, N.; Kietzmann, T.; Behrmann, I.; Haan, C. Hypoxia-inducible factor 1 alpha is upregulated by oncostatin M and participates in oncostatin M signalling. Eur. J. Cell Biol. 2010, 89, 6–7. [Google Scholar]
- Al Taleb, Z.; Petry, A.; Chi, T.F.; Mennerich, D.; Gorlach, A.; Dimova, E.Y.; Kietzmann, T. Differential transcriptional regulation of hypoxia-inducible factor-1 alpha by arsenite under normoxia and hypoxia: Involvement of Nrf2. J. Mol. Med. 2016, 94, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Lacher, S.E.; Levings, D.C.; Freeman, S.; Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018, 19, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, A.I.; Paraskeva, E.; Peidis, P.; Muaddi, H.; Li, S.; Raptis, L.; Pantopoulos, K.; Simos, G.; Koromilas, A.E. eIF2{alpha} Kinase PKR modulates the hypoxic response by Stat3-dependent transcriptional suppression of HIF-1{alpha}. Cancer Res. 2010, 70, 7820–7829. [Google Scholar] [CrossRef] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1 alpha (HIF-1 alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Arnesen, T.; Kong, X.; Evjenth, R.; Gromyko, D.; Varhaug, J.E.; Lin, Z.; Sang, N.; Caro, J.; Lillehaug, J.R. Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS Lett. 2005, 579, 6428–6432. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Kang, J.; Chun, Y.S.; Huh, J.; Park, J.W. FIH permits NAA10 to catalyze the oxygen-dependent lysyl-acetylation of HIF-1alpha. Redox Biol. 2018, 19, 364–374. [Google Scholar] [CrossRef]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.S.; Qian, D.Z. HIF1alpha protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.S.; Park, J.H.; Heo, J.Y.; Jing, K.; Han, J.; Min, K.N.; Kim, C.; Koh, G.Y.; Lim, K.; Kang, G.Y.; et al. SIRT2 regulates tumour hypoxia response by promoting HIF-1alpha hydroxylation. Oncogene 2015, 34, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef] [PubMed]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef]
- Kim, Y.; Nam, H.J.; Lee, J.; Park, D.Y.; Kim, C.; Yu, Y.S.; Kim, D.; Park, S.W.; Bhin, J.; Hwang, D.; et al. Methylation-dependent regulation of HIF-1 alpha stability restricts retinal and tumour angiogenesis. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, J.H.; Choi, H.J.; Won, H.Y.; Joo, H.S.; Shin, D.H.; Park, M.K.; Han, B.; Kim, K.P.; Lee, T.J.; et al. LSD1 demethylates HIF1 alpha to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene 2017, 36, 5512–5521. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Z.; Xu, C.; Leng, X.; Cao, H.; Ouyang, G.; Xiao, W. Repression of hypoxia-inducible factor alpha signaling by Set7-mediated methylation. Nucleic Acids Res. 2015, 43, 5081–5098. [Google Scholar] [CrossRef]
- Bao, L.; Chen, Y.; Lai, H.T.; Wu, S.Y.; Wang, J.E.; Hatanpaa, K.J.; Raisanen, J.M.; Fontenot, M.; Lega, B.; Chiang, C.M.; et al. Methylation of hypoxia-inducible factor (HIF)-1alpha by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res. 2018, 46, 6576–6591. [Google Scholar] [CrossRef]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef]
- Bae, S.H.; Jeong, J.W.; Park, J.A.; Kim, S.H.; Bae, M.K.; Choi, S.J.; Kim, K.W. Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem. Biophys. Res. Commun. 2004, 324, 394–400. [Google Scholar] [CrossRef]
- Berta, M.A.; Mazure, N.; Hattab, M.; Pouyssegur, J.; Brahimi-Horn, M.C. SUMOylation of hypoxia-inducible factor-1alpha reduces its transcriptional activity. Biochem. Biophys. Res. Commun. 2007, 360, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Li, J.; Zou, Y.; Yi, J.; Zhang, H.; Cao, M.; Yeh, E.T.; Cheng, J. PIASy stimulates HIF1alpha SUMOylation and negatively regulates HIF1alpha activity in response to hypoxia. Oncogene 2010, 29, 5568–5578. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, Y.; Long, X.D.; Wang, W.; Jiao, H.K.; Mei, Z.; Yin, Q.Q.; Ma, L.N.; Zhou, A.W.; Wang, L.S.; et al. Cbx4 governs HIF-1alpha to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 2014, 25, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Van Hagen, M.; Overmeer, R.M.; Abolvardi, S.S.; Vertegaal, A.C. RNF4 and VHL regulate the proteasomal degradation of SUMO-conjugated hypoxia-inducible factor-2alpha. Nucleic Acids Res. 2010, 38, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Yasinska, I.M.; Sumbayev, V.V. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003, 549, 105–109. [Google Scholar] [CrossRef]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.L.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.Y. Regulation of HIF-1 alpha stability through S-nitrosylation. Molecular Cell 2007, 26, 63–74. [Google Scholar] [CrossRef]
- Koike, T.; Kimura, N.; Miyazaki, K.; Yabuta, T.; Kumamoto, K.; Takenoshita, S.; Chen, H.; Kobayashi, M.; Hosokawa, M.; Taniguchi, A.; et al. Hypoxia induces adhesion molecules on cancer cells: A missing link between Warburg effect and induction of selectin-ligand carbohydrates. Proc. Natl. Acad. Sci. USA 2004, 101, 10494. [Google Scholar] [CrossRef]
- Nonaka, M.; Fukuda, M.N.; Gao, C.; Li, Z.; Zhang, H.T.; Greene, M.I.; Peehl, D.M.; Ten, F.Z.; Fukuda, M. Determination of carbohydrate structure recognized by prostate-specific F77 monoclonal antibody through expression analysis of glycosyltransferase genes. J. Biol. Chem. 2014, 289, 16478–16486. [Google Scholar] [CrossRef]
- Jeon, D.; Park, H.J.; Kim, H.S. Protein S-glutathionylation induced by hypoxia increases hypoxia-inducible factor-1 alpha in human colon cancer cells. Biochem. Biophys. Res. Commun. 2018, 495, 212–216. [Google Scholar] [CrossRef]
- Tajima, M.; Kurashima, Y.; Sugiyama, K.; Ogura, T.; Sakagami, H. The redox state of glutathione regulates the hypoxic induction of HIF-1. Eur. J. Pharmacol. 2009, 606, 45–49. [Google Scholar] [CrossRef]
- Watanabe, Y.; Murdoch, C.E.; Sano, S.; Ido, Y.; Bachschmid, M.M.; Cohen, R.A.; Matsui, R. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1 alpha and improve limb revascularization. Proc. Natl. Acad. Sci. USA 2016, 113, 6011–6016. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Li, S.H.; Park, H.S.; Park, J.W.; Lee, B.; Chun, Y.S. Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J. Biol. Chem. 2011, 286, 6963–6970. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Foglia, B.; Villano, G.; Turato, C.; Delgado, T.C.; Morello, E.; Pin, F.; Novo, E.; Napione, L.; Quarta, S.; et al. SerpinB3 differently up-regulates hypoxia inducible factors-1 alpha and-2 alpha in hepatocellular carcinoma: Mechanisms revealing novel potential therapeutic targets. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albanese, A.; Daly, L.A.; Mennerich, D.; Kietzmann, T.; Sée, V. The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity. Int. J. Mol. Sci. 2021, 22, 268. https://doi.org/10.3390/ijms22010268
Albanese A, Daly LA, Mennerich D, Kietzmann T, Sée V. The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity. International Journal of Molecular Sciences. 2021; 22(1):268. https://doi.org/10.3390/ijms22010268
Chicago/Turabian StyleAlbanese, Adam, Leonard A. Daly, Daniela Mennerich, Thomas Kietzmann, and Violaine Sée. 2021. "The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity" International Journal of Molecular Sciences 22, no. 1: 268. https://doi.org/10.3390/ijms22010268
APA StyleAlbanese, A., Daly, L. A., Mennerich, D., Kietzmann, T., & Sée, V. (2021). The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity. International Journal of Molecular Sciences, 22(1), 268. https://doi.org/10.3390/ijms22010268