Discovery of Potential Inhibitors for RNA-Dependent RNA Polymerase of Norovirus: Virtual Screening, and Molecular Dynamics


Abstract
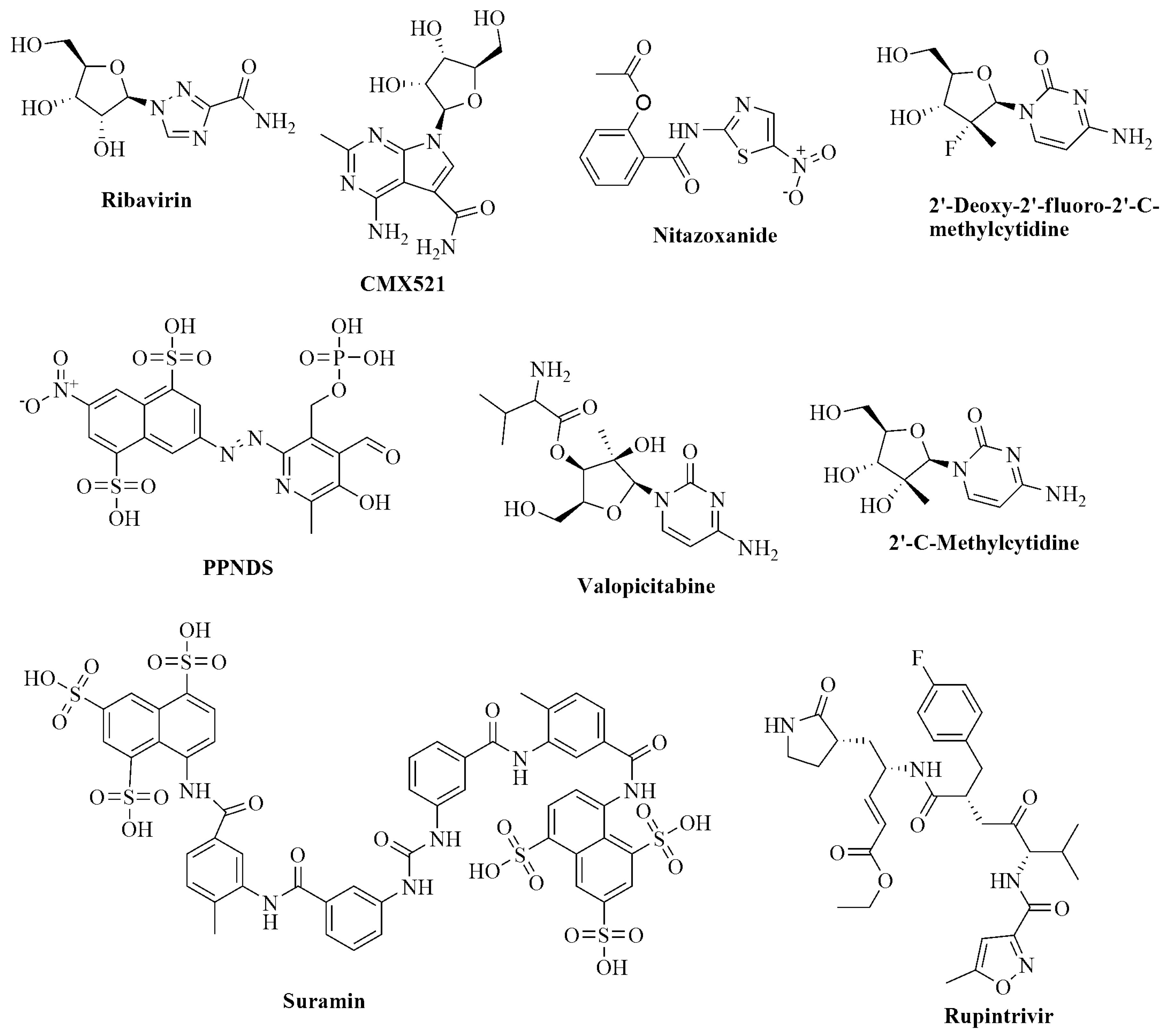
1. Introduction
2. Results and Discussion
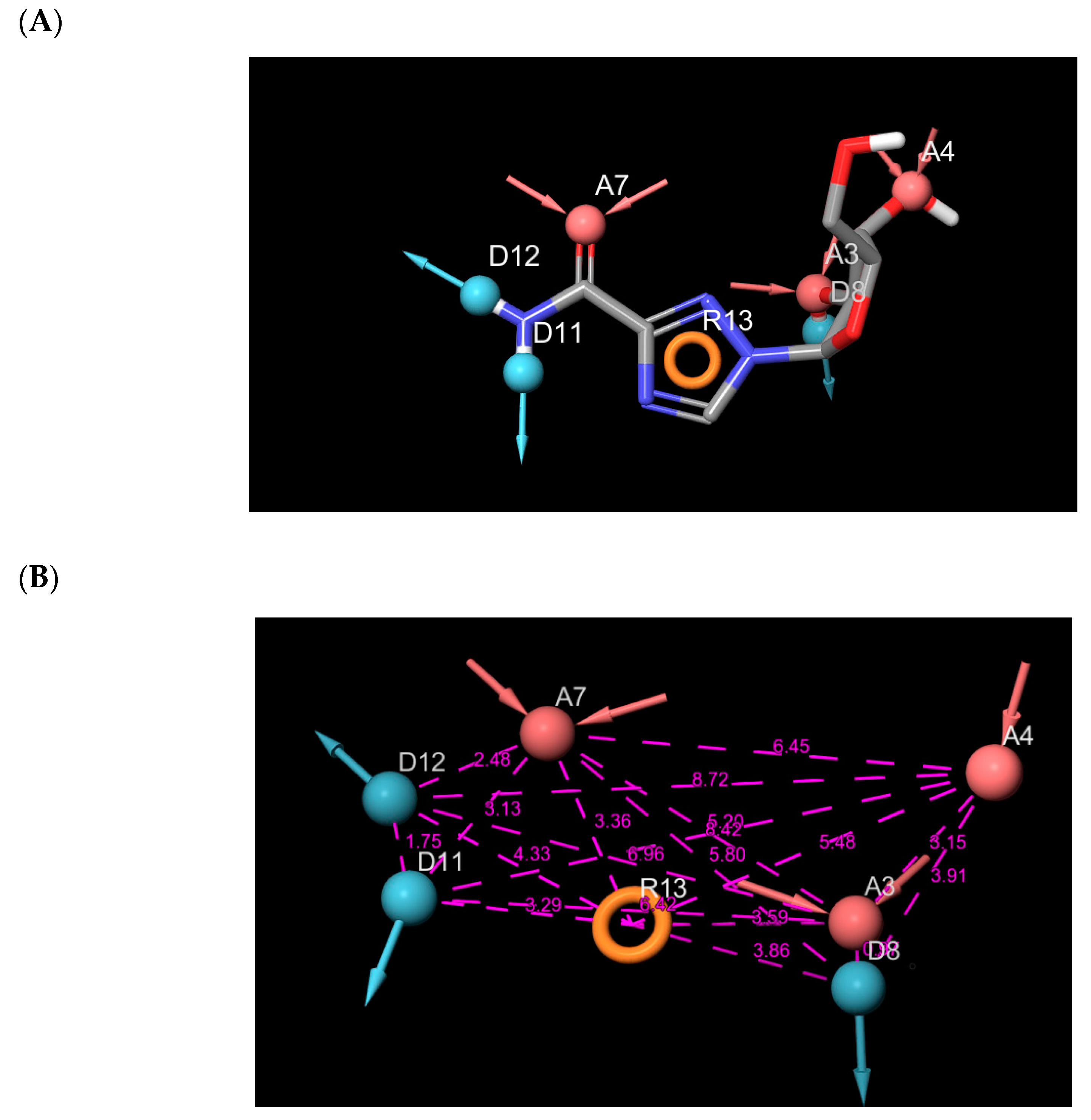
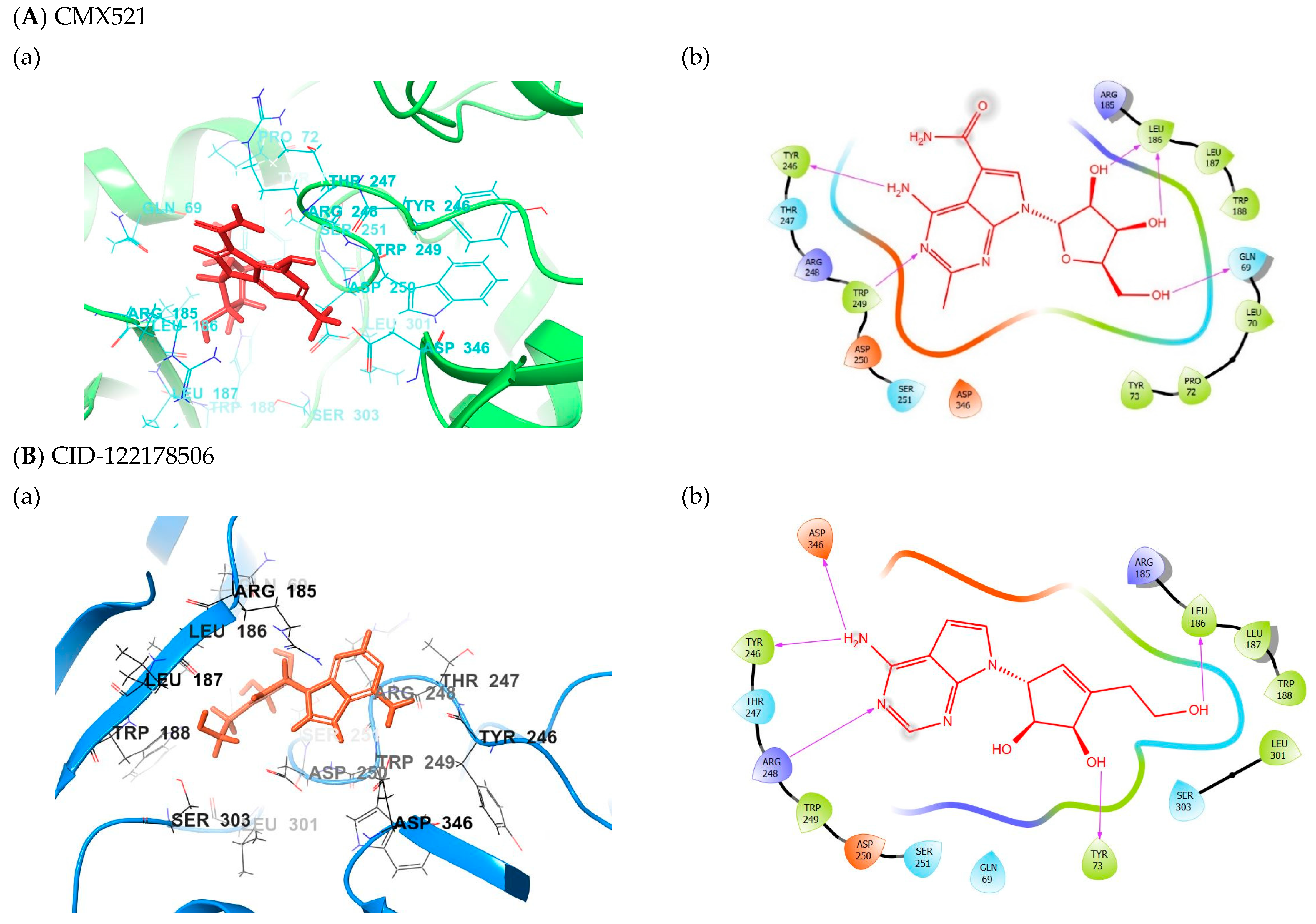
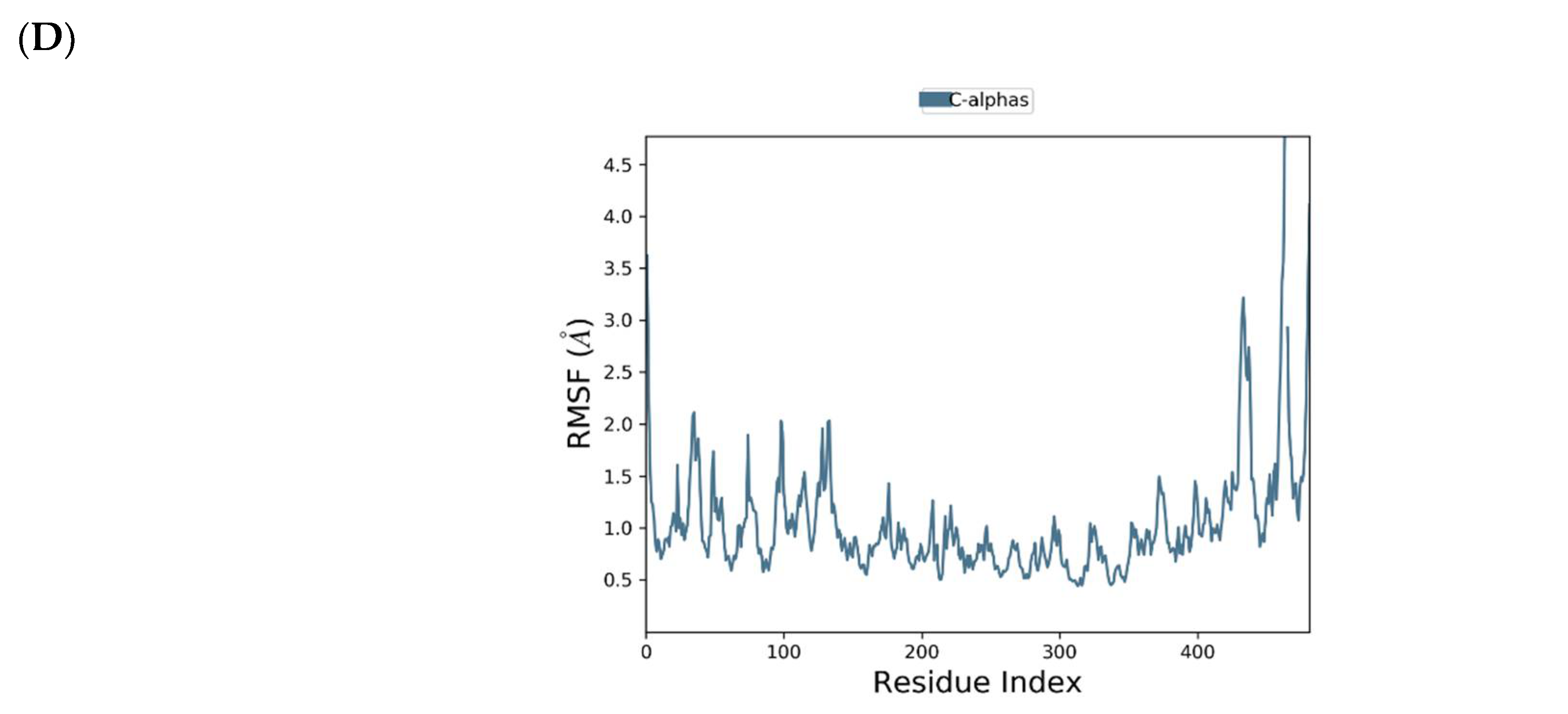
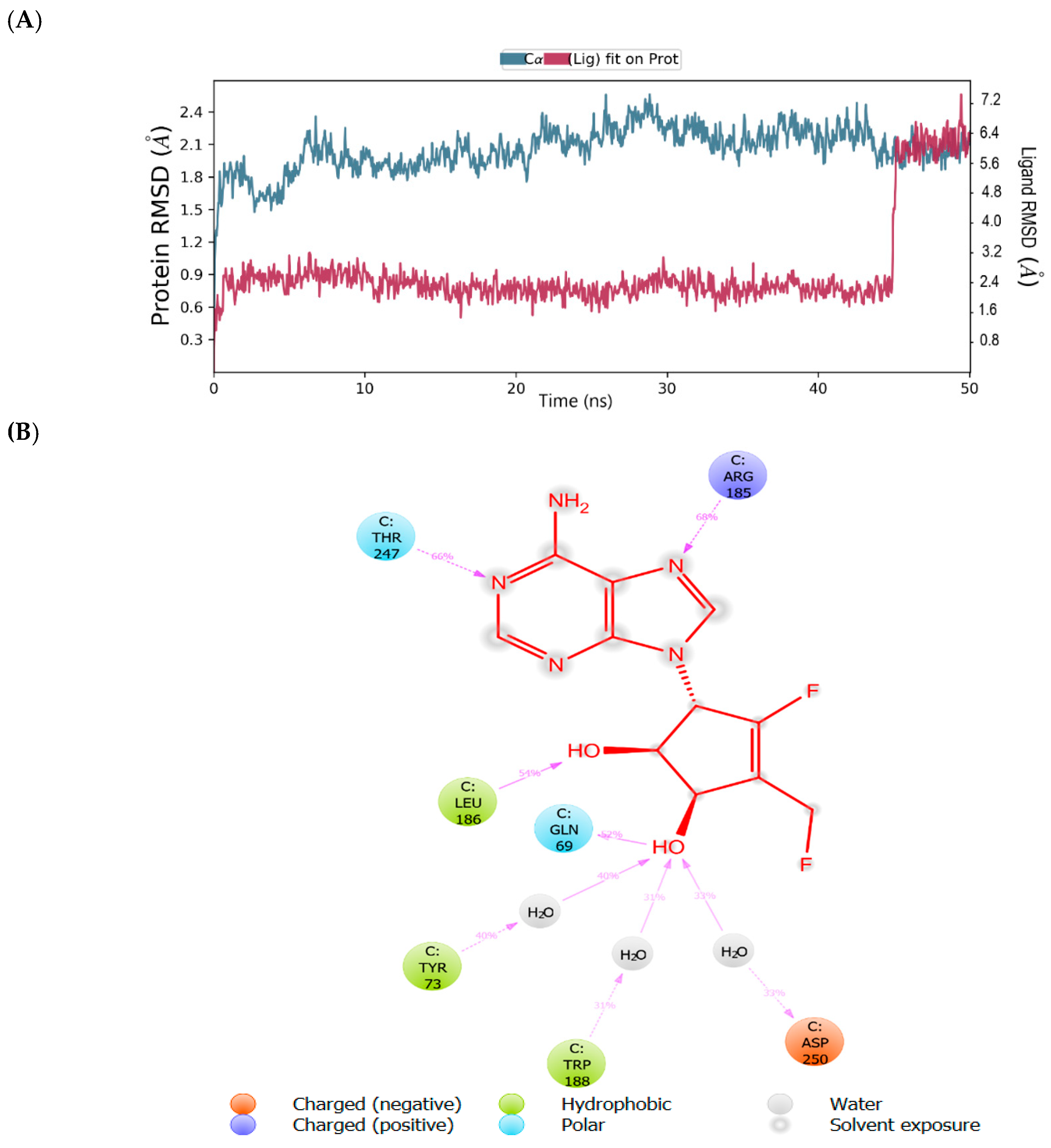
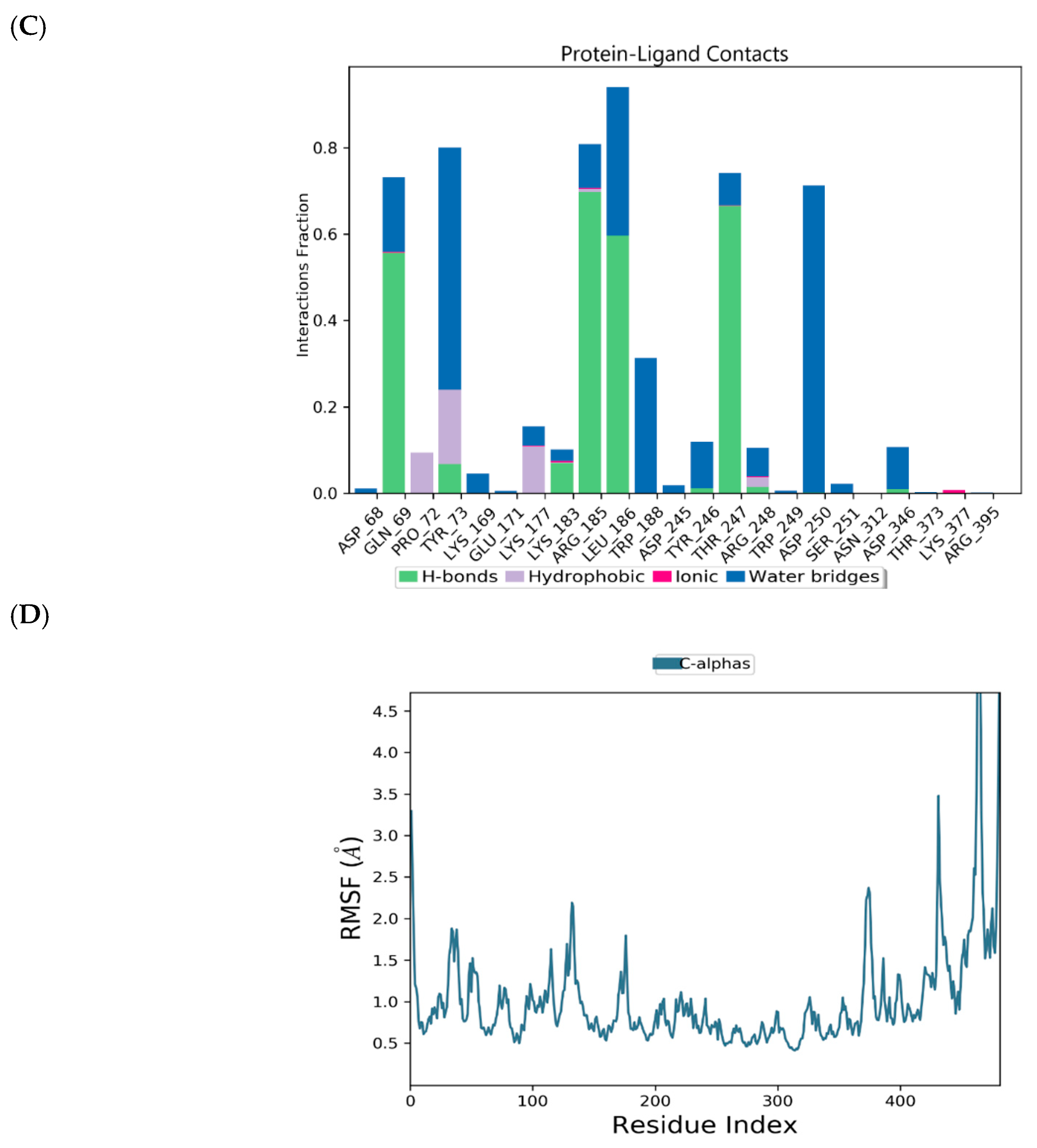
2.1. Binding Mode of the CMX521 and Ribavirin—MNV RdRp Complex
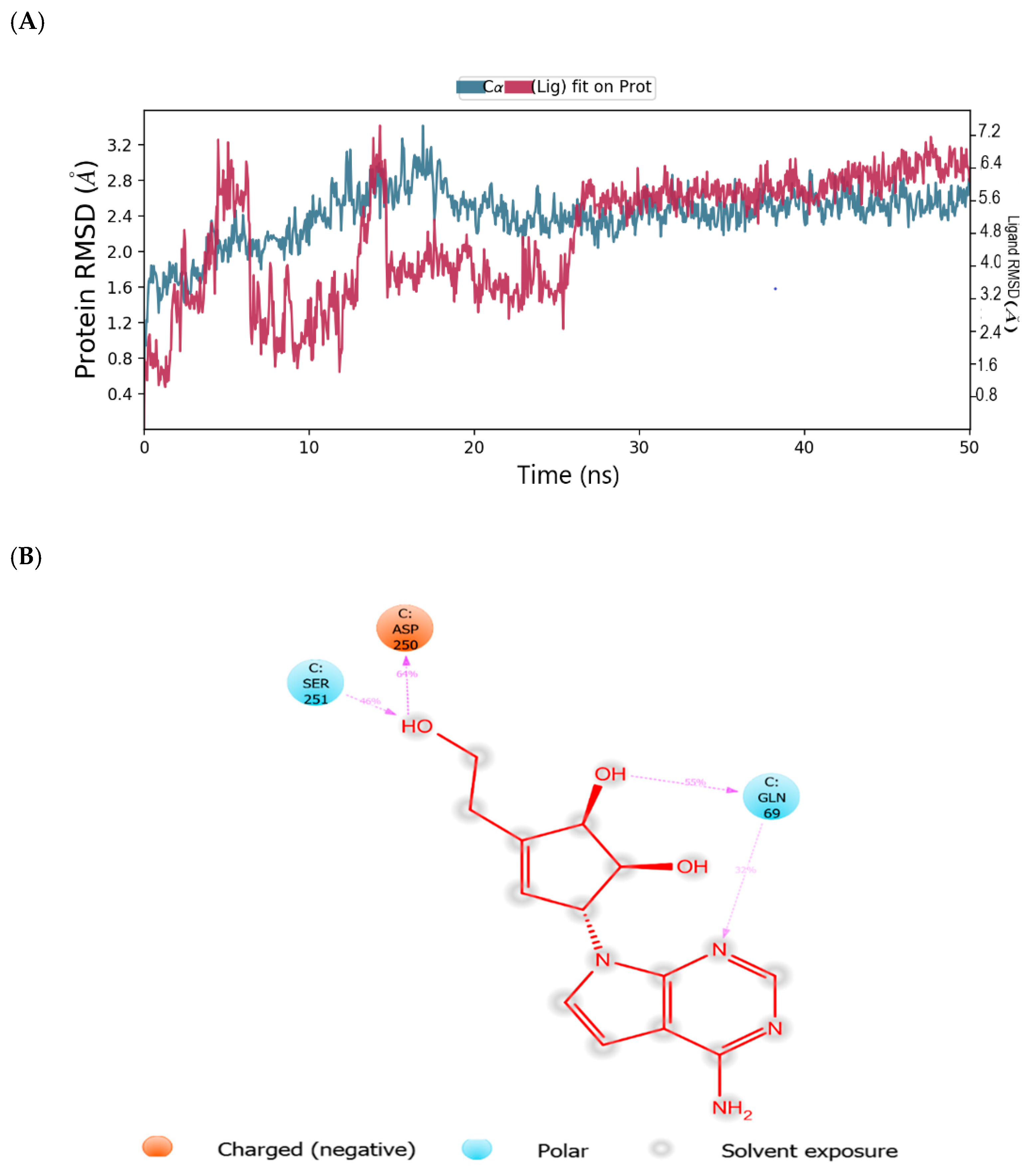
2.2. Binding Mode of the CID-122178506—MNV RdRp Complex
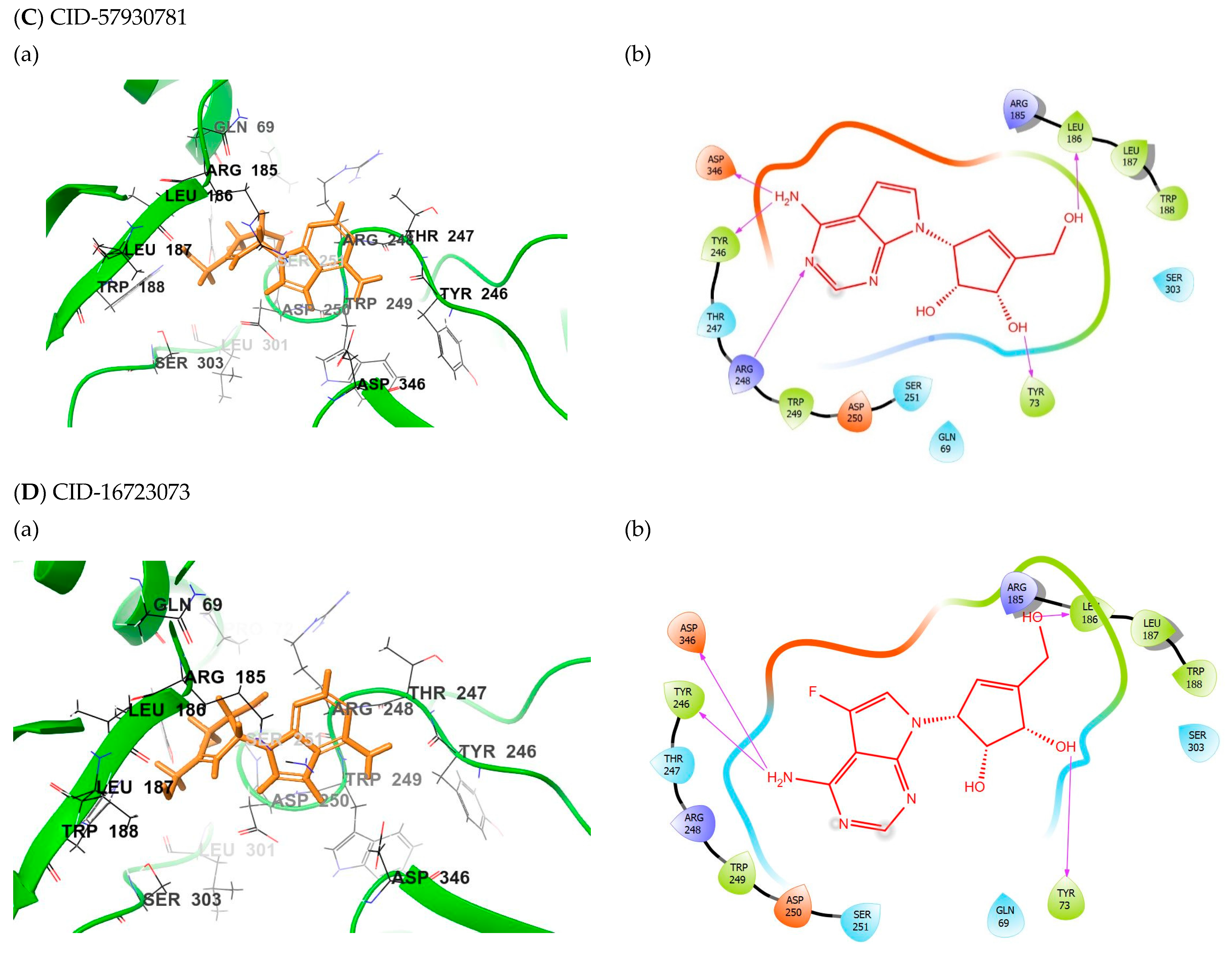
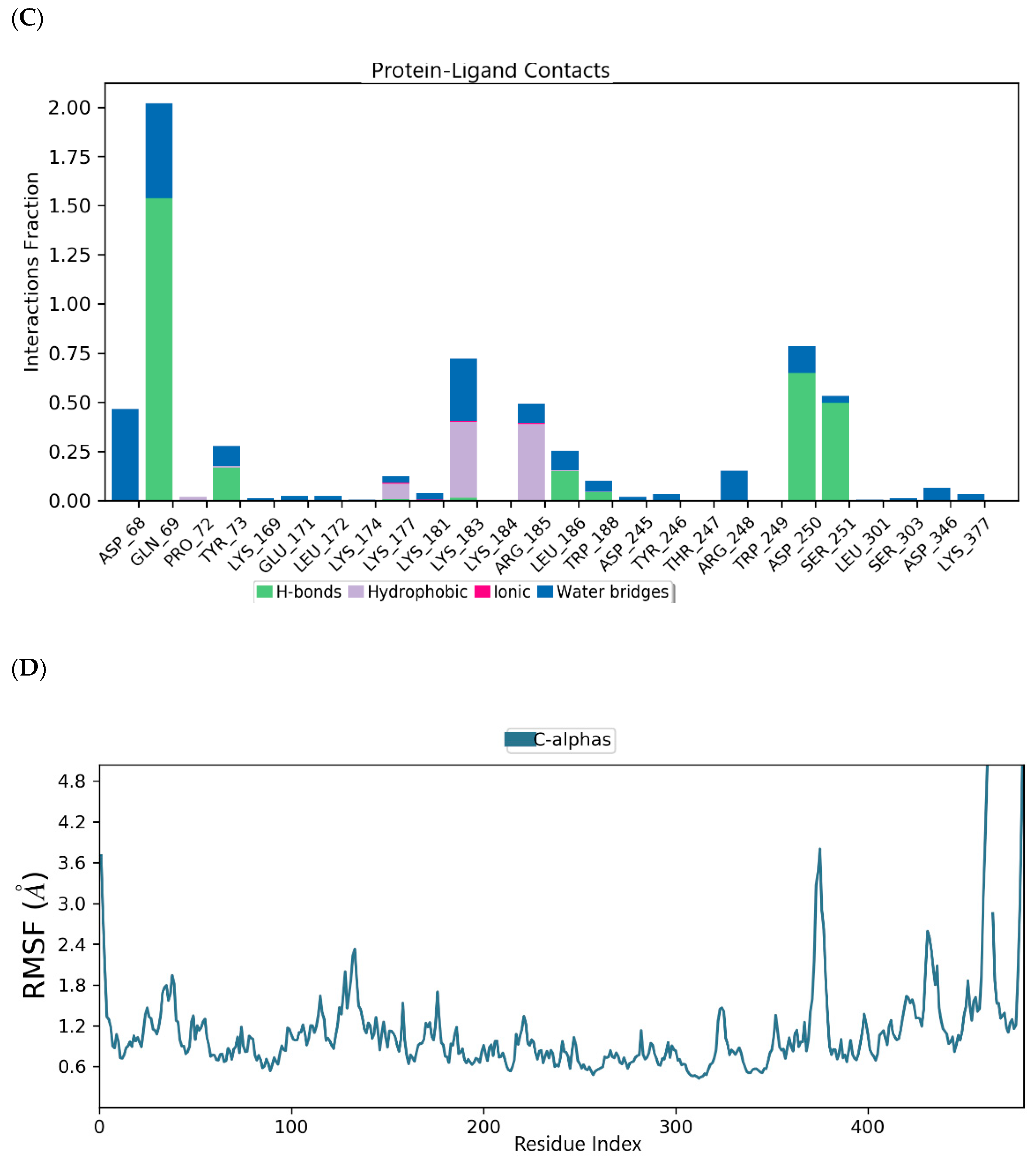
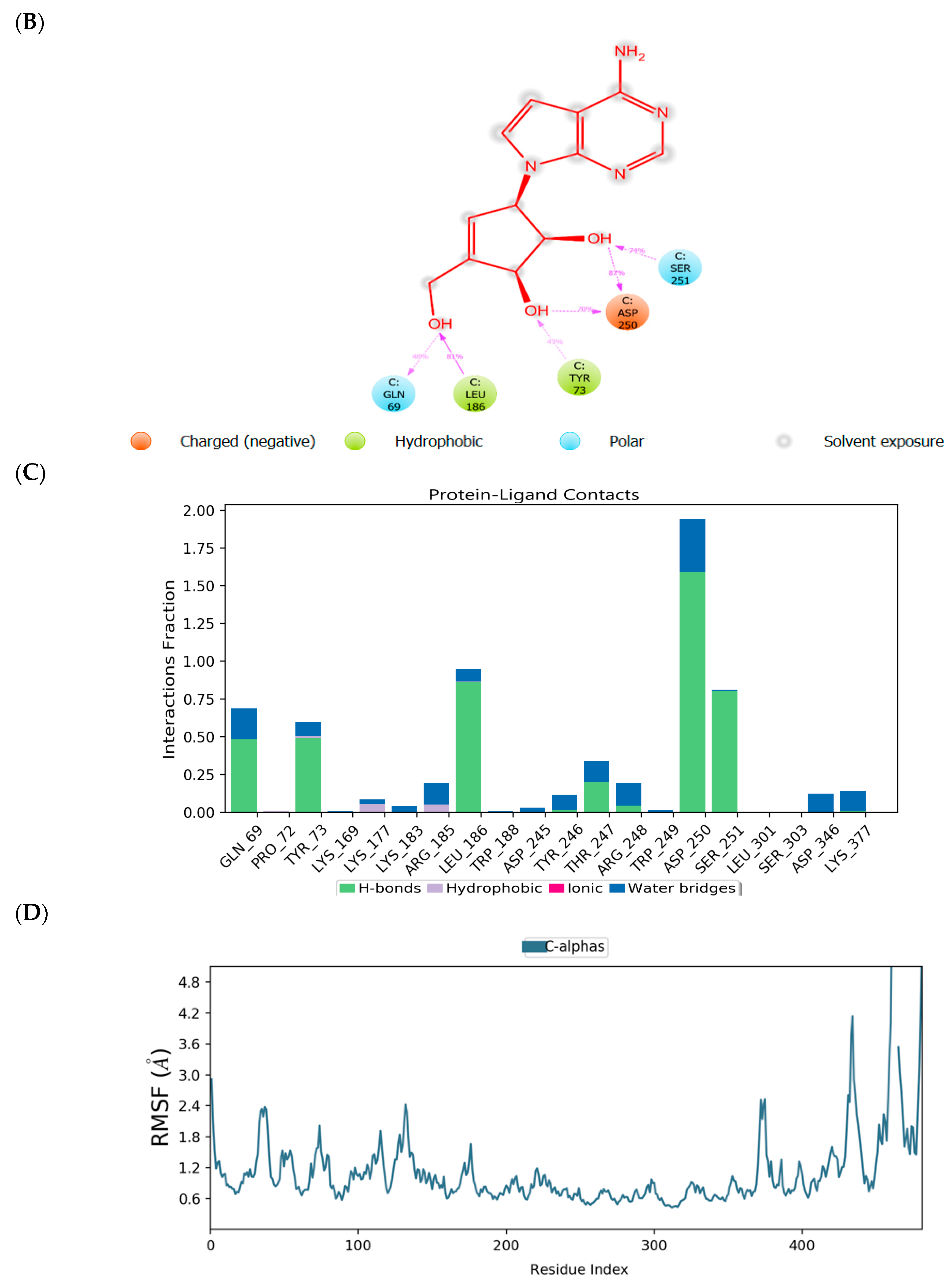
2.3. Binding Mode of the CID-57930781—MNV RdRp Complex
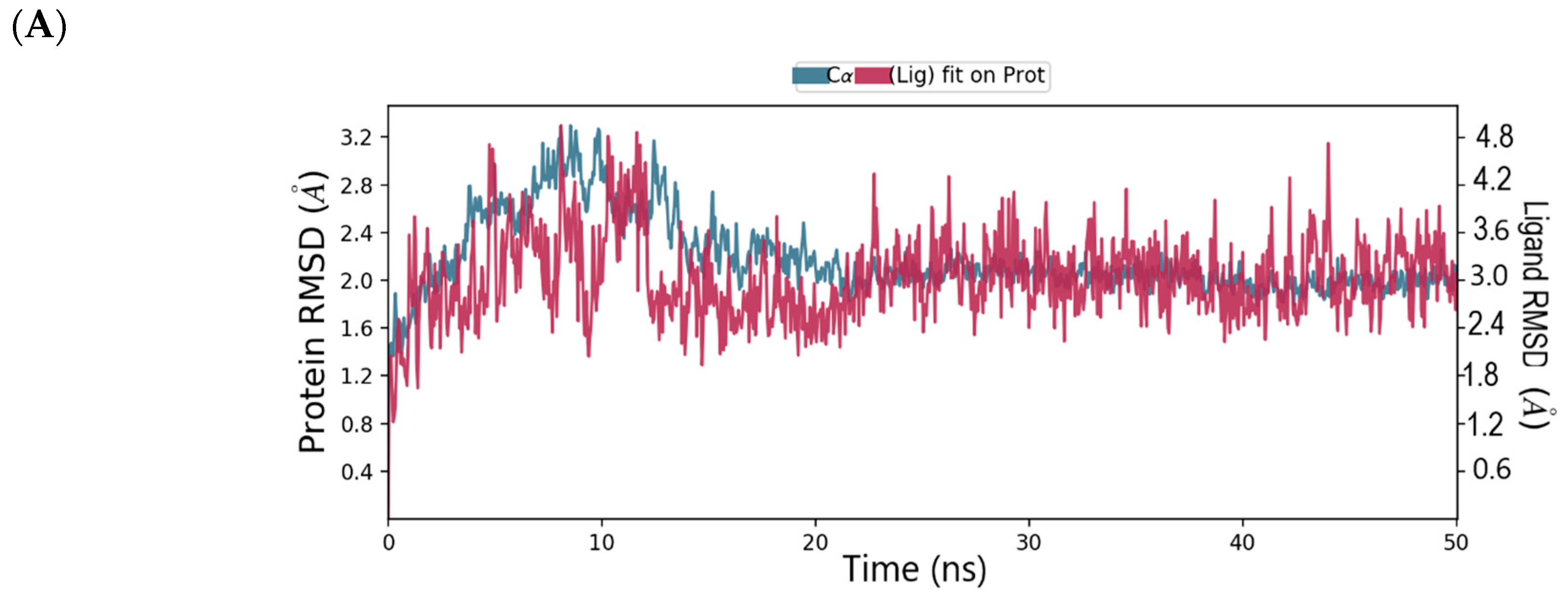
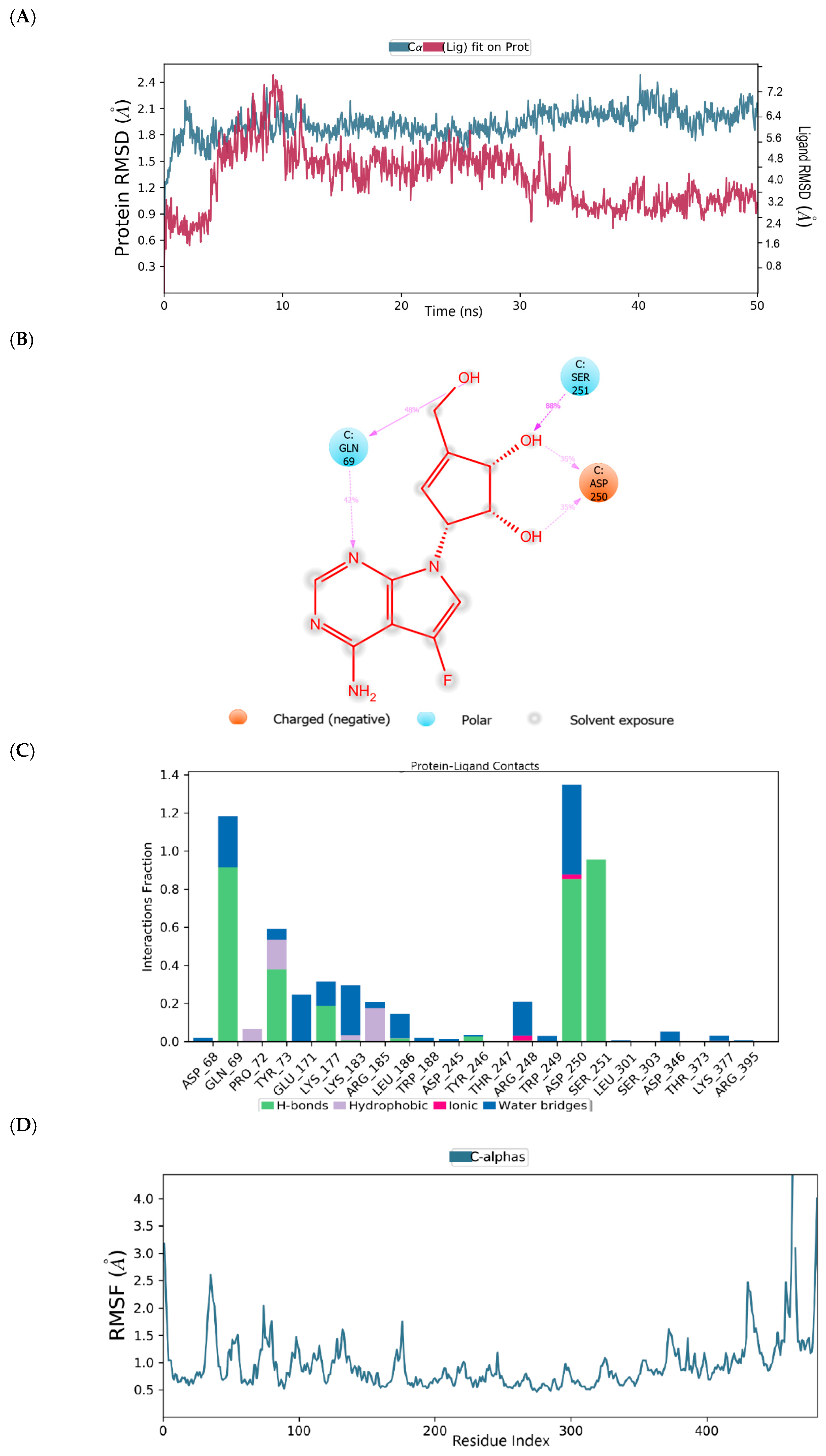
2.4. Binding Mode of the CID-16723073—MNV RdRp Complex
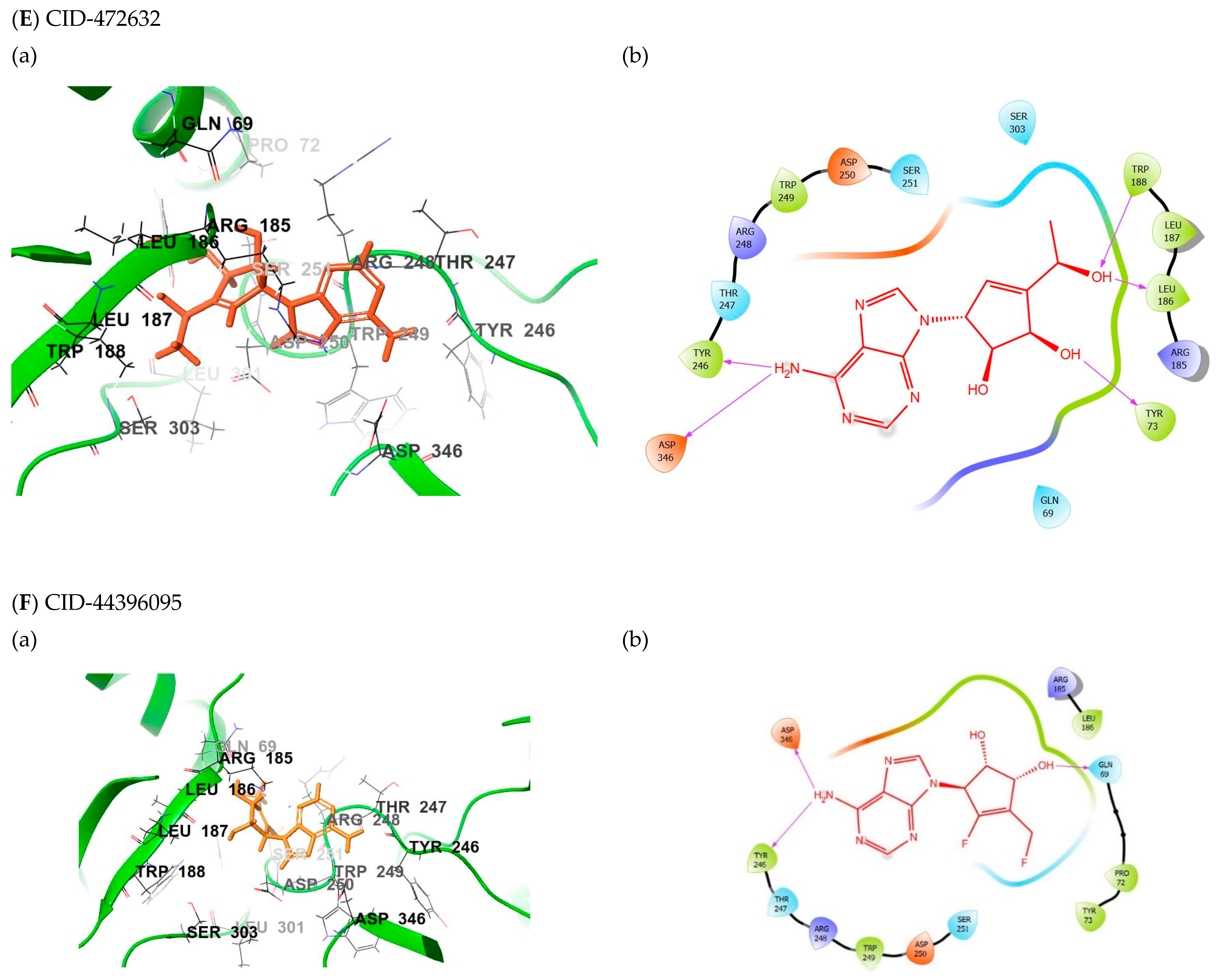
2.5. Binding Mode of the CID-472632—MNV RdRp Complex
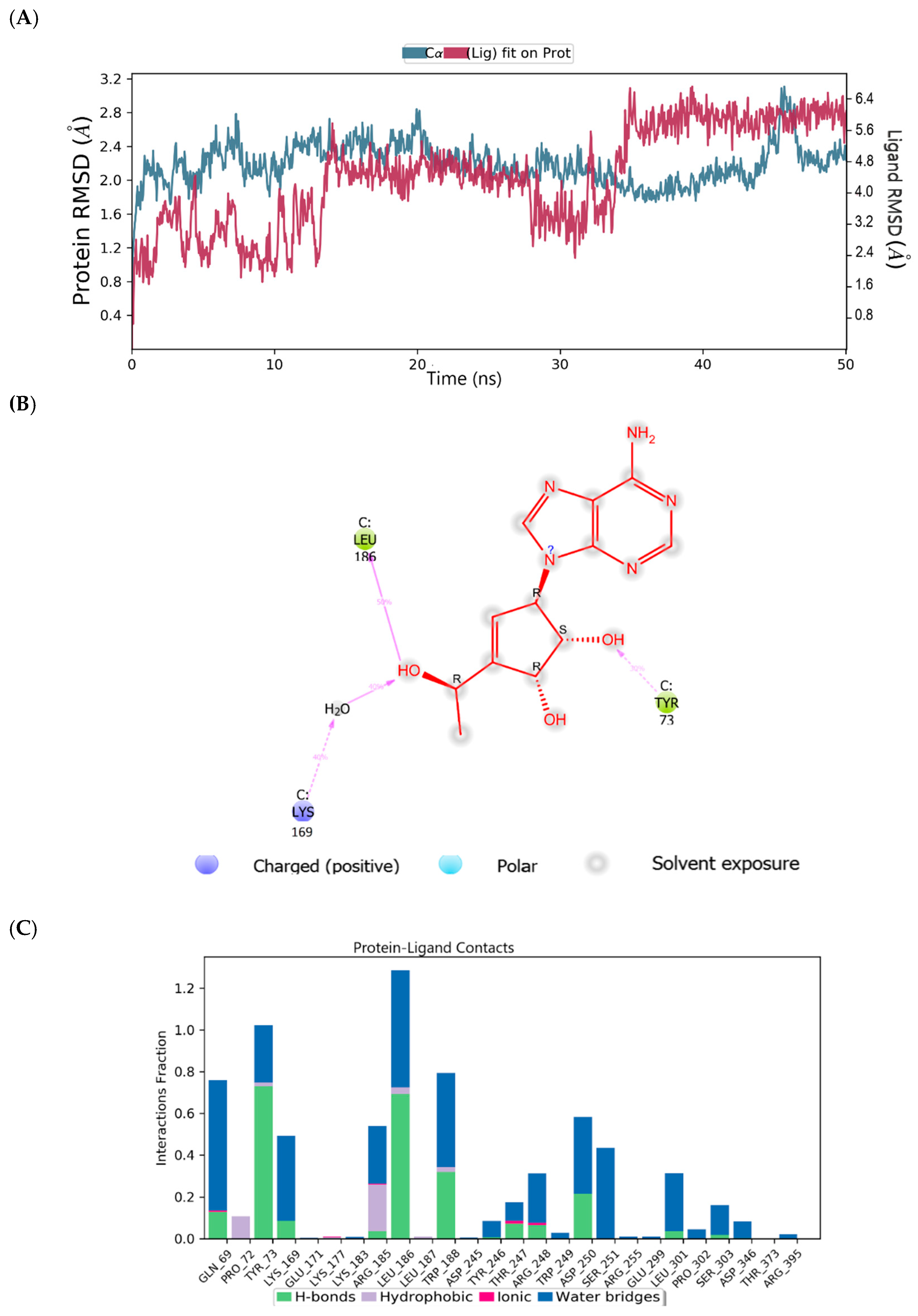
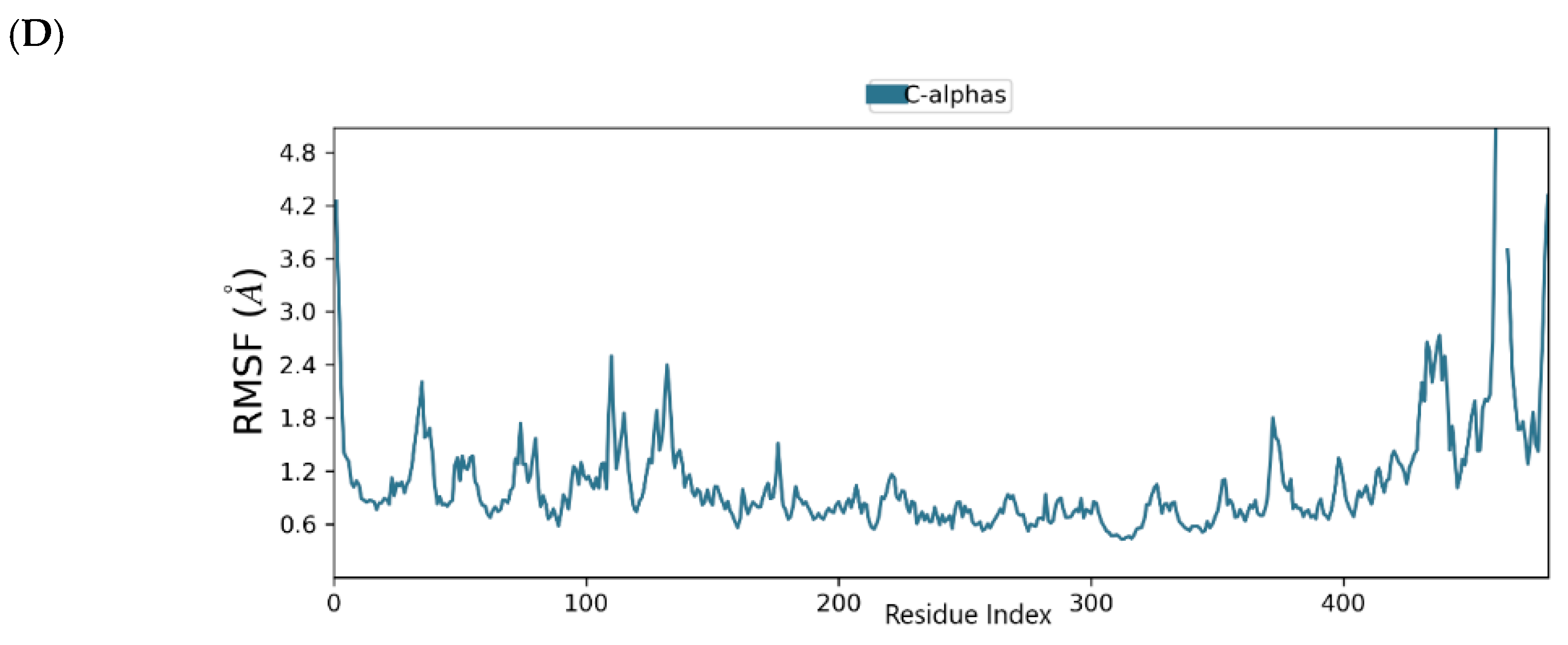
2.6. Binding Mode of the CID-44396095—MNV RdRp Complex
3. Material and Methods
3.1. Data Software and Visualization
3.2. Dataset and Compound Library Preparation
3.3. Ligand Preparation
3.4. Receptor Preparation and Grid Generation
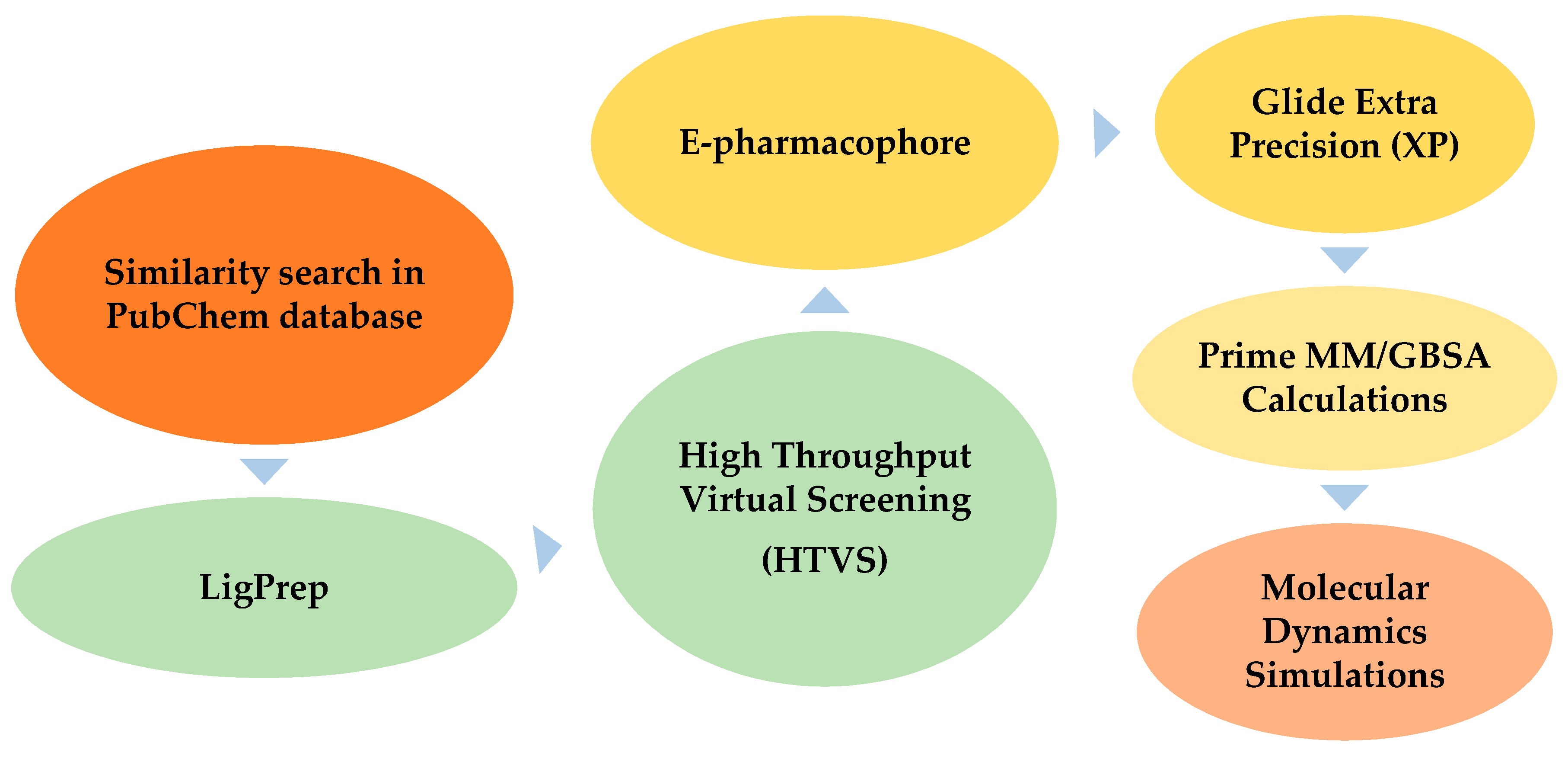
3.5. Structure-Based Virtual Screening
3.5.1. High Throughput Virtual Screening
3.5.2. E-Pharmacophore Hypothesis Generation and Database Screening
3.5.3. Extra Precision Docking (XP)
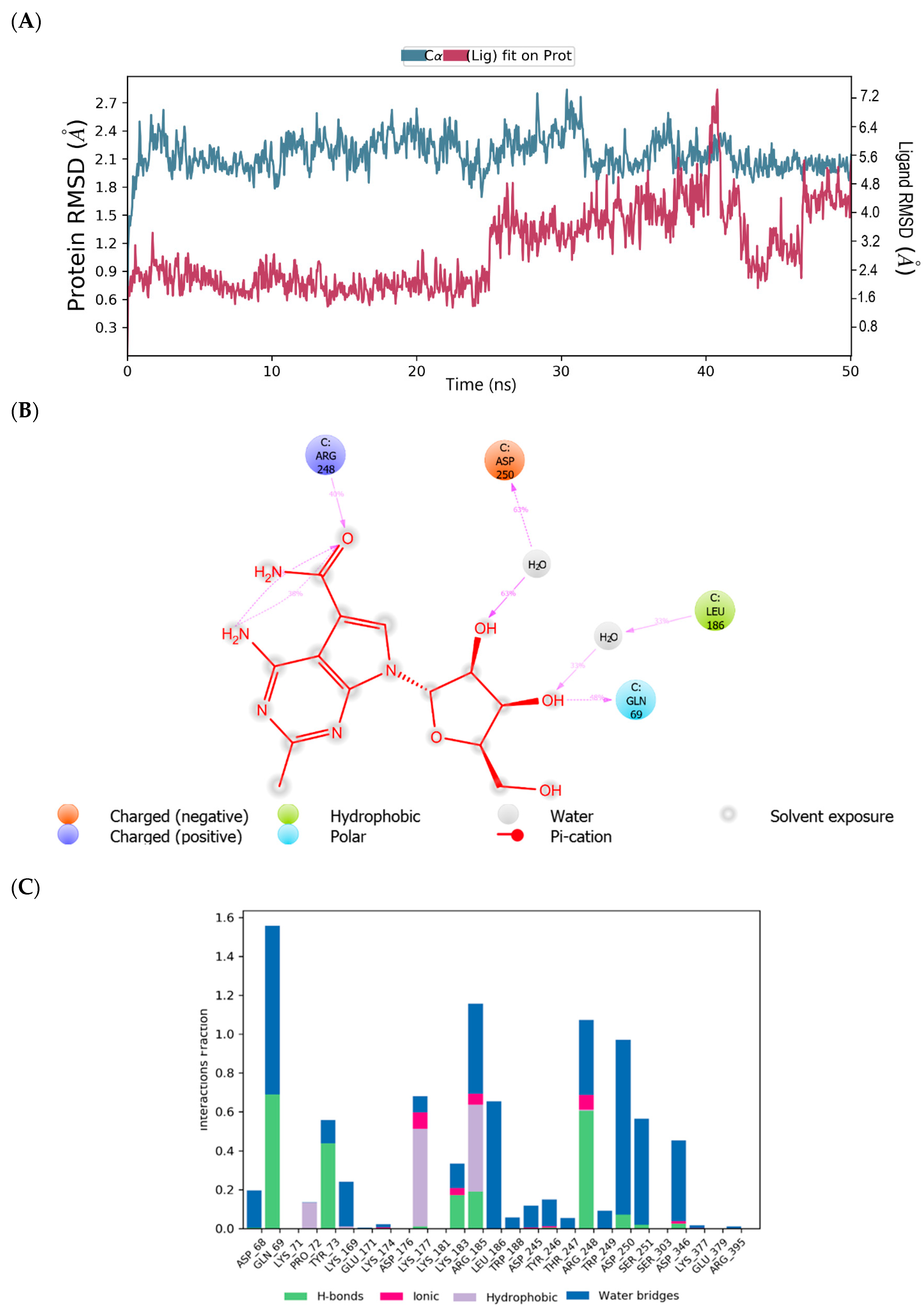
3.6. Molecular Dynamics Simulations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Estrada, E.; Uriarte, E.; Montero, A.; Teijeira, M.; Santana, L.; De Clercq, E. A novel approach for the virtual screening and rational design of anticancer compounds. J. Med. Chem. 2000, 43, 1975–1985. [Google Scholar] [CrossRef] [PubMed]
- Maia, E.H.B.; Assis, L.C.; De Oliveira, T.A.; Da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-F.; Tan, M.; Chhabra, M.; Dai, Y.-C.; Meller, J.; Jiang, X. Inhibition of Histo-blood Group Antigen Binding as a Novel Strategy to Block Norovirus Infections. PLoS ONE 2013, 8, e69379. [Google Scholar] [CrossRef] [PubMed]
- Koromyslova, A.D.; Tripathi, S.; Morozov, V.; Schroten, H.; Hansman, G.S. Human norovirus inhibition by a human milk oligosaccharide. Virology 2017, 508, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ko, G. Antiviral effect of vitamin A on norovirus infection via modulation of the gut microbiome. Sci. Rep. 2017, 6, 1–9. [Google Scholar] [CrossRef]
- Netzler, N.E.; Tuipulotu, D.E.; A White, P. Norovirus antivirals: Where are we now? Med. Res. Rev. 2019, 39, 860–886. [Google Scholar] [CrossRef]
- Zheng, D.-P.; Ando, T.; Fankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S.S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef]
- Teunis, P.F.M.; Moe, C.L.; Liu, P.; Miller, S.E.; Lindesmith, L.; Baric, R.S.; Le Pendu, J.; Calderon, R.L. Norwalk virus: How infectious is it? J. Med. Virol. 2008, 80, 1468–1476. [Google Scholar] [CrossRef]
- Glass, R.I.; Parashar, U.D.; Estes, M.K. Norovirus Gastroenteritis. N. Engl. J. Med. 2009, 361, 1776–1785. [Google Scholar] [CrossRef]
- Robilotti, E.; Deresinski, S.; Pinsky, B. Norovirus. Clin. Microbiol. Rev. 2015, 28, 134–164. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.L.; Ajami, N.; Atmar, R.L.; Dupont, H.L. Noroviruses: The leading cause of gastroenteritis worldwide. Discov. Med. 2010, 10, 61–70. [Google Scholar] [PubMed]
- Green, K.Y.; Belliot, G.; Taylor, J.L.; Valdesuso, J.; Lew, J.F.; Kapikian, A.Z.; Lin, F.-Y.C. A Predominant Role for Norwalk-like Viruses as Agents of Epidemic Gastroenteritis in Maryland Nursing Homes for the Elderly. J. Infect. Dis. 2002, 185, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Available online: www.cdc.gov/norovirus (accessed on 24 July 2020).
- Chong, P.P.; Atmar, R.L. Norovirus in health care and implications for the immunocompromised host. Curr. Opin. Infect. Dis. 2019, 32, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Weerathunge, P.; Ramanathan, R.; Torok, V.A.; Hodgson, K.; Xu, Y.; Goodacre, R.; Behera, B.K.; Bansal, V. Ultrasensitive Colorimetric Detection of Murine Norovirus Using NanoZyme Aptasensor. Anal. Chem. 2019, 91, 3270–3276. [Google Scholar] [CrossRef] [PubMed]
- Thornton, A.C.; Jennings-Conklin, K.S.; McCormick, M.I. Noroviruses: Agents in outbreaks of acute gastroenteritis. Disaster Manag. Response 2004, 2, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Rispens, J.R.; Freeland, A.; Wittry, B.; Kramer, A.; Barclay, L.; Vinjé, J.; Treffiletti, A.; Houston, K. Notes from the Field: Multiple Cruise Ship Outbreaks of Norovirus Associated with Frozen Fruits and Berries—USA, 2019. MMWR. Morb. Mortal. Wkly. Rep. 2020, 69, 501–502. [Google Scholar] [CrossRef]
- Patel, M.M.; Hall, A.J.; Vinjé, J.; Parashar, U.D. Noroviruses: A comprehensive review. J. Clin. Virol. 2009, 44, 1–8. [Google Scholar] [CrossRef]
- Aoki, Y.; Suto, A.; Mizuta, K.; Ahiko, T.; Osaka, K.; Matsuzaki, Y. Duration of norovirus excretion and the longitudinal course of viral load in norovirus-infected elderly patients. J. Hosp. Infect. 2010, 75, 42–46. [Google Scholar] [CrossRef]
- Graham, D.Y.; Jiang, X.; Tanaka, T.; Opekun, A.R.; Madore, H.P.; Estes, M.K. Norwalk Virus Infection of Volunteers: New Insights Based on Improved Assays. J. Infect. Dis. 1994, 170, 34–43. [Google Scholar] [CrossRef]
- Lopman, B.; Reacher, M.H.; Vipond, I.B.; Sarangi, J.; Brown, D.W.G. Clinical Manifestation of Norovirus Gastroenteritis in Health Care Settings. Clin. Infect. Dis. 2004, 39, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Mattner, F.; Sohr, D.; Heim, A.; Gastmeier, P.; Vennema, H.; Koopmans, M. Risk groups for clinical complications of norovirus infections: An outbreak investigation. Clin. Microbiol. Infect. 2006, 12, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Tu, E.T.-V.; Bull, R.A.; Kim, M.-J.; McIver, C.J.; Heron, L.; Rawlinson, W.D.; White, P.A. Norovirus Excretion in an Aged-Care Setting. J. Clin. Microbiol. 2008, 46, 2119–2121. [Google Scholar] [CrossRef] [PubMed]
- Bassetto, M.; Van Dycke, J.; Neyts, J.; Brancale, A.; Rocha-Pereira, J. Targeting the Viral Polymerase of Diarrhea-Causing Viruses as a Strategy to Develop a Single Broad-Spectrum Antiviral Therapy. Viruses 2019, 11, 173. [Google Scholar] [CrossRef]
- Ferla, S.; Netzler, N.E.; Ferla, S.; Veronese, S.; Tuipulotu, D.E.; Guccione, S.; Brancale, A.; White, P.A.; Bassetto, M. In silico screening for human norovirus antivirals reveals a novel non-nucleoside inhibitor of the viral polymerase. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef]
- Harmalkar, D.S.; Lee, S.-J.; Lu, Q.; Kim, M.I.; Park, J.; Lee, H.; Park, M.; Lee, A.; Lee, C.; Lee, K. Identification of novel non-nucleoside vinyl-stilbene analogs as potent norovirus replication inhibitors with a potential host-targeting mechanism. Eur. J. Med. Chem. 2019, 184, 111733. [Google Scholar] [CrossRef]
- Rossignol, J.-F.; El-Gohary, Y.M. Nitazoxanide in the treatment of viral gastroenteritis: A randomized double-blind placebo-controlled clinical trial. Aliment. Pharmacol. Ther. 2006, 24, 1423–1430. [Google Scholar] [CrossRef]
- Salam, N.K.; Nuti, R.; Sherman, W. Novel Method for Generating Structure-Based Pharmacophores Using Energetic Analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef]
- Loving, K.; Salam, N.K.; Sherman, W. Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Mol. Des. 2009, 23, 541–554. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Ntie-Kang, F. An in silico evaluation of the ADMET profile of the StreptomeDB database. SpringerPlus 2013, 2, 353. [Google Scholar] [CrossRef] [PubMed]
- Available online: www.pubchem.ncbi.nlm.nih.gov (accessed on 24 July 2020).
- Xie, X.-Q. Exploiting PubChem for virtual screening. Expert Opin. Drug Discov. 2010, 5, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Foloppe, N. Drug-like Bioactive Structures and Conformational Coverage with the LigPrep/ConfGen Suite: Comparison to Programs MOE and Catalyst. J. Chem. Inf. Model. 2010, 50, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Henderson, K.S. Murine norovirus, a recently discovered and highly prevalent viral agent of mice. Lab. Anim. 2008, 37, 314–320. [Google Scholar] [CrossRef]
- Lochridge, V.P.; Hardy, M.E. A Single-Amino-Acid Substitution in the P2 Domain of VP1 of Murine Norovirus Is Sufficient for Escape from Antibody Neutralization. J. Virol. 2007, 81, 12316–12322. [Google Scholar] [CrossRef]
- Alam, I.; Lee, J.-H.; Cho, K.J.; Han, K.R.; Yang, J.M.; Chung, M.S.; Kim, K.H. Crystal structures of murine norovirus-1 RNA-dependent RNA polymerase in complex with 2-thiouridine or ribavirin. J. Virol. 2012, 426, 143–151. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef]
- Deval, J.; Jin, Z.; Chuang, Y.-C.; Kao, C.C. Structure(s), function(s), and inhibition of the RNA-dependent RNA polymerase of noroviruses. Virus Res. 2016, 234, 21–33. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Kollman, P.A. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem. 2000, 43, 3786–3791. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Lyne, P.; Lamb, A.M.L.; Saeh, J.C. Accurate Prediction of the Relative Potencies of Members of a Series of Kinase Inhibitors Using Molecular Docking and MM-GBSA Scoring. J. Med. Chem. 2006, 49, 4805–4808. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 1–9. [Google Scholar] [CrossRef]
- Sirous, H.; Chemi, G.; Campiani, G.; Brogi, S. An integrated in silico screening strategy for identifying promising disruptors of p53-MDM2 interaction. Comput. Biol. Chem. 2019, 83, 107105. [Google Scholar] [CrossRef]
- Desmond Molecular Dynamics System; Version 4.1; Maestro-Desmond Interoperability Tools Schrödinger; D.E. Shaw Research: New York, NY, USA, 2015.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Glide Score(kcal/mol) | Fitness Score | Prime MM-GBSA (dG bind kcal/mol) |
|---|---|---|---|
| CID-122178506 | −7.073 | 2.356 | −62.554 |
| CID-57930781 | −7.587 | 2.336 | −62.528 |
| CID-16723073 | −6.901 | 2.326 | −53.900 |
| CID-472632 | −7.720 | 2.201 | −59.285 |
| CID-44396095 | −7.390 | 2.118 | −61.969 |
| Compound Identifier | MW | SASA | HBA | HBD | PSA | RO5 |
|---|---|---|---|---|---|---|
| CID-122178506 | 276.29 | 492.310 | 7.6 | 5 | 111.360 | 0 |
| CID-57930781 | 262.27 | 478.545 | 7.6 | 5 | 111.298 | 0 |
| CID-16723073 | 280.258 | 482.612 | 7.6 | 5 | 112.376 | 0 |
| CID-472632 | 277.28 | 489.838 | 9.1 | 5 | 128.212 | 0 |
| CID-44396095 | 283.24 | 484.955 | 7.4 | 4 | 109.498 | 0 |
| CMX521 | 323.308 | 542.358 | 11.8 | 7 | 169.66 | 1 |
| Entry | QPpolrz | QPlogPw | QPlogPo/w | QPlogS | QPlogHERG | QPPCaco | QPlogBB | QPlogKp | QPlogKhsa | HOR | QPPMDCK |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 25.008 | 16.812 | −0.252 | −2.033 | −4.173 | 78.500 | −1.721 | −4.552 | −0.650 | 2 | 31.611 |
| 2 | 24.349 | 17.097 | −0.366 | −2.034 | −4.319 | 95.715 | −1.591 | −4.399 | −0.675 | 2 | 39.166 |
| 3 | 24.727 | 16.87 | −0.128 | −2.264 | −4.069 | 100.678 | −1.446 | -4.478 | −0.636 | 3 | 70.187 |
| 4 | 25.164 | 18.530 | −0.953 | −2.065 | −4.036 | 39.974 | −1.927 | −5.314 | −0.720 | 2 | 15.242 |
| 5 | 25.376 | 15.542 | 0.081 | −2.815 | −4.172 | 97.571 | −1.308 | −4.766 | −0.549 | 3 | 102.922 |
| 6 | 28.005 | 24.005 | −1.897 | −2.282 | −4.182 | 14.282 | −2.542 | −6.261 | −0.873 | 2 | 5.011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebenezer, O.; Jordaan, M.A.; Damoyi, N.; Shapi, M. Discovery of Potential Inhibitors for RNA-Dependent RNA Polymerase of Norovirus: Virtual Screening, and Molecular Dynamics. Int. J. Mol. Sci. 2021, 22, 171. https://doi.org/10.3390/ijms22010171
Ebenezer O, Jordaan MA, Damoyi N, Shapi M. Discovery of Potential Inhibitors for RNA-Dependent RNA Polymerase of Norovirus: Virtual Screening, and Molecular Dynamics. International Journal of Molecular Sciences. 2021; 22(1):171. https://doi.org/10.3390/ijms22010171
Chicago/Turabian StyleEbenezer, Oluwakemi, Maryam A. Jordaan, Nkululeko Damoyi, and Michael Shapi. 2021. "Discovery of Potential Inhibitors for RNA-Dependent RNA Polymerase of Norovirus: Virtual Screening, and Molecular Dynamics" International Journal of Molecular Sciences 22, no. 1: 171. https://doi.org/10.3390/ijms22010171
APA StyleEbenezer, O., Jordaan, M. A., Damoyi, N., & Shapi, M. (2021). Discovery of Potential Inhibitors for RNA-Dependent RNA Polymerase of Norovirus: Virtual Screening, and Molecular Dynamics. International Journal of Molecular Sciences, 22(1), 171. https://doi.org/10.3390/ijms22010171