Neuroprotective Effects of Coffee Bioactive Compounds: A Review




Abstract
1. Introduction
2. Bioavailability and Pharmacokinetics of Coffee Bioactive Compounds
2.1. Caffeine
2.2. Chlorogenic Acids
2.3. Caffeic Acid
2.4. Trigonelline
2.5. Kahweol and Cafestol
3. Neurodegenerative Diseases
3.1. Dementias, Including Alzheimer’s Disease
3.2. Parkinson’s Disease
3.3. Ischemic Stroke
3.4. Epilepsy
4. Neuroprotective Effects of Coffee Bioactive Compounds
4.1. Neuroprotective Effects of Caffeine
4.2. Neuroprotective Effects of Chlorogenic Acid
4.3. Neuroprotective Effects of Caffeic Acid
4.4. Neuroprotective Effects of Trigonelline
4.5. Neuroprotective Effects of Kahweol and Cafestol
5. Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid beta |
| AChE | Acetylcholinesterase |
| AGEs | Advanced glycation end products |
| Akt | Protein kinase B |
| APP | Amyloid precursor protein |
| APPsw | Swedish mutation mice, mice carrying the mutant APPK670N, M671L gene |
| ATP | Adenosine-5′-triphosphate |
| Bax | Bcl-2-associated X protein |
| BBB | Blood brain barrier |
| BChE | Butyrylcholinesterase |
| Bcl2 | B-cell lymphoma protein 2 |
| BDNF | Brain-derived neurotrophic factor |
| CAPE | Caffeic acid phenyl ester |
| CBF | Cerebral blood flow |
| CD31 | Platelet/endothelial cell adhesion molecule-1 |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase 2 |
| CSF | Cerebrospinal fluid |
| CYP | Cytochrome P450 |
| DAT | Dopamine transporter |
| ER | Endoplasmic reticulum |
| ERK1/2 | Extracellular signal-regulated kinase-1 and -2 |
| GABA | Gamma-aminobutyric acid |
| GDNF | Glial cell line-derived neurotrophic factor |
| GFAP | Glial fibrillary acidic protein |
| GSK3β | Glycogen synthase kinase 3 beta |
| GSH | Reduced glutathione |
| GSH-Px | Glutathione peroxidase |
| GST | Glutathione-S-transferase |
| HI | Hypoxia-ischemia |
| HIF1α | Hypoxia-inducible factor 1 alpha |
| HO-1 | Heme oxygenase 1 |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IL-1β | Interleukin 1 beta |
| IL-2 | Interleukin 2 |
| IL-4 | Interleukin 4 |
| IL-6 | Interleukin 6 |
| IL-13 | Interleukin 13 |
| i.n. | Intranasal |
| iNOS | Inducible nitric oxide synthase |
| i.p. | Intraperitoneally |
| i.v. | Intravenously |
| LDH | Lactate dehydrogenase |
| 5-LOX | 5-Lipoxygenase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MB | Manganese bisethylenedithiocarbamate |
| MDA | Malondialdehyde |
| mGluR1 | Metabotropic glutamate receptor type 1 |
| mGluR5 | Metabotropic glutamate receptor type 5 |
| MMP-2, -9 | Metallomatrixprotease-2,-9 |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MRI | Magnetic resonance imaging |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGF | Nerve growth factor |
| NMDA | N-methyl-d-aspartate |
| nNOS | Neuronal nitric oxide synthase |
| NO | Nitric oxide |
| NOS-2 | Nitric oxide synthase-2 |
| NQO-1 | NAD(P)H quinone oxidoreductase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| 6-OHDA | 6-Hydroxydopamine |
| p53 | Tumor protein p53 |
| p65 | Transcription factor p65 |
| PARP-1 | Poly [ADP-ribose] polymerase 1 |
| p-JNK | C-Jun N-terminal kinases |
| p.o. | Orally |
| PQ | 1,1′-Dimethyl-4,4′-bipyridinium dichloride hydrate |
| RNS | Nitrogen reactive species |
| ROS | Reactive oxygen species |
| rpS3 | Ribosomal protein |
| S100b | S100 calcium-binding protein B |
| SNP | Sodium nitroprusside |
| SOD | Superoxide dismutase |
| SOD2 | Superoxide dismutase 2 |
| TFEB | Transcription factor EB |
| TH+ | Tyrosine hydroxylase immunoreactivity |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor α |
| TrkB | Tirosine kinase receptor |
| UDP | Uridine 5′-diphosphate |
| VCAM-1 | Vascular cell adhesion protein 1 |
References
- Clifford, M.N.; Gibson, C.L.; Rakotomalala, J.-J.R.; Cross, E.; Charrier, A. Caffeine from green beans of Mascarocoffea. Phytochemistry 1991, 30, 4039–4040. [Google Scholar] [CrossRef]
- Cheek, M.; Csiba, L.; Bridson, D.M. A new species of Coffea (Rubiaceae) from western Cameroon. Kew Bull. 2002, 57, 675–680. [Google Scholar] [CrossRef]
- Davis, A.P.; Rakotonasolo, F. New species of Coffea L. (Rubiaceae) from Madagascar. Bot. J. Linn. Soc. 2003, 142, 111–118. [Google Scholar] [CrossRef][Green Version]
- Davis, A.P.; Mvungi, E.F. Two new and endangered species of Coffea (Rubiaceae) from the Eastern Arc Mountains (Tanzania) and notes on associated conservation issues. Bot. J. Linn. Soc. 2004, 146, 111–118. [Google Scholar] [CrossRef][Green Version]
- Davis, A.P.; Govaerts, R.; Bridson, D.M.; Stoffelen, P. An annotated taxonomic conspectus of the genus Coffea (Rubiaceae). Bot. J. Linn. Soc. 2006, 152, 465–512. [Google Scholar] [CrossRef]
- Sonké, B.; Nguembou, C.K.; Davis, A.P. A new dwarf Coffea (Rubiaceae) from southern Cameroon. Bot. J. Linn. Soc. 2006, 151, 425–430. [Google Scholar] [CrossRef][Green Version]
- Higdon, J.V.; Frei, B. Coffee and health: A review of recent human research. Crit. Rev. Food Sci. Nutr. 2006, 46, 101–123. [Google Scholar] [CrossRef]
- Lee, D.R.; Lee, J.; Rota, M.; Lee, J.; Ahn, H.S.; Park, S.M.; Shin, D. Coffee consumption and risk of fractures: A systematic review and dose-response meta-analysis. Bone 2014, 63, 20–28. [Google Scholar] [CrossRef]
- Bae, J.H.; Park, J.H.; Im, S.S.; Song, D.K. Coffee and health. Integr. Med. Res. 2014, 3, 189–191. [Google Scholar] [CrossRef]
- International Coffee Organization. Coffee Trade Statistics; International Coffee Organization: London, UK, 2020. [Google Scholar]
- Bizzo, M.L.G.; Farah, A.; Kempa, J.A.; Scancetti, L.B. Highlight in the history of coffee science related to health. In Coffee in Health and Disease Prevention; Preedy, V.R., Ed.; Elsevier: London, UK, 2015; pp. 11–17. [Google Scholar]
- Butt, M.S.; Sultan, M.T. Coffee and its consumption: Benefits and risks. Crit. Rev. Food Sci. Nutr. 2011, 51, 363–373. [Google Scholar] [CrossRef]
- Ciaramelli, C.; Palmioli, A.; Airoldi, C. Coffee variety, origin and extraction procedure: Implications for coffee beneficial effects on human health. Food Chem. 2019, 278, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Hwang, E.; Park, J.B. Association between consumption of coffee and the prevalence of periodontitis: The 2008–2010 Korea National Health and Nutrition Examination Survey. PLoS ONE 2016, 11, e0158845. [Google Scholar] [CrossRef] [PubMed]
- Horrigan, L.A.; Kelly, J.P.; Connor, T.J. Immunomodulatory effects of caffeine: Friend or foe? Pharmacol. Ther. 2006, 111, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Kim, K.; Ahn, Y.; Yang, M.; Lee, J.E. Habitual coffee intake, genetic polymorphisms, and type 2 diabetes. Eur. J. Endocrinol. 2015, 172, 595–601. [Google Scholar] [CrossRef]
- Ludwig, I.A.; Clifford, M.N.; Lean, M.E.; Ashihara, H.; Crozier, A. Coffee: Biochemistry and potential impact on health. Food Funct. 2014, 5, 1695–1717. [Google Scholar] [CrossRef]
- Poole, R.; Kennedy, O.J.; Roderick, P.; Fallowfield, J.A.; Hayes, P.C.; Parkes, J. Coffee consumption and health: Umbrella review of meta-analyses of multiple health outcomes. BMJ 2017, 359, j5024. [Google Scholar] [CrossRef]
- Santos, R.M.; Lima, D.R. Coffee consumption, obesity and type 2 diabetes: A mini-review. Eur. J. Nutr. 2016, 55, 1345–1358. [Google Scholar] [CrossRef]
- Dórea, J.G.; da Costa, T.H. Is coffee a functional food? Br. J. Nutr. 2005, 93, 773–782. [Google Scholar] [CrossRef]
- Cornelis, M.C.; El-Sohemy, A. Coffee, caffeine, and coronary heart disease. Curr. Opin. Lipidol. 2007, 18, 13–19. [Google Scholar] [CrossRef]
- Gökcen, B.B.; Sanlier, N. Coffee consumption and disease correlations. Crit. Rev. Food Sci. Nutr. 2019, 59, 336–348. [Google Scholar] [CrossRef]
- Nieber, K. The impact of coffee on health. Planta Med. 2017, 83, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Cano-Marquina, A.; Tarín, J.J.; Cano, A. The impact of coffee on health. Maturitas 2013, 75, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Oleaga, C.; Ciudad, C.J.; Noé, V.; Izquierdo-Pulido, M. Coffee polyphenols change the expression of STAT5B and ATF-2 modifying cyclin D1 levels in cancer cells. Oxid. Med. Cell Longev. 2012, 2012, 390385. [Google Scholar] [CrossRef] [PubMed]
- Mineharu, Y.; Koizumi, A.; Wada, Y.; Iso, H.; Watanabe, Y.; Date, C.; Yamamoto, A.; Kikuchi, S.; Inaba, Y.; Toyoshima, H.; et al. Coffee, green tea, black tea and oolong tea consumption and risk of mortality from cardiovascular disease in Japanese men and women. J. Epidemiol. Community Health 2011, 65, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Sääksjärvi, K.; Knekt, P.; Rissanen, H.; Laaksonen, M.A.; Reunanen, A.; Männistö, S. Prospective study of coffee consumption and risk of Parkinson’s disease. Eur. J. Clin. Nutr. 2008, 62, 908–915. [Google Scholar] [CrossRef]
- Trevitt, J.; Kawa, K.; Jalali, A.; Larsen, C. Differential effects of adenosine antagonists in two models of parkinsonian tremor. Pharmacol. Biochem. Behav. 2009, 94, 24–29. [Google Scholar] [CrossRef]
- Arendash, G.W.; Cao, C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J. Alzheimers. Dis. 2010, 20 (Suppl. 1), S117–S126. [Google Scholar] [CrossRef]
- de Mendonça, A.; Cunha, R.A. Therapeutic opportunities for caffeine in Alzheimer’s disease and other neurodegenerative disorders. J. Alzheimers. Dis. 2010, 20 (Suppl. 1), S1–S2. [Google Scholar] [CrossRef]
- Hermansen, K.; Krogholm, K.S.; Bech, B.H.; Dragsted, L.O.; Hyldstrup, L.; Jorgensen, K.; Larsen, M.L.; Tjonneland, A.M. Coffee can protect against disease. Ugeskr. Laeger 2012, 174, 2293–2297. [Google Scholar]
- Kawachi, I.; Willett, W.C.; Colditz, G.A.; Stampfer, M.J.; Speizer, F.E. A prospective study of coffee drinking and suicide in women. Arch. Intern. Med. 1996, 156, 521–525. [Google Scholar] [CrossRef]
- Homan, D.J.; Mobarhan, S. Coffee: Good, bad, or just fun? A critical review of coffee’s effects on liver enzymes. Nutr. Rev. 2006, 64, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Jee, S.H.; He, J.; Appel, L.J.; Whelton, P.K.; Suh, I.; Klag, M.J. Coffee consumption and serum lipids: A meta-analysis of randomized controlled clinical trials. Am. J. Epidemiol. 2001, 153, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Farah, A. Nutritional and health effects of coffee. In Achieving Sustainable Cultivaiton of Coffee, 1st ed.; Lashermes, P., Ed.; Burleigh Dodds Science Publishing: Cambridge, UK, 2018; pp. 259–290. [Google Scholar]
- Diviš, P.; Pořízka, J.; Kříkala, J. The effect of coffee beans roasting on its chemical composition. Potr. S. J. F. Sci 2019, 13, 344–350. [Google Scholar] [CrossRef]
- Farah, A.; Duarte, G. Bioavailability and metabolism of chlorogenic acids from coffee. In Coffee in Health and Disease Prevention; Preedy, V.R., Ed.; Elsevier: London, UK, 2015; pp. 789–801. [Google Scholar]
- Moreira, A.S.; Nunes, F.M.; Domingues, M.R.; Coimbra, M.A. Coffee melanoidins: Structures, mechanisms of formation and potential health impacts. Food Funct. 2012, 3, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Nunes, F.M.; Coimbra, M.A. Role of hydroxycinnamates in coffee melanoidin formation. Phytochem. Rev. 2010, 9, 171–185. [Google Scholar] [CrossRef]
- Arnaud, M.J. Metabolism of caffeine and other components of coffee. In Caffeine, Coffee and Health; Garattini, S., Ed.; Raven: New York, NY, USA, 1993; pp. 43–95. [Google Scholar]
- Callahan, M.M.; Robertson, R.S.; Arnaud, M.J.; Branfman, A.R.; McComish, M.F.; Yesair, D.W. Human metabolism of [1-methyl-14C]- and [2-14C]caffeine after oral administration. Drug Metab. Dispos. 1982, 10, 417–423. [Google Scholar]
- Grosso, L.M.; Triche, E.; Benowitz, N.L.; Bracken, M.B. Prenatal caffeine assessment: Fetal and maternal biomarkers or self-reported intake? Ann. Epidemiol. 2008, 18, 172–178. [Google Scholar] [CrossRef]
- Sachse, K.T.; Jackson, E.K.; Wisniewski, S.R.; Gillespie, D.G.; Puccio, A.M.; Clark, R.S.; Dixon, C.E.; Kochanek, P.M. Increases in cerebrospinal fluid caffeine concentration are associated with favorable outcome after severe traumatic brain injury in humans. J. Cereb. Blood Flow Metab. 2008, 28, 395–401. [Google Scholar] [CrossRef]
- Scott, N.R.; Chakraborty, J.; Marks, V. Determination of caffeine, theophylline and theobromine in serum and saliva using high-performance liquid chromatography. Ann. Clin. Biochem. 1984, 21 Pt 2, 120–124. [Google Scholar] [CrossRef]
- Blanchard, J.; Sawers, S.J. The absolute bioavailability of caffeine in man. Eur. J. Clin. Pharmacol. 1983, 24, 93–98. [Google Scholar] [CrossRef]
- Bonati, M.; Latini, R.; Galletti, F.; Young, J.F.; Tognoni, G.; Garattini, S. Caffeine disposition after oral doses. Clin. Pharmacol. Ther. 1982, 32, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.; Broughton, L.J.; Lind, M.J.; Morrison, P.J.; Rogers, H.J.; Bradbrook, I.D. Plasma and salivary pharmacokinetics of caffeine in man. Eur. J. Clin. Pharmacol. 1981, 21, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Brachtel, D.; Richter, E. Effect of altered gastric emptying on caffeine absorption. Z. Gastroenterol. 1988, 26, 245–251. [Google Scholar] [PubMed]
- Trang, J.M.; Blanchard, J.; Conrad, K.A.; Harrison, G.G. Relationship between total body clearance of caffeine and urine flow rate in elderly men. Biopharm. Drug Dispos. 1985, 6, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.A.; Dirix, L.Y.; Hayllar, K.M.; Preisig, R.; Tredger, J.M.; Williams, R. Use of quantitative liver function tests—Caffeine clearance and galactose elimination capacity—After orthotopic liver transplantation. J. Hepatol. 1990, 10, 149–157. [Google Scholar] [CrossRef]
- Levy, M.; Granit, L.; Zylber-Katz, E. Chronopharmacokinetics of caffeine in healthy volunteers. Annu. Rev. Chronopharmacol. 1984, 1, 97–100. [Google Scholar]
- Grant, D.M.; Tang, B.K.; Kalow, W. Polymorphic N-acetylation of a caffeine metabolite. Clin. Pharmacol. Ther. 1983, 33, 355–359. [Google Scholar] [CrossRef]
- Ha, H.R.; Chen, J.; Krahenbuhl, S.; Follath, F. Biotransformation of caffeine by cDNA-expressed human cytochromes P-450. Eur. J. Clin. Pharmacol. 1996, 49, 309–315. [Google Scholar] [CrossRef]
- Rasmussen, B.B.; Brix, T.H.; Kyvik, K.O.; Brøsen, K. The interindividual differences in the 3-demthylation of caffeine alias CYP1A2 is determined by both genetic and environmental factors. Pharmacogenetics 2002, 12, 473–478. [Google Scholar] [CrossRef]
- Vistisen, K.; Loft, S.; Poulsen, H.E. Cytochrome P450 IA2 activity in man measured by caffeine metabolism: Effect of smoking, broccoli and exercise. Adv. Exp. Med. Biol. 1991, 283, 407–411. [Google Scholar]
- Sasaki, S.; Limpar, M.; Sata, F.; Kobayashi, S.; Kishi, R. Interaction between maternal caffeine intake during pregnancy and CYP1A2 C164A polymorphism affects infant birth size in the Hokkaido study. Pediatr. Res. 2017, 82, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, W.H.; Chen, B.L.; Fan, L.; Han, Y.; Wang, G.; Hu, D.L.; Tan, Z.R.; Zhou, G.; Cao, S.; et al. Plant polyphenol curcumin significantly affects CYP1A2 and CYP2A6 activity in healthy, male Chinese volunteers. Ann. Pharmacother. 2010, 44, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xiao, P.; Ou-Yang, D.S.; Fan, L.; Guo, D.; Wang, Y.N.; Han, Y.; Tu, J.H.; Zhou, G.; Huang, Y.F.; et al. Simultaneous action of the flavonoid quercetin on cytochrome P450 (CYP) 1A2, CYP2A6, N-acetyltransferase and xanthine oxidase activity in healthy volunteers. Clin. Exp. Pharmacol. Physiol. 2009, 36, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, U.; Klittich, K.; Staib, A.H. Inhibitory effect of grapefruit juice and its bitter principal, naringenin, on CYP1A2 dependent metabolism of caffeine in man. Br. J. Clin. Pharmacol. 1993, 35, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, U.; Maier, A.; Keller, A.; Steinijans, V.W.; Sauter, R.; Staib, A.H. Lacking effect of grapefruit juice on theophylline pharmacokinetics. Int. J. Clin. Pharmacol. Ther. 1995, 33, 311–314. [Google Scholar]
- Lampe, J.W.; King, I.B.; Li, S.; Grate, M.T.; Barale, K.V.; Chen, C.; Feng, Z.; Potter, J.D. Brassica vegetables increase and apiaceous vegetables decrease cytochrome P450 1A2 activity in humans: Changes in caffeine metabolite ratios in response to controlled vegetable diets. Carcinogenesis 2000, 21, 1157–1162. [Google Scholar] [CrossRef]
- Faber, M.S.; Fuhr, U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin. Pharmacol. Ther. 2004, 76, 178–184. [Google Scholar] [CrossRef]
- Jodynis-Liebert, J.; Flieger, J.; Matuszewska, A.; Juszczyk, J. Serum metabolite/caffeine ratios as a test for liver function. J. Clin. Pharmacol. 2004, 44, 338–347. [Google Scholar] [CrossRef]
- Park, G.J.; Katelaris, P.H.; Jones, D.B.; Seow, F.; Le Couteur, D.G.; Ngu, M.C. Validity of the 13C-caffeine breath test as a noninvasive, quantitative test of liver function. Hepatology 2003, 38, 1227–1236. [Google Scholar] [CrossRef]
- Backman, J.T.; Karjalainen, M.J.; Neuvonen, M.; Laitila, J.; Neuvonen, P.J. Rofecoxib is a potent inhibitor of cytochrome P450 1A2: Studies with tizanidine and caffeine in healthy subjects. Br. J. Clin. Pharmacol. 2006, 62, 345–357. [Google Scholar] [CrossRef]
- Carrillo, J.A.; Benitez, J. Clinically significant pharmacokinetic interactions between dietary caffeine and medications. Clin. Pharmacokinet. 2000, 39, 127–153. [Google Scholar] [CrossRef] [PubMed]
- Doude van Troostwijk, L.J.; Koopmans, R.P.; Vermeulen, H.D.; Guchelaar, H.J. CYP1A2 activity is an important determinant of clozapine dosage in schizophrenic patients. Eur. J. Pharm. Sci. 2003, 20, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, U.; Anders, E.M.; Mahr, G.; Sörgel, F.; Staib, A.H. Inhibitory potency of quinolone antibacterial agents against cytochrome P450IA2 activity in vivo and in vitro. Antimicrob. Agents Chemother. 1992, 36, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.; Sawers, S.J. Comparative pharmacokinetics of caffeine in young and elderly men. J. Pharmacokinet. Biopharm. 1983, 11, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.; Sawers, S.J. Relationship between urine flow rate and renal clearance of caffeine in man. J. Clin. Pharmacol. 1983, 23, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Tang-Liu, D.D.; Williams, R.L.; Riegelman, S. Disposition of caffeine and its metabolites in man. J. Pharmacol. Exp. Ther. 1983, 224, 180–185. [Google Scholar]
- Clifford, M.N. Chlorogenic acids and other cinnamates—nature, occurrence and dietary burden. J. Sci. Food Agric. 1999, 79, 362–372. [Google Scholar] [CrossRef]
- Naveed, M.; Hejazi, V.; Abbas, M.; Kamboh, A.A.; Khan, G.J.; Shumzaid, M.; Ahmad, F.; Babazadeh, D.; FangFang, X.; Modarresi-Ghazani, F.; et al. Chlorogenic acid (CGA): A pharmacological review and call for further research. Biomed. Pharmacother. 2018, 97, 67–74. [Google Scholar] [CrossRef]
- Clifford, M.N.; Jaganath, I.B.; Ludwig, I.A.; Crozier, A. Chlorogenic acids and the acyl-quinic acids: Discovery, biosynthesis, bioavailability and bioactivity. Nat. Prod. Rep. 2017, 34, 1391–1421. [Google Scholar] [CrossRef]
- Renouf, M.; Guy, P.A.; Marmet, C.; Fraering, A.L.; Longet, K.; Moulin, J.; Enslen, M.; Barron, D.; Dionisi, F.; Cavin, C.; et al. Measurement of caffeic and ferulic acid equivalents in plasma after coffee consumption: Small intestine and colon are key sites for coffee metabolism. Mol. Nutr. Food Res. 2010, 54, 760–766. [Google Scholar] [CrossRef]
- Stalmach, A.; Mullen, W.; Barron, D.; Uchida, K.; Yokota, T.; Cavin, C.; Steiling, H.; Williamson, G.; Crozier, A. Metabolite profiling of hydroxycinnamate derivatives in plasma and urine after the ingestion of coffee by humans: Identification of biomarkers of coffee consumption. Drug Metab. Dispos. 2009, 37, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Farrell, T.L.; Dew, T.P.; Poquet, L.; Hanson, P.; Williamson, G. Absorption and metabolism of chlorogenic acids in cultured gastric epithelial monolayers. Drug Metab. Dispos. 2011, 39, 2338–2346. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Kobayashi, S. Transepithelial transport of chlorogenic acid, caffeic acid, and their colonic metabolites in intestinal caco-2 cell monolayers. J. Agric. Food Chem. 2004, 52, 2518–2526. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Moghadasian, M.H. Bioavailability of hydroxycinnamates: A brief review of in vivo and in vitro studies. Phytochem. Rev. Vol. 2010, 9, 133–145. [Google Scholar] [CrossRef]
- Erk, T.; Williamson, G.; Renouf, M.; Marmet, C.; Steiling, H.; Dionisi, F.; Barron, D.; Melcher, R.; Richling, E. Dose-dependent absorption of chlorogenic acids in the small intestine assessed by coffee consumption in ileostomists. Mol. Nutr. Food Res. 2012, 56, 1488–1500. [Google Scholar] [CrossRef]
- Clifford, M.N.; Kerimi, A.; Williamson, G. Bioavailability and metabolism of chlorogenic acids (acyl-quinic acids) in humans. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1299–1352. [Google Scholar] [CrossRef]
- Farah, A.; Monteiro, M.; Donangelo, C.M.; Lafay, S. Chlorogenic acids from green coffee extract are highly bioavailable in humans. J. Nutr. 2008, 138, 2309–2315. [Google Scholar] [CrossRef]
- Matsui, Y.; Nakamura, S.; Kondou, N.; Takasu, Y.; Ochiai, R.; Masukawa, Y. Liquid chromatography-electrospray ionization-tandem mass spectrometry for simultaneous analysis of chlorogenic acids and their metabolites in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 858, 96–105. [Google Scholar] [CrossRef]
- Stalmach, A.; Steiling, H.; Williamson, G.; Crozier, A. Bioavailability of chlorogenic acids following acute ingestion of coffee by humans with an ileostomy. Arch. Biochem. Biophys. 2010, 501, 98–105. [Google Scholar] [CrossRef]
- Wong, C.C.; Meinl, W.; Glatt, H.R.; Barron, D.; Stalmach, A.; Steiling, H.; Crozier, A.; Williamson, G. In vitro and in vivo conjugation of dietary hydroxycinnamic acids by UDP-glucuronosyltransferases and sulfotransferases in humans. J. Nutr. Biochem. 2010, 21, 1060–1068. [Google Scholar] [CrossRef]
- Olthof, M.R.; Hollman, P.C.; Katan, M.B. Chlorogenic acid and caffeic acid are absorbed in humans. J. Nutr. 2001, 131, 66–71. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Williamson, G. Chocolate: Modern science investigates an ancient medicine. J. Med. Food. 2000, 3, 121–125. [Google Scholar]
- Oliveira, D.M.; Bastos, D.H.M. Phenolic acids bioavailability. Quim. Nova 2011, 34, 1051–1056. [Google Scholar] [CrossRef]
- Rechner, A.R.; Smith, M.A.; Kuhnle, G.; Gibson, G.R.; Debnam, E.S.; Srai, S.K.; Moore, K.P.; Rice-Evans, C.A. Colonic metabolism of dietary polyphenols: Influence of structure on microbial fermentation products. Free Radic. Biol. Med. 2004, 36, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, L.; Tassotti, M.; Rosi, A.; Martini, D.; Antonini, M.; Dei Cas, A.; Bonadonna, R.; Brighenti, F.; Del Rio, D.; Mena, P. Absorption, pharmacokinetics, and urinary excretion of pyridines after consumption of coffee and cocoa-based products containing coffee in a repeated dose, crossover human intervention study. Mol. Nutr. Food Res. 2020, 64, e2000489. [Google Scholar] [CrossRef] [PubMed]
- Midttun, Ø.; Ulvik, A.; Nygård, O.; Ueland, P.M. Performance of plasma trigonelline as a marker of coffee consumption in an epidemiologic setting. Am. J. Clin. Nutr. 2018, 107, 941–947. [Google Scholar] [CrossRef]
- Lang, R.; Wahl, A.; Skurk, T.; Yagar, E.F.; Schmiech, L.; Eggers, R.; Hauner, H.; Hofmann, T. Development of a hydrophilic liquid interaction chromatography-high-performance liquid chromatography-tandem mass spectrometry based stable isotope dilution analysis and pharmacokinetic studies on bioactive pyridines in human plasma and urine after coffee consumption. Anal. Chem. 2010, 82, 1486–1497. [Google Scholar]
- Lang, R.; Dieminger, N.; Beusch, A.; Lee, Y.M.; Dunkel, A.; Suess, B.; Skurk, T.; Wahl, A.; Hauner, H.; Hofmann, T. Bioappearance and pharmacokinetics of bioactives upon coffee consumption. Anal. Bioanal. Chem. 2013, 405, 8487–8503. [Google Scholar] [CrossRef]
- Yuyama, S.; Kawano, Y. Urinary excretion of N1-methyl-2-pyridone-5-carboxylic acid and the fate of remaining of trigonelline. Adv. Exp. Med. Biol. 1996, 398, 599–603. [Google Scholar]
- Yuyama, S.; Suzuki, T. The excretion of N1-methyl-2-pyridone-5-carboxylic acid and related compounds in human subjects after oral administration of nicotinic acid, trigonelline and N1-methyl-2-pyridone-5-carboxylic acid. Adv. Exp. Med. Biol. 1991, 294, 475–479. [Google Scholar] [PubMed]
- De Roos, B.; Meyboom, S.; Kosmeijer-Schuil, T.G.; Katan, M.B. Absorption and urinary excretion of the coffee diterpenes cafestol and kahweol in healthy ileostomy volunteers. J. Intern. Med. 1998, 244, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Prince, M. World Alzheimer Report 2015: The Global Impact of Dementia. Available online: https://www.alz.co.uk/research/world-report-2015 (accessed on 8 April 2018).
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef]
- Dening, T.; Sandilyan, M.B. Dementia: Definitions and types. Nurs. Stand. 2015, 29, 37–42. [Google Scholar] [CrossRef]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 2011, 3, 1–13. [Google Scholar] [CrossRef]
- Terry, R.D.; Peck, A.; DeTeresa, R.; Schechter, R.; Horoupian, D.S. Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann. Neurol. 1981, 10, 184–192. [Google Scholar] [CrossRef]
- Teipel, S.J.; Flatz, W.H.; Heinsen, H.; Bokde, A.L.; Schoenberg, S.O.; Stöckel, S.; Dietrich, O.; Reiser, M.F.; Möller, H.J.; Hampel, H. Measurement of basal forebrain atrophy in Alzheimer’s disease using MRI. Brain 2005, 128 Pt 11, 2626–2644. [Google Scholar] [CrossRef]
- Klucken, J.; McLean, P.J.; Gomez-Tortosa, E.; Ingelsson, M.; Hyman, B.T. Neuritic alterations and neural system dysfunction in Alzheimer’s disease and dementia with Lewy bodies. Neurochem. Res. 2003, 28, 1683–1691. [Google Scholar] [CrossRef]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef]
- Grutzendler, J.; Helmin, K.; Tsai, J.; Gan, W.B. Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2007, 1097, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Lippa, C.F.; Hamos, J.E.; Pulaski-Salo, D.; DeGennaro, L.J.; Drachman, D.A. Alzheimer’s disease and aging: Effects on perforant pathway perikarya and synapses. Neurobiol. Aging 1992, 13, 405–411. [Google Scholar] [CrossRef]
- Perlson, E.; Maday, S.; Fu, M.M.; Moughamian, A.J.; Holzbaur, E.L. Retrograde axonal transport: Pathways to cell death? Trends Neurosci. 2010, 33, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Ingelsson, M.; Fukumoto, H.; Newell, K.L.; Growdon, J.H.; Hedley-Whyte, E.T.; Frosch, M.P.; Albert, M.S.; Hyman, B.T.; Irizarry, M.C. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 2004, 62, 925–931. [Google Scholar] [CrossRef]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Lashley, T.; Rohrer, J.D.; Mead, S.; Revesz, T. Review: An update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol. 2015, 41, 858–881. [Google Scholar] [CrossRef]
- Contestabile, A. The history of the cholinergic hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Cervellati, C.; Wood, P.L.; Romani, A.; Valacchi, G.; Squerzanti, M.; Sanz, J.M.; Ortolani, B.; Zuliani, G. Oxidative challenge in Alzheimer’s disease: State of knowledge and future needs. J. Investig. Med. 2016, 64, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, L.Y.; Venneri, A.; Farkas, E.; Evans, P.C.; Marzo, A.; Frangi, A.F. Vascular dysfunction in the pathogenesis of Alzheimer’s disease—A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol. Dis. 2015, 82, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.; Pike, C.J. Menopause, obesity and inflammation: Interactive risk factors for Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Patraca, I.; Martínez, N.; Pedrós, I.; Petrov, D.; Ettcheto, M.; Abad, S.; Marin, M.; Beas-Zarate, C.; Camins, A. The role of leptin in the sporadic form of Alzheimer’s disease. Interactions with the adipokines amylin, ghrelin and the pituitary hormone prolactin. Life Sci. 2015, 140, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Carrillo-de Sauvage, M.A.; Ceyzériat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Bagheri, H.; Damase-Michel, C.; Lapeyre-Mestre, M.; Cismondo, S.; O’Connell, D.; Senard, J.M.; Rascol, O.; Montastruc, J.L. A study of salivary secretion in Parkinson’s disease. Clin. Neuropharmacol. 1999, 22, 213–215. [Google Scholar]
- Park, J.H.; Kang, Y.J.; Horak, F.B. What is wrong with balance in Parkinson’s disease? J. Mov. Disord. 2015, 8, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.G.; Landau, S.; Hindle, J.V.; Playfer, J.; Samuel, M.; Wilson, K.C.; Hurt, C.S.; Anderson, R.J.; Carnell, J.; Dickinson, L.; et al. Depression and anxiety related subtypes in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2011, 82, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Hardoff, R.; Sula, M.; Tamir, A.; Soil, A.; Front, A.; Badarna, S.; Honigman, S.; Giladi, N. Gastric emptying time and gastric motility in patients with Parkinson’s disease. Mov. Disord. 2001, 16, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15 (Suppl. 1), 14–20. [Google Scholar] [CrossRef]
- Senard, J.M.; Raï, S.; Lapeyre-Mestre, M.; Brefel, C.; Rascol, O.; Rascol, A.; Montastruc, J.L. Prevalence of orthostatic hypotension in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 63, 584–589. [Google Scholar] [CrossRef]
- Uchiyama, T.; Sakakibara, R.; Yamamoto, T.; Ito, T.; Yamaguchi, C.; Awa, Y.; Yanagisawa, M.; Higuchi, Y.; Sato, Y.; Ichikawa, T.; et al. Urinary dysfunction in early and untreated Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1382–1386. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The alpha-synuclein burden hypothesis of Parkinson disease and its relationship to Alzheimer disease. Exp. Neurol. 2008, 212, 235–238. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson disease in 2015: Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Mullin, S.; Schapira, A.H. Pathogenic mechanisms of neurodegeneration in Parkinson disease. Neurol. Clin. 2015, 33, 1–17. [Google Scholar] [CrossRef]
- Hornykiewicz, O.; Kish, S.J. Biochemical pathophysiology of Parkinson’s disease. Adv. Neurol. 1987, 45, 19–34. [Google Scholar] [PubMed]
- Hayes, M.T. Parkinson’s disease and parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.G.; Savitt, J.M. Parkinson’s disease. Med. Clin. N. Am. 2019, 103, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.; et al. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef]
- Cereda, C.; Carrera, E. Posterior cerebral artery territory infarctions. Front. Neurol. Neurosci. 2012, 30, 128–131. [Google Scholar]
- Jensen, M.B.; St Louis, E.K. Management of acute cerebellar stroke. Arch. Neurol. 2005, 62, 537–544. [Google Scholar] [CrossRef]
- Kumral, E.; Bayulkem, G.; Evyapan, D.; Yunten, N. Spectrum of anterior cerebral artery territory infarction: Clinical and MRI findings. Eur. J. Neurol. 2002, 9, 615–624. [Google Scholar] [CrossRef]
- Wardlaw, J.M. What causes lacunar stroke? J. Neurol. Neurosurg. Psychiatry 2005, 76, 617–619. [Google Scholar] [CrossRef] [PubMed]
- White, H.; Boden-Albala, B.; Wang, C.; Elkind, M.S.; Rundek, T.; Wright, C.B.; Sacco, R.L. Ischemic stroke subtype incidence among whites, blacks, and Hispanics: The Northern Manhattan Study. Circulation 2005, 111, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A.; et al. Thrombectomy 6 to 24 h after stroke with a mismatch between deficit and infarct. N. Engl. J. Med. 2018, 378, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. 2018 Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2018, 49, e46–e110. [Google Scholar] [CrossRef] [PubMed]
- Sandercock, P.A.; Counsell, C.; Tseng, M.C.; Cecconi, E. Oral antiplatelet therapy for acute ischaemic stroke. Cochrane Database Syst. Rev. 2014, 2014, Cd000029. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Siesjö, B.K.; Katsura, K.I.; Kristián, T.; Li, P.A.; Siesjö, P. Molecular mechanisms of acidosis-mediated damage. Acta Neurochir. Suppl. 1996, 66, 8–14. [Google Scholar]
- Tombaugh, G.C.; Sapolsky, R.M. Evolving concepts about the role of acidosis in ischemic neuropathology. J. Neurochem. 1993, 61, 793–803. [Google Scholar] [CrossRef]
- Xiong, Z.G.; Zhu, X.M.; Chu, X.P.; Minami, M.; Hey, J.; Wei, W.L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. NeuroInflamm. 2019, 16, 142. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. Epigenetic mechanisms in stroke and epilepsy. Neuropsychopharmacology 2013, 38, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Xing, S.; Liang, Z.; Zeng, J. Secondary neurodegeneration in remote regions after focal cerebral infarction: A new target for stroke management? Stroke 2012, 43, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.A.; Galecki, A.T.; Langa, K.M.; Unverzagt, F.W.; Kabeto, M.U.; Giordani, B.; Wadley, V.G. Trajectory of cognitive decline after incident stroke. JAMA 2015, 314, 41–51. [Google Scholar] [CrossRef]
- Hervé, D.; Molko, N.; Pappata, S.; Buffon, F.; LeBihan, D.; Bousser, M.G.; Chabriat, H. Longitudinal thalamic diffusion changes after middle cerebral artery infarcts. J. Neurol. Neurosurg. Psychiatry 2005, 76, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ling, X.; Liu, S.; Xu, A.; Zhang, Y.; Xing, S.; Pei, Z.; Zeng, J. Early detection of secondary damage in ipsilateral thalamus after acute infarction at unilateral corona radiata by diffusion tensor imaging and magnetic resonance spectroscopy. BMC Neurol. 2011, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Nakane, M.; Tamura, A.; Sasaki, Y.; Teraoka, A. MRI of secondary changes in the thalamus following a cerebral infarct. Neuroradiology 2002, 44, 915–920. [Google Scholar] [PubMed]
- Aho, L.; Jolkkonen, J.; Alafuzoff, I. Beta-amyloid aggregation in human brains with cerebrovascular lesions. Stroke 2006, 37, 2940–2945. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.K.; Zhao, Z.; Kluge, M.; Walker, F.R.; Nilsson, M. Chronic stress exposure following photothrombotic stroke is associated with increased levels of Amyloid beta accumulation and altered oligomerisation at sites of thalamic secondary neurodegeneration in mice. J. Cereb. Blood Flow Metab. 2017, 37, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Mitkari, B.; Kerkelä, E.; Nystedt, J.; Korhonen, M.; Jolkkonen, J. Unexpected complication in a rat stroke model: Exacerbation of secondary pathology in the thalamus by subacute intraarterial administration of human bone marrow-derived mesenchymal stem cells. J. Cereb. Blood Flow Metab. 2015, 35, 363–366. [Google Scholar] [CrossRef]
- Zhang, Y.; Xing, S.; Zhang, J.; Li, J.; Li, C.; Pei, Z.; Zeng, J. Reduction of β-amyloid deposits by γ-secretase inhibitor is associated with the attenuation of secondary damage in the ipsilateral thalamus and sensory functional improvement after focal cortical infarction in hypertensive rats. J. Cereb. Blood Flow Metab. 2011, 31, 572–579. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Li, J.; Xing, S.; Li, C.; Li, Y.; Dang, C.; Fan, Y.; Yu, J.; Pei, Z.; et al. Autophagosomes accumulation is associated with β-amyloid deposits and secondary damage in the thalamus after focal cortical infarction in hypertensive rats. J. Neurochem. 2012, 120, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Sarajärvi, T.; Lipsanen, A.; Mäkinen, P.; Peräniemi, S.; Soininen, H.; Haapasalo, A.; Jolkkonen, J.; Hiltunen, M. Bepridil decreases Aβ and calcium levels in the thalamus after middle cerebral artery occlusion in rats. J. Cell Mol. Med. 2012, 16, 2754–2767. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E. The Epidemiology of Epilepsy. Neuroepidemiology 2020, 54, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Slater, N.; Perkins, A. Epilepsy: Treatment options. Am. Fam. Physician 2017, 96, 87–96. [Google Scholar] [PubMed]
- Blümcke, I.; Thom, M.; Aronica, E.; Armstrong, D.D.; Bartolomei, F.; Bernasconi, A.; Bernasconi, N.; Bien, C.G.; Cendes, F.; Coras, R.; et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: A Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia 2013, 54, 1315–1329. [Google Scholar] [CrossRef] [PubMed]
- De Lanerolle, N.C.; Lee, T.S.; Spencer, D.D. Histopathology of human epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology: Bethesda, MD, USA, 2010. [Google Scholar]
- Thom, M. Review: Hippocampal sclerosis in epilepsy: A neuropathology review. Neuropathol. Appl. Neurobiol. 2014, 40, 520–543. [Google Scholar] [CrossRef] [PubMed]
- Briellmann, R.S.; Berkovic, S.F.; Syngeniotis, A.; King, M.A.; Jackson, G.D. Seizure-associated hippocampal volume loss: A longitudinal magnetic resonance study of temporal lobe epilepsy. Ann. Neurol. 2002, 51, 641–644. [Google Scholar] [CrossRef]
- Jackson, G.D.; Berkovic, S.F.; Tress, B.M.; Kalnins, R.M.; Fabinyi, G.C.; Bladin, P.F. Hippocampal sclerosis can be reliably detected by magnetic resonance imaging. Neurology 1990, 40, 1869–1875. [Google Scholar] [CrossRef]
- Henshall, D.C. Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem. Soc. Trans. 2007, 35 Pt 2, 421–423. [Google Scholar] [CrossRef]
- Peltola, J.; Palmio, J.; Korhonen, L.; Suhonen, J.; Miettinen, A.; Hurme, M.; Lindholm, D.; Keränen, T. Interleukin-6 and interleukin-1 receptor antagonist in cerebrospinal fluid from patients with recent tonic-clonic seizures. Epilepsy Res. 2000, 41, 205–211. [Google Scholar] [CrossRef]
- Ravizza, T.; Gagliardi, B.; Noé, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef]
- De Lanerolle, N.C.; Lee, T.S. New facets of the neuropathology and molecular profile of human temporal lobe epilepsy. Epilepsy Behav. 2005, 7, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.A.; da Costa Araújo, S.; Redeker, S.; van Schaik, R.; Aronica, E.; Gorter, J.A. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007, 130 Pt 2, 521–534. [Google Scholar] [CrossRef]
- Bae, J.M. History of coffee consumption and risk of Alzheimer’s disease: A meta-epidemiological study of population-based cohort studies. Dement. Neurocogn. Disord. 2020, 19, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Kolahdouzan, M.; Hamadeh, M.J. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 272–290. [Google Scholar] [CrossRef] [PubMed]
- Wasim, S.; Kukkar, V.; Awad, V.M.; Sakhamuru, S.; Malik, B.H. Neuroprotective and neurodegenerative aspects of coffee and its active ingredients in view of scientific literature. Cureus 2020, 12, e9578. [Google Scholar] [PubMed]
- Colombo, R.; Papetti, A. An outlook on the role of decaffeinated coffee in neurodegenerative diseases. Crit. Rev. Food Sci. Nutr. 2020, 60, 760–779. [Google Scholar] [CrossRef] [PubMed]
- Patil, H.; Lavie, C.J.; O’Keefe, J.H. Cuppa joe: Friend or foe? Effects of chronic coffee consumption on cardiovascular and brain health. Mo. Med. 2011, 108, 431–438. [Google Scholar]
- Fredholm, B.B.; Bättig, K.; Holmén, J.; Nehlig, A.; Zvartau, E.E. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol. Rev. 1999, 51, 83–133. [Google Scholar]
- McCusker, R.R.; Goldberger, B.A.; Cone, E.J. Caffeine content of specialty coffees. J. Anal. Toxicol. 2003, 27, 520–522. [Google Scholar] [CrossRef]
- Smith, A. Effects of caffeine on human behavior. Food Chem. Toxicol. 2002, 40, 1243–1255. [Google Scholar] [CrossRef]
- van Gelder, B.M.; Buijsse, B.; Tijhuis, M.; Kalmijn, S.; Giampaoli, S.; Nissinen, A.; Kromhout, D. Coffee consumption is inversely associated with cognitive decline in elderly European men: The FINE Study. Eur. J. Clin. Nutr. 2007, 61, 226–232. [Google Scholar] [CrossRef]
- Devasagayam, T.P.; Kamat, J.P.; Mohan, H.; Kesavan, P.C. Caffeine as an antioxidant: Inhibition of lipid peroxidation induced by reactive oxygen species. Biochim. Biophys. Acta 1996, 1282, 63–70. [Google Scholar] [CrossRef]
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine prevents d-galactose-induced cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem. Int. 2015, 90, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Molinengo, L.; Scordo, I.; Pastorello, B. Action of caffeine, L-PIA and their combination on memory retention in the rat. Life Sci. 1994, 54, 1247–1250. [Google Scholar] [CrossRef]
- Costa, M.S.; Botton, P.H.; Mioranzza, S.; Souza, D.O.; Porciúncula, L.O. Caffeine prevents age-associated recognition memory decline and changes brain-derived neurotrophic factor and tirosine kinase receptor (TrkB) content in mice. Neuroscience 2008, 153, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 2006, 142, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Mori, T.; Cao, C.; Mamcarz, M.; Runfeldt, M.; Dickson, A.; Rezai-Zadeh, K.; Tane, J.; Citron, B.A.; Lin, X.; et al. Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer’s disease mice. J. Alzheimers Dis. 2009, 17, 661–680. [Google Scholar] [CrossRef]
- Dall’Igna, O.P.; Porciúncula, L.O.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Neuroprotection by caffeine and adenosine A2A receptor blockade of beta-amyloid neurotoxicity. Br. J. Pharmacol. 2003, 138, 1207–1209. [Google Scholar] [CrossRef] [PubMed]
- Dall’Igna, O.P.; Fett, P.; Gomes, M.W.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25-35)-induced cognitive deficits in mice. Exp. Neurol. 2007, 203, 241–245. [Google Scholar] [CrossRef]
- Giunta, S.; Andriolo, V.; Castorina, A. Dual blockade of the A1 and A2A adenosine receptor prevents amyloid beta toxicity in neuroblastoma cells exposed to aluminum chloride. Int. J. Biochem. Cell Biol. 2014, 54, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.R.; Wilhelm, E.A.; Jesse, C.R.; Brandăo, R.; Nogueira, C.W. Protective effect of caffeine and a selective A2A receptor antagonist on impairment of memory and oxidative stress of aged rats. Exp. Gerontol. 2011, 46, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Müller, C.E.; Buée, L.; et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 2014, 35, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Prasanthi, J.R.; Dasari, B.; Marwarha, G.; Larson, T.; Chen, X.; Geiger, J.D.; Ghribi, O. Caffeine protects against oxidative stress and Alzheimer’s disease-like pathology in rabbit hippocampus induced by cholesterol-enriched diet. Free Radic. Biol. Med. 2010, 49, 1212–1220. [Google Scholar] [CrossRef]
- Han, M.E.; Kim, H.J.; Lee, Y.S.; Kim, D.H.; Choi, J.T.; Pan, C.S.; Yoon, S.; Baek, S.Y.; Kim, B.S.; Kim, J.B.; et al. Regulation of cerebrospinal fluid production by caffeine consumption. BMC Neurosci. 2009, 10, 1–12. [Google Scholar] [CrossRef]
- Wostyn, P.; Van, D.D.; Audenaert, K.; De Deyn, P.P. Increased cerebrospinal fluid production as a possible mechanism underlying caffeine’s protective effect against Alzheimer’s disease. Int. J. Alzheimers Dis. 2011, 2011, 617420. [Google Scholar] [CrossRef]
- Bagga, P.; Chugani, A.N.; Patel, A.B. Neuroprotective effects of caffeine in MPTP model of Parkinson’s disease: A (13)C NMR study. Neurochem. Int. 2016, 92, 25–34. [Google Scholar] [CrossRef]
- Chen, X.; Lan, X.; Roche, I.; Liu, R.; Geiger, J.D. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J. Neurochem. 2008, 107, 1147–1157. [Google Scholar] [CrossRef]
- Gevaerd, M.S.; Takahashi, R.N.; Silveira, R.; Da, C.C. Caffeine reverses the memory disruption induced by intra-nigral MPTP-injection in rats. Brain Res. Bull. 2001, 55, 101–106. [Google Scholar] [CrossRef]
- Singh, S.; Singh, K.; Gupta, S.P.; Patel, D.K.; Singh, V.K.; Singh, R.K.; Singh, M.P. Effect of caffeine on the expression of cytochrome P450 1A2, adenosine A2A receptor and dopamine transporter in control and 1-methyl 4-phenyl 1, 2, 3, 6-tetrahydropyridine treated mouse striatum. Brain Res. 2009, 1283, 115–126. [Google Scholar] [CrossRef]
- Xu, K.; Xu, Y.; Brown-Jermyn, D.; Chen, J.F.; Ascherio, A.; Dluzen, D.E.; Schwarzschild, M.A. Estrogen prevents neuroprotection by caffeine in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J. Neurosci. 2006, 26, 535–541. [Google Scholar] [CrossRef]
- Xu, K.; Xu, Y.H.; Chen, J.F.; Schwarzschild, M.A. Neuroprotection by caffeine: Time course and role of its metabolites in the MPTP model of Parkinson’s disease. Neuroscience 2010, 167, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, L.M.; Nobre, H.V., Jr.; Macedo, D.S.; Oliveira, A.A.; Freitas, R.M.; Vasconcelos, S.M.; Cunha, G.M.; Sousa, F.C.; Viana, G.S. Neuroprotective effects of caffeine in the model of 6-hydroxydopamine lesion in rats. Pharmacol. Biochem. Behav. 2006, 84, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Joghataie, M.T.; Roghani, M.; Negahdar, F.; Hashemi, L. Protective effect of caffeine against neurodegeneration in a model of Parkinson’s disease in rat: Behavioral and histochemical evidence. Parkinsonism. Relat. Disord. 2004, 10, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.; Irizarry, M.C.; Schwarzschild, M.A. Caffeine protects against combined paraquat and maneb-induced dopaminergic neuron degeneration. Exp. Neurol. 2010, 223, 657–661. [Google Scholar] [CrossRef]
- Sonsalla, P.K.; Wong, L.Y.; Harris, S.L.; Richardson, J.R.; Khobahy, I.; Li, W.; Gadad, B.S.; German, D.C. Delayed caffeine treatment prevents nigral dopamine neuron loss in a progressive rat model of Parkinson’s disease. Exp. Neurol. 2012, 234, 482–487. [Google Scholar] [CrossRef]
- Machado-Filho, J.A.; Correia, A.O.; Montenegro, A.B.; Nobre, M.E.; Cerqueira, G.S.; Neves, K.R.; Naffah-Mazzacoratti, M.G.; Cavalheiro, E.A.; de Castro Brito, G.A.; de Barros Viana, G.S. Caffeine neuroprotective effects on 6-OHDA-lesioned rats are mediated by several factors, including pro-inflammatory cytokines and histone deacetylase inhibitions. Behav. Brain Res. 2014, 264, 116–125. [Google Scholar] [CrossRef]
- Chen, J.F.; Xu, K.; Petzer, J.P.; Staal, R.; Xu, Y.H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N., Jr.; Schwarzschild, M.A. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. 2001, 21, RC143. [Google Scholar] [CrossRef]
- Ikeda, K.; Kurokawa, M.; Aoyama, S.; Kuwana, Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J. Neurochem. 2002, 80, 262–270. [Google Scholar] [CrossRef]
- Pierri, M.; Vaudano, E.; Sager, T.; Englund, U. KW-6002 protects from MPTP induced dopaminergic toxicity in the mouse. Neuropharmacology 2005, 48, 517–524. [Google Scholar] [CrossRef]
- Xu, K.; Bastia, E.; Schwarzschild, M. Therapeutic potential of adenosine A(2A) receptor antagonists in Parkinson’s disease. Pharmacol. Ther. 2005, 105, 267–310. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Shen, H.Y.; Coelho, J.E.; Araújo, I.M.; Huang, Q.Y.; Day, Y.J.; Rebola, N.; Canas, P.M.; Rapp, E.K.; Ferrara, J.; et al. Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann. Neurol. 2008, 63, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Popoli, P.; Betto, P.; Reggio, R.; Ricciarello, G. Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur. J. Pharmacol. 1995, 287, 215–217. [Google Scholar] [CrossRef]
- Prediger, R.D. Effects of caffeine in Parkinson’s disease: From neuroprotection to the management of motor and non-motor symptoms. J. Alzheimers. Dis. 2010, 20 (Suppl. 1), S205–S220. [Google Scholar] [CrossRef]
- Morelli, M.; Carta, A.R.; Jenner, P. Adenosine A2A receptors and Parkinson’s disease. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 589–615. [Google Scholar]
- Carta, A.R.; Kachroo, A.; Schintu, N.; Xu, K.; Schwarzschild, M.A.; Wardas, J.; Morelli, M. Inactivation of neuronal forebrain A receptors protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurochem. 2009, 111, 1478–1489. [Google Scholar] [CrossRef]
- Rudolphi, K.A.; Keil, M.; Fastbom, J.; Fredholm, B.B. Ischaemic damage in gerbil hippocampus is reduced following upregulation of adenosine (A1) receptors by caffeine treatment. Neurosci. Lett. 1989, 103, 275–280. [Google Scholar] [CrossRef]
- Georgiev, V.; Johansson, B.; Fredholm, B.B. Long-term caffeine treatment leads to a decreased susceptibility to NMDA-induced clonic seizures in mice without changes in adenosine A1 receptor number. Brain Res. 1993, 612, 271–277. [Google Scholar] [CrossRef]
- Evans, S.M.; Pinto Pereira, L.M.; Addae, J.I. Neuroprotection by caffeine and pentoxifylline during experimental cerebral ischaemia. West Indian Med. J. 1999, 48, 23–25. [Google Scholar]
- Sutherland, G.R.; Peeling, J.; Lesiuk, H.J.; Brownstone, R.M.; Rydzy, M.; Saunders, J.K.; Geiger, J.D. The effects of caffeine on ischemic neuronal injury as determined by magnetic resonance imaging and histopathology. Neuroscience 1991, 42, 171–182. [Google Scholar] [CrossRef]
- Alexander, M.; Smith, A.L.; Rosenkrantz, T.S.; Fitch, R.H. Therapeutic effect of caffeine treatment immediately following neonatal hypoxic-ischemic injury on spatial memory in male rats. Brain Sci. 2013, 3, 177–190. [Google Scholar] [CrossRef]
- Kilicdag, H.; Daglioglu, Y.K.; Erdogan, S.; Zorludemir, S. Effects of caffeine on neuronal apoptosis in neonatal hypoxic-ischemic brain injury. J. Matern. Fetal Neonatal Med. 2014, 27, 1470–1475. [Google Scholar] [CrossRef]
- Potter, M.; Rosenkrantz, T.; Fitch, R.H. Behavioral and neuroanatomical outcomes in a rat model of preterm hypoxic-ischemic brain Injury: Effects of caffeine and hypothermia. Int. J. Dev. Neurosci. 2018, 70, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.; de Mendonça, A. Does caffeine intake protect from Alzheimer’s disease? Eur. J. Neurol. 2002, 9, 377–382. [Google Scholar] [CrossRef]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hébert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk factors for Alzheimer’s disease: A prospective analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife coffee and tea drinking and the risk of late-life dementia: A population-based CAIDE study. J. Alzheimers Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S167–S174. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, M.A.; Beydoun, H.A.; Gamaldo, A.A.; Teel, A.; Zonderman, A.B.; Wang, Y. Epidemiologic studies of modifiable factors associated with cognition and dementia: Systematic review and meta-analysis. BMC. Public Health 2014, 14, 643. [Google Scholar] [CrossRef]
- Santos, C.; Costa, J.; Santos, J.; Vaz-Carneiro, A.; Lunet, N. Caffeine intake and dementia: Systematic review and meta-analysis. J. Alzheimers. Dis. 2010, 20 (Suppl. 1), S187–S204. [Google Scholar] [CrossRef]
- Barranco Quintana, J.L.; Allam, M.F.; Serrano Del Castillo, A.; Fernández-Crehuet Navajas, R. Alzheimer’s disease and coffee: A quantitative review. Neurol. Res. 2007, 29, 91–95. [Google Scholar] [CrossRef]
- Crichton, G.E.; Bryan, J.; Murphy, K.J. Dietary antioxidants, cognitive function and dementia—A systematic review. Plant. Foods Hum. Nutr. 2013, 68, 279–292. [Google Scholar] [CrossRef]
- Di Marco, L.Y.; Marzo, A.; Munoz-Ruiz, M.; Ikram, M.A.; Kivipelto, M.; Ruefenacht, D.; Venneri, A.; Soininen, H.; Wanke, I.; Ventikos, Y.A.; et al. Modifiable lifestyle factors in dementia: A systematic review of longitudinal observational cohort studies. J. Alzheimers Dis. 2014, 42, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.; Carriére, I.; de Mendonca, A.; Portet, F.; Dartigues, J.F.; Rouaud, O.; Barberger-Gateau, P.; Ancelin, M.L. The neuroprotective effects of caffeine: A prospective population study (the Three City Study). Neurology 2007, 69, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kwak, S.M.; Myung, S.K. Caffeine intake from coffee or tea and cognitive disorders: A meta-analysis of observational studies. Neuroepidemiology 2015, 44, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; Lunet, N.; Santos, C.; Santos, J.; Vaz-Carneiro, A. Caffeine exposure and the risk of Parkinson’s disease: A systematic review and meta-analysis of observational studies. J. Alzheimers. Dis. 2010, 20 (Suppl. 1), S221–S238. [Google Scholar] [CrossRef] [PubMed]
- Hernán, M.A.; Takkouche, B.; Caamano-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson’s disease. Geriatr. Gerontol. Int. 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Hu, G.; Bidel, S.; Jousilahti, P.; Antikainen, R.; Tuomilehto, J. Coffee and tea consumption and the risk of Parkinson’s disease. Mov. Disord. 2007, 22, 2242–2248. [Google Scholar] [CrossRef]
- Liu, R.; Guo, X.; Park, Y.; Huang, X.; Sinha, R.; Freedman, N.D.; Hollenbeck, A.R.; Blair, A.; Chen, H. Caffeine intake, smoking, and risk of Parkinson disease in men and women. Am. J. Epidemiol. 2012, 175, 1200–1207. [Google Scholar] [CrossRef]
- Palacios, N.; Gao, X.; McCullough, M.L.; Schwarzschild, M.A.; Shah, R.; Gapstur, S.; Ascherio, A. Caffeine and risk of Parkinson’s disease in a large cohort of men and women. Mov. Disord. 2012, 27, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.D.; Lang, A.E.; Postuma, R.B. Caffeine in Parkinson’s disease: A pilot open-label, dose-escalation study. Mov. Disord. 2011, 26, 2427–2431. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Rios, R.S.; Altman, R.; et al. Caffeine for treatment of Parkinson disease: A randomized controlled trial. Neurology 2012, 79, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Anang, J.; Pelletier, A.; Joseph, L.; Moscovich, M.; Grimes, D.; Furtado, S.; Munhoz, R.P.; Appel-Cresswell, S.; Moro, A.; et al. Caffeine as symptomatic treatment for Parkinson disease (Café-PD): A randomized trial. Neurology 2017, 89, 1795–1803. [Google Scholar] [CrossRef]
- Tan, E.K.; Chua, E.; Fook-Chong, S.M.; Teo, Y.Y.; Yuen, Y.; Tan, L.; Zhao, Y. Association between caffeine intake and risk of Parkinson’s disease among fast and slow metabolizers. Pharmacogenet. Genom. 2007, 17, 1001–1005. [Google Scholar] [CrossRef]
- Fondell, E.; O’Reilly, É.I.J.; Fitzgerald, K.C.; Falcone, G.J.; Kolonel, L.N.; Park, Y.; Gapstur, S.M.; Ascherio, A. Intakes of caffeine, coffee and tea and risk of amyotrophic lateral sclerosis: Results from five cohort studies. Amyotroph. Lateral. Scler. Front. Degener. 2015, 16, 366–371. [Google Scholar] [CrossRef]
- Larsson, S.C. Coffee, tea, and cocoa and risk of stroke. Stroke 2014, 45, 309–314. [Google Scholar] [CrossRef]
- Selb, S.J.; Selb, K. Coffee and alcohol consumption as triggering factors for sudden cardiac death: Case-crossover study. Croat. Med. J. 2004, 45, 775–780. [Google Scholar]
- Tavani, A.; Bertuzzi, M.; Negri, E.; Sorbara, L.; La, V.C. Alcohol, smoking, coffee and risk of non-fatal acute myocardial infarction in Italy. Eur. J. Epidemiol. 2001, 17, 1131–1137. [Google Scholar] [CrossRef]
- Smits, P.; Thien, T.; van’t Laar, A. Circulatory effects of coffee in relation to the pharmacokinetics of caffeine. Am. J. Cardiol. 1985, 56, 958–963. [Google Scholar] [CrossRef]
- Prineas, R.J.; Jacobs, D.R., Jr.; Crow, R.S.; Blackburn, H. Coffee, tea and VPB. J. Chronic. Dis. 1980, 33, 67–72. [Google Scholar] [CrossRef]
- Hartley, T.R.; Lovallo, W.R.; Whitsett, T.L. Cardiovascular effects of caffeine in men and women. Am. J. Cardiol. 2004, 93, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, E.; Rodriguez-Artalejo, F.; Rexrode, K.M.; Logroscino, G.; Hu, F.B.; van Dam, R.M. Coffee consumption and risk of stroke in women. Circulation 2009, 119, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Silletta, M.G.; Marfisi, R.; Levantesi, G.; Boccanelli, A.; Chieffo, C.; Franzosi, M.; Geraci, E.; Maggioni, A.P.; Nicolosi, G.; Schweiger, C.; et al. Coffee consumption and risk of cardiovascular events after acute myocardial infarction: Results from the GISSI (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico)-Prevenzione trial. Circulation 2007, 116, 2944–2951. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.A.; Chow, G.; Ziegelstein, R.C. Caffeinated coffee consumption, cardiovascular disease, and heart valve disease in the elderly (from the Framingham Study). Am. J. Cardiol. 2008, 102, 1502–1508. [Google Scholar] [CrossRef]
- Zhang, W.; Lopez-Garcia, E.; Li, T.Y.; Hu, F.B.; van Dam, R.M. Coffee consumption and risk of cardiovascular diseases and all-cause mortality among men with type 2 diabetes. Diabetes Care 2009, 32, 1043–1045. [Google Scholar] [CrossRef]
- Mukamal, K.J.; Hallqvist, J.; Hammar, N.; Ljung, R.; Gémes, K.; Ahlbom, A.; Ahnve, S.; Janszky, I. Coffee consumption and mortality after acute myocardial infarction: The Stockholm Heart Epidemiology Program. Am. Heart J. 2009, 157, 495–501. [Google Scholar] [CrossRef]
- De Koning Gans, J.M.; Uiterwaal, C.S.; van der Schouw, Y.T.; Boer, J.M.; Grobbee, D.E.; Verschuren, W.M.; Beulens, J.W. Tea and coffee consumption and cardiovascular morbidity and mortality. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1665–1671. [Google Scholar] [CrossRef]
- Ding, M.; Bhupathiraju, S.N.; Satija, A.; van Dam, R.M.; Hu, F.B. Long-term coffee consumption and risk of cardiovascular disease: A systematic review and a dose-response meta-analysis of prospective cohort studies. Circulation 2014, 129, 643–659. [Google Scholar] [CrossRef]
- Larsson, S.C.; Männistö, S.; Virtanen, M.J.; Kontto, J.; Albanes, D.; Virtamo, J. Coffee and tea consumption and risk of stroke subtypes in male smokers. Stroke 2008, 39, 1681–1687. [Google Scholar] [CrossRef]
- Kim, B.; Nam, Y.; Kim, J.; Choi, H.; Won, C. Coffee consumption and stroke risk: A meta-analysis of epidemiologic studies. Korean J. Fam. Med. 2012, 33, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Mostofsky, E.; Schlaug, G.; Mukamal, K.J.; Rosamond, W.D.; Mittleman, M.A. Coffee and acute ischemic stroke onset: The Stroke Onset Study. Neurology 2010, 75, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Washio, T.; Sasaki, H.; Ogoh, S. Acute impact of drinking coffee on the cerebral and systemic vasculature. Physiol. Rep. 2017, 5, e13288. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Orsini, N. Coffee consumption and risk of stroke: A dose-response meta-analysis of prospective studies. Am. J. Epidemiol. 2011, 174, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Strong, R.; Grotta, J.C.; Aronowski, J. Combination of low dose ethanol and caffeine protects brain from damage produced by focal ischemia in rats. Neuropharmacology 2000, 39, 515–522. [Google Scholar] [CrossRef]
- Aronowski, J.; Strong, R.; Shirzadi, A.; Grotta, J.C. Ethanol plus caffeine (caffeinol) for treatment of ischemic stroke: Preclinical experience. Stroke 2003, 34, 1246–1251. [Google Scholar] [CrossRef]
- Piriyawat, P.; Labiche, L.A.; Burgin, W.S.; Aronowski, J.A.; Grotta, J.C. Pilot dose-escalation study of caffeine plus ethanol (caffeinol) in acute ischemic stroke. Stroke 2003, 34, 1242–1245. [Google Scholar] [CrossRef]
- Martin-Schild, S.; Hallevi, H.; Shaltoni, H.; Barreto, A.D.; Gonzales, N.R.; Aronowski, J.; Savitz, S.I.; Grotta, J.C. Combined neuroprotective modalities coupled with thrombolysis in acute ischemic stroke: A pilot study of caffeinol and mild hypothermia. J. Stroke Cerebrovasc. Dis. 2009, 18, 86–96. [Google Scholar] [CrossRef]
- Zhao, X.; Strong, R.; Piriyawat, P.; Palusinski, R.; Grotta, J.C.; Aronowski, J. Caffeinol at the receptor level: Anti-ischemic effect of N-methyl-D-aspartate receptor blockade is potentiated by caffeine. Stroke 2010, 41, 363–367. [Google Scholar] [CrossRef]
- Heitman, E.; Ingram, D.K. Cognitive and neuroprotective effects of chlorogenic acid. Nutr. Neurosci. 2017, 20, 32–39. [Google Scholar] [CrossRef]
- Tajik, N.; Tajik, M.; Mack, I.; Enck, P. The potential effects of chlorogenic acid, the main phenolic components in coffee, on health: A comprehensive review of the literature. Eur. J. Nutr. 2017, 56, 2215–2244. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Tejada, S.; Setzer, W.N.; Gortzi, O.; Sureda, A.; Braidy, N.; Daglia, M.; Manayi, A.; Nabavi, S.M. Chlorogenic acid and mental diseases: From hhemistry to medicine. Curr. Neuropharmacol. 2017, 15, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Jang, Y.J.; Hwang, M.K.; Kang, N.J.; Lee, K.W.; Lee, H.J. Attenuation of oxidative neuronal cell death by coffee phenolic phytochemicals. Mutat. Res. 2009, 661, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Shim, J.; Kim, H.W.; Kim, J.; Jang, Y.J.; Yang, H.; Park, J.; Choi, S.H.; Yoon, J.H.; et al. Caffeinated coffee, decaffeinated coffee, and the phenolic phytochemical chlorogenic acid up-regulate NQO1 expression and prevent H₂O₂-induced apoptosis in primary cortical neurons. Neurochem. Int. 2012, 60, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.F.; Brown, P.H.; Lyle, B.J.; Chen, Y.; Black, R.M.; Williams, C.E.; Lin, Y.C.; Hsu, C.W.; Cheng, I.H. Roasted coffees high in lipophilic antioxidants and chlorogenic acid lactones are more neuroprotective than green coffees. J. Agric. Food Chem. 2009, 57, 9801–9808. [Google Scholar] [CrossRef] [PubMed]
- Mira, A.; Yamashita, S.; Katakura, Y.; Shimizu, K. In vitro neuroprotective activities of compounds from Angelica shikokiana Makino. Molecules 2015, 20, 4813–4832. [Google Scholar] [CrossRef] [PubMed]
- Gul, Z.; Demircan, C.; Bagdas, D.; Buyukuysal, R.L. Protective effects of chlorogenic acid and its metabolites on hydrogen peroxide-induced alterations in rat brain slices: A comparative study with resveratrol. Neurochem. Res. 2016, 41, 2075–2085. [Google Scholar] [CrossRef]
- Yao, J.; Peng, S.; Xu, J.; Fang, J. Reversing ROS-mediated neurotoxicity by chlorogenic acid involves its direct antioxidant activity and activation of Nrf2-ARE signaling pathway. Biofactors 2019, 45, 616–626. [Google Scholar] [CrossRef]
- Wang, X.; Fan, X.; Yuan, S.; Jiao, W.; Liu, B.; Cao, J.; Jiang, W. Chlorogenic acid protects against aluminium-induced cytotoxicity through chelation and antioxidant actions in primary hippocampal neuronal cells. Food Funct. 2017, 8, 2924–2934. [Google Scholar] [CrossRef]
- Nakajima, Y.; Shimazawa, M.; Mishima, S.; Hara, H. Water extract of propolis and its main constituents, caffeoylquinic acid derivatives, exert neuroprotective effects via antioxidant actions. Life Sci. 2007, 80, 370–377. [Google Scholar] [CrossRef]
- Li, Y.; Shi, W.; Li, Y.; Zhou, Y.; Hu, X.; Song, C.; Ma, H.; Wang, C.; Li, Y. Neuroprotective effects of chlorogenic acid against apoptosis of PC12 cells induced by methylmercury. Environ. Toxicol. Pharmacol. 2008, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Oboh, G.; Agunloye, O.M.; Akinyemi, A.J.; Ademiluyi, A.O.; Adefegha, S.A. Comparative study on the inhibitory effect of caffeic and chlorogenic acids on key enzymes linked to Alzheimer’s disease and some pro-oxidant induced oxidative stress in rats’ brain-in vitro. Neurochem. Res. 2013, 38, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Taram, F.; Winter, A.N.; Linseman, D.A. Neuroprotection comparison of chlorogenic acid and its metabolites against mechanistically distinct cell death-inducing agents in cultured cerebellar granule neurons. Brain Res. 2016, 1648 Pt A, 69–80. [Google Scholar] [CrossRef]
- Lee, M.; McGeer, E.G.; McGeer, P.L. Quercetin, not caffeine, is a major neuroprotective component in coffee. Neurobiol. Aging 2016, 46, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases – What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef]
- Mikami, Y.; Yamazawa, T. Chlorogenic acid, a polyphenol in coffee, protects neurons against glutamate neurotoxicity. Life Sci. 2015, 139, 69–74. [Google Scholar] [CrossRef]
- Rebai, O.; Belkhir, M.; Sanchez-Gomez, M.V.; Matute, C.; Fattouch, S.; Amri, M. Differential molecular targets for neuroprotective effect of chlorogenic acid and its related compounds against glutamate induced excitotoxicity and oxidative stress in rat cortical neurons. Neurochem. Res. 2017, 42, 3559–3572. [Google Scholar] [CrossRef]
- Rebai, O.; Amri, M. Chlorogenic Acid Prevents AMPA-mediated excitotoxicity in optic nerve oligodendrocytes through a PKC and caspase-dependent pathways. Neurotox. Res. 2018, 34, 559–573. [Google Scholar] [CrossRef]
- Liu, Q.F.; Jeon, Y.; Sung, Y.W.; Lee, J.H.; Jeong, H.; Kim, Y.M.; Yun, H.S.; Chin, Y.W.; Jeon, S.; Cho, K.S.; et al. Nardostachys jatamansi ethanol extract ameliorates Aβ42 cytotoxicity. Biol. Pharm. Bull. 2018, 41, 470–477. [Google Scholar] [CrossRef]
- Gao, L.; Li, X.; Meng, S.; Ma, T.; Wan, L.; Xu, S. Chlorogenic acid alleviates Aβ(25-35)-induced autophagy and cognitive impairment via the mTOR/TFEB signaling pathway. Drug Des. Dev. Ther. 2020, 14, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Chen, L.; Liu, J.; Zhao, J.; Liu, W.; Feng, F. Protective effects of a Chotosan Fraction and its active components on β-amyloid-induced neurotoxicity. Neurosci. Lett. 2016, 617, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.S.; Wang, Y.; Weaver, D.F. Phenylindanes in brewed coffee inhibit amyloid-beta and tau aggregation. Front. Neurosci. 2018, 12, 735. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Lee, H.K.; Kim, J.A.; Hong, S.I.; Kim, H.C.; Jo, T.H.; Park, Y.I.; Lee, C.K.; Kim, Y.B.; Lee, S.Y.; et al. Neuroprotective effects of chlorogenic acid on scopolamine-induced amnesia via anti-acetylcholinesterase and anti-oxidative activities in mice. Eur. J. Pharmacol. 2010, 649, 210–217. [Google Scholar] [CrossRef]
- Szwajgier, D. Anticholinesterase activity of selected phenolic acids and flavonoids—Interaction testing in model solutions. Ann. Agric. Environ. Med. 2015, 22, 690–694. [Google Scholar] [CrossRef]
- Nazir, N.; Zahoor, M.; Nisar, M.; Karim, N.; Latif, A.; Ahmad, S.; Uddin, Z. Evaluation of neuroprotective and anti-amnesic effects of Elaeagnus umbellata Thunb. On scopolamine-induced memory impairment in mice. BMC Complementary Med. Ther. 2020, 20, 1–17. [Google Scholar] [CrossRef]
- Shan, S.; Tian, L.; Fang, R. Chlorogenic acid exerts beneficial effects in 6-hydroxydopamine-induced neurotoxicity by inhibition of endoplasmic reticulum stress. Med. Sci. Monit. 2019, 25, 453–459. [Google Scholar] [CrossRef]
- Kwon, S.H.; Ma, S.X.; Hong, S.I.; Kim, S.Y.; Lee, S.Y.; Jang, C.G. Eucommia ulmoides Oliv. bark. attenuates 6-hydroxydopamine-induced neuronal cell death through inhibition of oxidative stress in SH-SY5Y cells. J. Ethnopharmacol. 2014, 152, 173–182. [Google Scholar] [CrossRef]
- Lin, C.M.; Lin, Y.T.; Lee, T.L.; Imtiyaz, Z.; Hou, W.C.; Lee, M.H. In vitro and in vivo evaluation of the neuroprotective activity of Uncaria hirsuta Haviland. J. Food Drug Anal. 2020, 28, 147–158. [Google Scholar] [CrossRef]
- Teraoka, M.; Nakaso, K.; Kusumoto, C.; Katano, S.; Tajima, N.; Yamashita, A.; Zushi, T.; Ito, S.; Matsura, T. Cytoprotective effect of chlorogenic acid against α-synuclein-related toxicity in catecholaminergic PC12 cells. J. Clin. Biochem. Nutr. 2012, 51, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Hasegawa, M.; Nonaka, T.; Oikawa, T.; Yonetani, M.; Yamaguchi, Y.; Kato, K.; Hisanaga, S.; Goedert, M. Inhibition of alpha-synuclein fibril assembly by small molecules: Analysis using epitope-specific antibodies. FEBS Lett. 2009, 583, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Vardi, N.; Parlakpinar, H.; Ates, B. Beneficial effects of chlorogenic acid on methotrexate-induced cerebellar Purkinje cell damage in rats. J. Chem. Neuroanat. 2012, 43, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.L.; Pan, N.; Zhang, Q.H.; Wang, X.H. Therapeutic efficacy of chlorogenic acid on cadmium-induced oxidative neuropathy in a murine model. Exp. Ther. Med. 2015, 9, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, J. Chlorogenic Acid Prevents Alcohol-induced Brain Damage in Neonatal Rat. Transl. Neurosci. 2017, 8, 176–181. [Google Scholar] [CrossRef][Green Version]
- Alarcón-Herrera, N.; Flores-Maya, S.; Bellido, B.; García-Bores, A.M.; Mendoza, E.; Ávila-Acevedo, G.; Hernández-Echeagaray, E. Protective effects of chlorogenic acid in 3-nitropropionic acid induced toxicity and genotoxicity. Food Chem. Toxicol. 2017, 109 Pt. 2, 1018–1025. [Google Scholar] [CrossRef]
- Miao, M.; Cao, L.; Li, R.; Fang, X.; Miao, Y. Protective effect of chlorogenic acid on the focal cerebral ischemia reperfusion rat models. Saudi Pharm. J. 2017, 25, 556–563. [Google Scholar] [CrossRef]
- Liu, D.; Wang, H.; Zhang, Y.; Zhang, Z. Protective effects of chlorogenic acid on cerebral ischemia/reperfusion injury rats by regulating oxidative stress-related Nrf2 pathway. Drug Des. Dev. Ther. 2020, 14, 51–60. [Google Scholar] [CrossRef]
- Lee, K.; Lee, J.S.; Jang, H.J.; Kim, S.M.; Chang, M.S.; Park, S.H.; Kim, K.S.; Bae, J.; Park, J.W.; Lee, B.; et al. Chlorogenic acid ameliorates brain damage and edema by inhibiting matrix metalloproteinase-2 and 9 in a rat model of focal cerebral ischemia. Eur. J. Pharmacol. 2012, 689, 89–95. [Google Scholar] [CrossRef]
- Kumar, G.; Mukherjee, S.; Paliwal, P.; Singh, S.S.; Birla, H.; Singh, S.P.; Krishnamurthy, S.; Patnaik, R. Neuroprotective effect of chlorogenic acid in global cerebral ischemia-reperfusion rat model. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 1293–1309. [Google Scholar] [CrossRef]
- Ahn, E.H.; Kim, D.W.; Shin, M.J.; Kwon, S.W.; Kim, Y.N.; Kim, D.S.; Lim, S.S.; Kim, J.; Park, J.; Eum, W.S.; et al. Chlorogenic acid improves neuroprotective effect of PEP-1-ribosomal protein S3 against ischemic insult. Exp. Neurobiol. 2011, 20, 169–175. [Google Scholar] [CrossRef]
- Hermawati, E.; Arfian, N.; Mustofa, M.; Partadiredja, G. Chlorogenic acid ameliorates memory loss and hippocampal cell death after transient global ischemia. Eur. J. Neurosci. 2020, 51, 651–669. [Google Scholar] [CrossRef]
- Lee, T.K.; Kang, I.J.; Kim, B.; Sim, H.J.; Kim, D.W.; Ahn, J.H.; Lee, J.C.; Ryoo, S.; Shin, M.C.; Cho, J.H.; et al. Experimental pretreatment with chlorogenic acid prevents transient ischemia-induced cognitive decline and neuronal damage in the hippocampus through anti-oxidative and anti-inflammatory effects. Molecules 2020, 25, 3578. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Isooka, N.; Wada, K.; Kikuoka, R.; Kitamura, Y.; Asanuma, M. Effects of enteric environmental modification by coffee components on neurodegeneration in rotenone-treated mice. Cells 2019, 8, 221. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Dilnashin, H.; Singh, R.; Singh, S.P. Neuroprotective effect of chlorogenic acid on mitochondrial dysfunction-mediated apoptotic death of DA neurons in a Parkinsonian mouse model. Oxid. Med. Cell Longev. 2020, 2020, 6571484. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Qi, R.; Zhang, J.; Wang, Z.; Wang, H.; Hu, C.; Zhao, Y.; Bie, M.; Wang, Y.; Fu, Y.; et al. Chlorogenic acid inhibits LPS-induced microglial activation and improves survival of dopaminergic neurons. Brain Res. Bull. 2012, 88, 487–494. [Google Scholar] [CrossRef]
- Tu, Q.; Tang, X.; Hu, Z. Chlorogenic acid protection of neuronal nitric oxide synthase-positive neurons in the hippocampus of mice with impaired learning and memory. Neural Regen. Res. 2008, 3, 1218–1221. [Google Scholar]
- Aseervatham, G.S.; Suryakala, U.; Sundaram, S.; Bose, P.C.; Sivasudha, T. Expression pattern of NMDA receptors reveals antiepileptic potential of apigenin 8-C-glucoside and chlorogenic acid in pilocarpine induced epileptic mice. Biomed. Pharmacother. 2016, 82, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Yamamoto, M.; Misawa, K.; Nishimura, H.; Misawa, K.; Ota, N.; Shimotoyodome, A. Coffee polyphenols prevent cognitive dysfunction and suppress amyloid β plaques in APP/PS2 transgenic mouse. Neurosci. Res. 2020, 154, 35–44. [Google Scholar] [CrossRef]
- Cropley, V.; Croft, R.; Silber, B.; Neale, C.; Scholey, A.; Stough, C.; Schmitt, J. Does coffee enriched with chlorogenic acids improve mood and cognition after acute administration in healthy elderly? A pilot study. Psychopharmacology 2012, 219, 737–749. [Google Scholar] [CrossRef]
- Camfield, D.A.; Silber, B.Y.; Scholey, A.B.; Nolidin, K.; Goh, A.; Stough, C. A randomised placebo-controlled trial to differentiate the acute cognitive and mood effects of chlorogenic acid from decaffeinated coffee. PLoS ONE 2013, 8, e82897. [Google Scholar] [CrossRef]
- Ochiai, R.; Saitou, K.; Suzukamo, C.; Osaki, N.; Asada, T. Effect of chlorogenic acids on cognitive function in mild cognitive impairment: A randomized controlled crossover trial. J. Alzheimers Dis. 2019, 72, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Saitou, K.; Ochiai, R.; Kozuma, K.; Sato, H.; Koikeda, T.; Osaki, N.; Katsuragi, Y. Effect of chlorogenic acids on cognitive function: A randomized, double-blind, placebo-controlled trial. Nutrients 2018, 10, 1337. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Ochiai, R.; Kozuma, K.; Sato, H.; Katsuragi, Y. Effect of chlorogenic acid intake on cognitive function in the elderly: A pilot study. Evid. Based Complementary Altern. Med. 2018, 2018, 8608497. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Protective effects of caffeic acid and the Alzheimer’s brain: An update. Mini Rev. Med. Chem 2017, 17, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Itagaki, S.; Kurokawa, T.; Ogura, J.; Kobayashi, M.; Hirano, T.; Sugawara, M.; Iseki, K. In vitro and in vivo antioxidant properties of chlorogenic acid and caffeic acid. Int. J. Pharm. 2011, 403, 136–138. [Google Scholar] [CrossRef]
- Espíndola, K.M.M.; Ferreira, R.G.; Narvaez, L.E.M.; Silva Rosario, A.C.R.; da Silva, A.H.M.; Silva, A.G.B.; Vieira, A.P.O.; Monteiro, M.C. Chemical and pharmacological aspects of caffeic acid and its activity in hepatocarcinoma. Front. Oncol. 2019, 9, 541. [Google Scholar] [CrossRef]
- Pereira, P.; de Oliveira, P.A.; Ardenghi, P.; Rotta, L.; Henriques, J.A.; Picada, J.N. Neuropharmacological analysis of caffeic acid in rats. Basic Clin. Pharmacol. Toxicol. 2006, 99, 374–378. [Google Scholar] [CrossRef]
- Silva, T.; Oliveira, C.; Borges, F. Caffeic acid derivatives, analogs and applications: A patent review (2009–2013). Expert Opin. Ther. Pat. 2014, 24, 1257–1270. [Google Scholar] [CrossRef]
- Im, S.E.; Yoon, H.; Nam, T.G.; Heo, H.J.; Lee, C.Y.; Kim, D.O. Antineurodegenerative effect of phenolic extracts and caffeic acid derivatives in romaine lettuce on neuron-like PC-12 cells. J. Med. Food 2010, 13, 779–784. [Google Scholar] [CrossRef]
- Jeong, C.H.; Jeong, H.R.; Choi, G.N.; Kim, D.O.; Lee, U.; Heo, H.J. Neuroprotective and anti-oxidant effects of caffeic acid isolated from Erigeron annuus leaf. Chin. Med. 2011, 6, 1–9. [Google Scholar] [CrossRef]
- Pavlica, S.; Gebhardt, R. Protective effects of ellagic and chlorogenic acids against oxidative stress in PC12 cells. Free Radic Res. 2005, 39, 1377–1390. [Google Scholar] [CrossRef] [PubMed]
- Omar, S.H.; Kerr, P.G.; Scott, C.J.; Hamlin, A.S.; Obied, H.K. Olive (Olea europaea L.) Biophenols: A nutriceutical against oxidative stress in SH-SY5Y cells. Molecules 2017, 22, 1858. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Kim, Y.C. Neuroprotective phenylpropanoid esters of rhamnose isolated from roots of Scrophularia buergeriana. Phytochemistry 2000, 54, 503–509. [Google Scholar] [CrossRef]
- Koo, K.A.; Kim, S.H.; Oh, T.H.; Kim, Y.C. Acteoside and its aglycones protect primary cultures of rat cortical cells from glutamate-induced excitotoxicity. Life Sci. 2006, 79, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wei, E.Q.; Zhang, W.P.; Ge, Q.F.; Liu, J.R.; Wang, M.L.; Huang, X.J.; Hu, X.; Chen, Z. Minocycline protects PC12 cells against NMDA-induced injury via inhibiting 5-lipoxygenase activation. Brain Res. 2006, 1085, 57–67. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Sánchez-Hernández, S.; De Lima, M.E.; Ali, S.F.; Chavarría, A.; Villeda, J.; Santamaría, A. Protective effects of caffeic acid on quinolinic acid-induced behavioral and oxidative alterations in rats. J. Drug Alcohol Res. 2015, 4, 1–5. [Google Scholar] [CrossRef]
- Izuta, H.; Shimazawa, M.; Tazawa, S.; Araki, Y.; Mishima, S.; Hara, H. Protective effects of Chinese propolis and its component, chrysin, against neuronal cell death via inhibition of mitochondrial apoptosis pathway in SH-SY5Y cells. J. Agric. Food Chem. 2008, 56, 8944–8953. [Google Scholar] [CrossRef]
- Huang, Y.; Jin, M.; Pi, R.; Zhang, J.; Chen, M.; Ouyang, Y.; Liu, A.; Chao, X.; Liu, P.; Liu, J.; et al. Protective effects of caffeic acid and caffeic acid phenethyl ester against acrolein-induced neurotoxicity in HT22 mouse hippocampal cells. Neurosci. Lett. 2013, 535, 146–151. [Google Scholar] [CrossRef]
- Omar, S.H.; Scott, C.J.; Hamlin, A.S.; Obied, H.K. Biophenols: Enzymes (β-secretase, Cholinesterases, histone deacetylase and tyrosinase) inhibitors from olive (Olea europaea L.). Fitoterapia 2018, 128, 118–129. [Google Scholar] [CrossRef]
- Sul, D.; Kim, H.S.; Lee, D.; Joo, S.S.; Hwang, K.W.; Park, S.Y. Protective effect of caffeic acid against beta-amyloid-induced neurotoxicity by the inhibition of calcium influx and tau phosphorylation. Life Sci. 2009, 84, 257–262. [Google Scholar] [CrossRef]
- Yang, J.Q.; Zhou, Q.X.; Liu, B.Z.; He, B.C. Protection of mouse brain from aluminum-induced damage by caffeic acid. CNS Neurosci. Ther. 2008, 14, 10–16. [Google Scholar] [CrossRef]
- Khan, K.A.; Kumar, N.; Nayak, P.G.; Nampoothiri, M.; Shenoy, R.R.; Krishnadas, N.; Rao, C.M.; Mudgal, J. Impact of caffeic acid on aluminium chloride-induced dementia in rats. J. Pharm. Pharmacol. 2013, 65, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, R.; Kaundal, M.; Bansal, V. Samardeep, Caffeic acid attenuates oxidative stress, learning and memory deficit in intra-cerebroventricular streptozotocin induced experimental dementia in rats. Biomed. Pharmacother. 2016, 81, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Li, J.; Hua, L.; Han, B.; Zhang, Y.; Yang, X.; Zeng, Z.; Bai, H.; Yin, H.; et al. Effects of caffeic acid on learning deficits in a model of Alzheimer’s disease. Int. J. Mol. Med. 2016, 38, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Anwar, J.; Spanevello, R.M.; Thomé, G.; Stefanello, N.; Schmatz, R.; Gutierres, J.; Vieira, J.; Baldissarelli, J.; Carvalho, F.B.; da Rosa, M.M.; et al. Effects of caffeic acid on behavioral parameters and on the activity of acetylcholinesterase in different tissues from adult rats. Pharmacol. Biochem. Behav. 2012, 103, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Akomolafe, S.F.; Akinyemi, A.J.; Ogunsuyi, O.B.; Oyeleye, S.I.; Oboh, G.; Adeoyo, O.O.; Allismith, Y.R. Effect of caffeine, caffeic acid and their various combinations on enzymes of cholinergic, monoaminergic and purinergic systems critical to neurodegeneration in rat brain-In vitro. Neurotoxicology 2017, 62, 6–13. [Google Scholar] [CrossRef]
- Dos Santos Sales, Í.M.; do Nascimento, K.G.; Feitosa, C.M.; Saldanha, G.B.; Feng, D.; de Freitas, R.M. Caffeic acid effects on oxidative stress in rat hippocampus after pilocarpine-induced seizures. Neurol. Sci. 2011, 32, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Kalonia, H.; Kumar, P.; Kumar, A.; Nehru, B. Effects of caffeic acid, rofecoxib, and their combination against quinolinic acid-induced behavioral alterations and disruption in glutathione redox status. Neurosci. Bull. 2009, 25, 343–352. [Google Scholar] [CrossRef]
- Uz, T.; Pesold, C.; Longone, P.; Manev, H. Aging-associated up-regulation of neuronal 5-lipoxygenase expression: Putative role in neuronal vulnerability. FASEB J. 1998, 12, 439–449. [Google Scholar] [CrossRef]
- Coelho, V.R.; Vieira, C.G.; de Souza, L.P.; da Silva, L.L.; Pflüger, P.; Regner, G.G.; Papke, D.K.; Picada, J.N.; Pereira, P. Behavioral and genotoxic evaluation of rosmarinic and caffeic acid in acute seizure models induced by pentylenetetrazole and pilocarpine in mice. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 1195–1203. [Google Scholar] [CrossRef]
- Coelho, V.R.; Vieira, C.G.; de Souza, L.P.; Moysés, F.; Basso, C.; Papke, D.K.; Pires, T.R.; Siqueira, I.R.; Picada, J.N.; Pereira, P. Antiepileptogenic, antioxidant and genotoxic evaluation of rosmarinic acid and its metabolite caffeic acid in mice. Life Sci. 2015, 122, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.H.; Yan, B.C.; Park, J.H.; Yeun, G.H.; Yim, Y.; Ahn, J.H.; Lee, J.C.; Hwang, I.K.; Cho, J.H.; Kim, Y.M.; et al. Neuroprotection of a novel synthetic caffeic acid-syringic acid hybrid compound against experimentally induced transient cerebral ischemic damage. Planta Med. 2013, 79, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro Fernandes, F.D.; Fontenele Menezes, A.P.; de Sousa Neves, J.C.; Fonteles, A.A.; da Silva, A.T.; de Araújo Rodrigues, P.; Santos do Carmo, M.R.; de Souza, C.M.; de Andrade, G.M. Caffeic acid protects mice from memory deficits induced by focal cerebral ischemia. Behav. Pharmacol. 2014, 25, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Bei, Y.; Xiao, Y.; Tong, H.D.; Wu, X.Q.; Chen, M.T. Edaravone, a free radical scavenger, protects neuronal cells’ mitochondria from ischemia by inactivating another new critical factor of the 5-lipoxygenase pathway affecting the arachidonic acid metabolism. Brain Res. 2018, 1690, 96–104. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, S.H.; Ye, Y.L.; Chu, L.S.; Zhang, W.P.; Wang, M.L.; Wei, E.Q. Caffeic acid ameliorates early and delayed brain injuries after focal cerebral ischemia in rats. Acta Pharmacol. Sin. 2006, 27, 1103–1110. [Google Scholar] [CrossRef]
- Liang, G.; Shi, B.; Luo, W.; Yang, J. The protective effect of caffeic acid on global cerebral ischemia-reperfusion injury in rats. Behav. Brain Funct. 2015, 11, 18. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, W.P.; Chen, K.D.; Qian, X.D.; Fang, S.H.; Wei, E.Q. Caffeic acid attenuates neuronal damage, astrogliosis and glial scar formation in mouse brain with cryoinjury. Life Sci. 2007, 80, 530–537. [Google Scholar] [CrossRef]
- Vauzour, D.; Corona, G.; Spencer, J.P. Caffeic acid, tyrosol and p-coumaric acid are potent inhibitors of 5-S-cysteinyl-dopamine induced neurotoxicity. Arch. Biochem. Biophys. 2010, 501, 106–111. [Google Scholar] [CrossRef]
- Li, Z.; Choi, D.Y.; Shin, E.J.; Hunter, R.L.; Jin, C.H.; Wie, M.B.; Kim, M.S.; Park, S.J.; Bing, G.; Kim, H.C. Phenidone protects the nigral dopaminergic neurons from LPS-induced neurotoxicity. Neurosci. Lett. 2008, 445, 1–6. [Google Scholar] [CrossRef]
- Jimenez-Del-Rio, M.; Guzman-Martinez, C.; Velez-Pardo, C. The effects of polyphenols on survival and locomotor activity in Drosophila melanogaster exposed to iron and paraquat. Neurochem. Res. 2010, 35, 227–238. [Google Scholar] [CrossRef]
- Dos Santos Nunes, R.G.; Pereira, P.S.; Elekofehinti, O.O.; Fidelis, K.R.; da Silva, C.S.; Ibrahim, M.; Barros, L.M.; da Cunha, F.A.B.; Lukong, K.E.; de Menezes, I.R.A.; et al. Possible involvement of transcriptional activation of nuclear factor erythroid 2-related factor 2 (Nrf2) in the protective effect of caffeic acid on paraquat-induced oxidative damage in Drosophila melanogaster. Pestic Biochem. Physiol. 2019, 157, 161–168. [Google Scholar] [CrossRef]
- Tsai, S.J.; Chao, C.Y.; Yin, M.C. Preventive and therapeutic effects of caffeic acid against inflammatory injury in striatum of MPTP-treated mice. Eur. J. Pharmacol. 2011, 670, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, Q.; Zhang, L.; Wang, Q.; Yang, Z.; Liu, J.; Feng, L. Caffeic acid reduces A53T α-synuclein by activating JNK/Bcl-2-mediated autophagy in vitro and improves behaviour and protects dopaminergic neurons in a mouse model of Parkinson’s disease. Pharmacol. Res. 2019, 150, 104538. [Google Scholar] [CrossRef]
- Colonnello, A.; Aguilera-Portillo, G.; Rubio-López, L.C.; Robles-Bañuelos, B.; Rangel-López, E.; Cortez-Núñez, S.; Evaristo-Priego, Y.; Silva-Palacios, A.; Galván-Arzate, S.; García-Contreras, R.; et al. Comparing the neuroprotective effects of caffeic acid in rat cortical slices and Caenorhabditis elegans: Involvement of Nrf2 and SKN-1 signaling pathways. Neurotox. Res. 2020, 37, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Ma, Z.; Fontanilla, C.V.; Zhao, L.; Xu, Z.C.; Taggliabraci, V.; Johnstone, B.H.; Dodel, R.C.; Farlow, M.R.; Du, Y. Caffeic acid phenethyl ester prevents cerebellar granule neurons (CGNs) against glutamate-induced neurotoxicity. Neuroscience 2008, 155, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Scapagnini, G.; Vasto, S.; Abraham, N.G.; Caruso, C.; Zella, D.; Fabio, G. Modulation of Nrf2/ARE pathway by food polyphenols: A nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol. Neurobiol. 2011, 44, 192–201. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Dua, P.; Hill, J.M.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. microRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 2012, 3, 365–373. [Google Scholar]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective effect of caffeic acid phenethyl ester in a mouse model of Alzheimer’s disease involves Nrf2/HO-1 pathway. Aging Dis. 2018, 9, 605–622. [Google Scholar] [CrossRef]
- Dos Santos, N.A.; Martins, N.M.; Silva Rde, B.; Ferreira, R.S.; Sisti, F.M.; dos Santos, A.C. Caffeic acid phenethyl ester (CAPE) protects PC12 cells from MPP+ toxicity by inducing the expression of neuron-typical proteins. Neurotoxicology 2014, 45, 131–138. [Google Scholar] [CrossRef]
- Fontanilla, C.V.; Ma, Z.; Wei, X.; Klotsche, J.; Zhao, L.; Wisniowski, P.; Dodel, R.C.; Farlow, M.R.; Oertel, W.H.; Du, Y. Caffeic acid phenethyl ester prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurodegeneration. Neuroscience 2011, 188, 135–141. [Google Scholar] [CrossRef]
- Noelker, C.; Bacher, M.; Gocke, P.; Wei, X.; Klockgether, T.; Du, Y.; Dodel, R. The flavanoide caffeic acid phenethyl ester blocks 6-hydroxydopamine-induced neurotoxicity. Neurosci. Lett. 2005, 383, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Turan, D.; Abdik, H.; Sahin, F.; Avşar Abdik, E. Evaluation of the neuroprotective potential of caffeic acid phenethyl ester in a cellular model of Parkinson’s disease. Eur. J. Pharmacol. 2020, 883, 173342. [Google Scholar] [CrossRef] [PubMed]
- Kurauchi, Y.; Hisatsune, A.; Isohama, Y.; Mishima, S.; Katsuki, H. Caffeic acid phenethyl ester protects nigral dopaminergic neurons via dual mechanisms involving haem oxygenase-1 and brain-derived neurotrophic factor. Br. J. Pharmacol. 2012, 166, 1151–1168. [Google Scholar] [CrossRef]
- Silva, R.B.; Santos, N.A.; Martins, N.M.; Ferreira, D.A.; Barbosa, F., Jr.; Oliveira Souza, V.C.; Kinoshita, A.; Baffa, O.; Del-Bel, E.; Santos, A.C. Caffeic acid phenethyl ester protects against the dopaminergic neuronal loss induced by 6-hydroxydopamine in rats. Neuroscience 2013, 233, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.K.; Lin, M.J.; Liao, P.H.; Yang, C.Y.; Lin, S.M.; Liu, S.M.; Lin, R.H.; Chih, C.L.; Huang, S.S. Caffeic acid phenethyl ester ameliorates cerebral infarction in rats subjected to focal cerebral ischemia. Life Sci. 2006, 78, 2758–2762. [Google Scholar] [CrossRef]
- Hwang, S.A.; Kim, C.D.; Lee, W.S. Caffeic acid phenethyl ester protects against photothrombotic cortical ischemic injury in mice. Korean J. Physiol. Pharmacol. 2018, 22, 101–110. [Google Scholar] [CrossRef]
- Khan, M.; Elango, C.; Ansari, M.A.; Singh, I.; Singh, A.K. Caffeic acid phenethyl ester reduces neurovascular inflammation and protects rat brain following transient focal cerebral ischemia. J. Neurochem. 2007, 102, 365–377. [Google Scholar] [CrossRef]
- Palaz, M.N.; Akcay, E. The impact of propolis factor caffeic acid phenethyl-ester on the cerebral vasospasm and early brain damage in the experimentally induced subarachnoid hemorrhage on rats. World Neurosurg. 2020, 138, e736–e742. [Google Scholar] [CrossRef]
- Kumar, M.; Kaur, D.; Bansal, N. Caffeic Acid Phenethyl Ester (CAPE) Prevents development of STZ-ICV induced dementia in rats. Pharmacogn. Mag. 2017, 13 (Suppl. 1), S10–S15. [Google Scholar]
- Fu, W.; Wang, H.; Ren, X.; Yu, H.; Lei, Y.; Chen, Q. Neuroprotective effect of three caffeic acid derivatives via ameliorate oxidative stress and enhance PKA/CREB signaling pathway. Behav. Brain Res. 2017, 328, 81–86. [Google Scholar] [CrossRef]
- Hao, R.; Song, X.; Li, F.; Tan, X.; Sun-Waterhouse, D.; Li, D. Caffeic acid phenethyl ester reversed cadmium-induced cell death in hippocampus and cortex and subsequent cognitive disorders in mice: Involvements of AMPK/SIRT1 pathway and amyloid-tau-neuroinflammation axis. Food Chem. Toxicol. 2020, 144, 111636. [Google Scholar] [CrossRef] [PubMed]
- Mohamadi, N.; Sharififar, F.; Pournamdari, M.; Ansari, M. A review on biosynthesis, analytical techniques, and pharmacological activities of trigonelline as a plant alkaloid. J. Diet. Suppl. 2018, 15, 207–222. [Google Scholar] [CrossRef]
- Qiu, Z.; Wang, K.; Jiang, C.; Su, Y.; Fan, X.; Li, J.; Xue, S.; Yao, L. Trigonelline protects hippocampal neurons from oxygen-glucose deprivation-induced injury through activating the PI3K/Akt pathway. Chem. Biol. Interact. 2020, 317, 108946. [Google Scholar] [CrossRef]
- Makowska, J.; Szczesny, D.; Lichucka, A.; Giełdoń, A.; Chmurzyński, L.; Kaliszan, R. Preliminary studies on trigonelline as potential anti-Alzheimer disease agent: Determination by hydrophilic interaction liquid chromatography and modeling of interactions with beta-amyloid. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 968, 101–104. [Google Scholar] [CrossRef]
- Tohda, C.; Kuboyama, T.; Komatsu, K. Search for natural products related to regeneration of the neuronal network. Neurosignals 2005, 14, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Fahanik-Babaei, J.; Baluchnejadmojarad, T.; Nikbakht, F.; Roghani, M. Trigonelline protects hippocampus against intracerebral Aβ(1-40) as a model of Alzheimer’s disease in the rat: Insights into underlying mechanisms. Metab. Brain Dis. 2019, 34, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.A.; Gawali, N.B.; Munshi, R.; Juvekar, A.R. Trigonelline insulates against oxidative stress, proinflammatory cytokines and restores BDNF levels in lipopolysaccharide induced cognitive impairment in adult mice. Metab. Brain Dis. 2018, 33, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Khalili, M.; Alavi, M.; Esmaeil-Jamaat, E.; Baluchnejadmojarad, T.; Roghani, M. Trigonelline mitigates lipopolysaccharide-induced learning and memory impairment in the rat due to its anti-oxidative and anti-inflammatory effect. Int. Immunopharmacol. 2018, 61, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.A.; Gawali, N.B.; Bulani, V.D.; Kothavade, P.S.; Mestry, S.N.; Deshpande, P.S.; Juvekar, A.R. In vitro antiglycating effect and in vivo neuroprotective activity of Trigonelline in d-galactose induced cognitive impairment. Pharmacol. Rep. 2018, 70, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Mirzaie, M.; Khalili, M.; Kiasalari, Z.; Roghani, M. Neuroprotective and antiapoptotic potential of trigonelline in a striatal 6-hydroxydopamine rat model of parkinson’s disease. Neurophysiology 2016, 48, 176–183. [Google Scholar] [CrossRef]
- Gaur, V.; Bodhankar, S.L.; Mohan, V.; Thakurdesai, P.A. Neurobehavioral assessment of hydroalcoholic extract of Trigonella foenum-graecum seeds in rodent models of Parkinson’s disease. Pharm. Biol. 2013, 51, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Pravalika, K.; Sarmah, D.; Kaur, H.; Vats, K.; Saraf, J.; Wanve, M.; Kalia, K.; Borah, A.; Yavagal, D.R.; Dave, K.R.; et al. Trigonelline therapy confers neuroprotection by reduced glutathione mediated myeloperoxidase expression in animal model of ischemic stroke. Life Sci. 2019, 216, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Anjomshoa, M.; Boroujeni, S.N.; Bagheri, E.; Lorigooini, Z.; Amini-Khoei, H. Possible involvement of N-methyl-D-aspartate receptor (NMDA-R) in the antidepressant-like effect of trigonelline in male mice. Curr. Pharm. Des. 2020, 26, 5067–5071. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Martin, S. Health effects of coffee: Mechanism unraveled? Nutrients 2020, 12, 1842. [Google Scholar] [CrossRef] [PubMed]
- Paur, I.; Balstad, T.R.; Blomhoff, R. Degree of roasting is the main determinant of the effects of coffee on NF-kappaB and EpRE. Free Radic. Biol. Med. 2010, 48, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Boettler, U.; Sommerfeld, K.; Volz, N.; Pahlke, G.; Teller, N.; Somoza, V.; Lang, R.; Hofmann, T.; Marko, D. Coffee constituents as modulators of Nrf2 nuclear translocation and ARE (EpRE)-dependent gene expression. J. Nutr. Biochem. 2011, 22, 426–440. [Google Scholar] [CrossRef]
- Farias-Pereira, R.; Park, C.S.; Park, Y. Mechanisms of action of coffee bioactive components on lipid metabolism. Food Sci. Biotechnol. 2019, 28, 1287–1296. [Google Scholar] [CrossRef]
- Ren, Y.; Wang, C.; Xu, J.; Wang, S. Cafestol and kahweol: A review on their bioactivities and pharmacological properties. Int. J. Mol. Sci. 2019, 20, 4238. [Google Scholar] [CrossRef]
- De Oliveira, M.R.; de Souza, I.C.C.; Fürstenau, C.R. Mitochondrial protection promoted by the coffee diterpene kahweol in methylglyoxal-treated human neuroblastoma SH-SY5Y cells. Neurotox. Res. 2020, 37, 100–110. [Google Scholar] [CrossRef]
- Lee, K.J.; Jeong, H.G. Protective effects of kahweol and cafestol against hydrogen peroxide-induced oxidative stress and DNA damage. Toxicol. Lett. 2007, 173, 80–87. [Google Scholar] [CrossRef]
- Hwang, Y.P.; Jeong, H.G. The coffee diterpene kahweol induces heme oxygenase-1 via the PI3K and p38/Nrf2 pathway to protect human dopaminergic neurons from 6-hydroxydopamine-derived oxidative stress. FEBS Lett. 2008, 582, 2655–2662. [Google Scholar] [CrossRef] [PubMed]
- Fürstenau, C.R.; de Souza, I.C.C.; de Oliveira, M.R. The effects of kahweol, a diterpene present in coffee, on the mitochondria of the human neuroblastoma SH-SY5Y cells exposed to hydrogen peroxide. Toxicol. In Vitro 2019, 61, 104601. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.F.; Lin, J.S.; Chang, C.F. Acute kahweol treatment attenuates traumatic brain injury neuroinflammation and functional deficits. Nutrients 2019, 11, 2301. [Google Scholar] [CrossRef] [PubMed]
- Esopenko, C.; Levine, B. Aging, neurodegenerative disease, and traumatic brain injury: The role of neuroimaging. J. Neurotrauma 2015, 32, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Trinh, K.; Andrews, L.; Krause, J.; Hanak, T.; Lee, D.; Gelb, M.; Pallanck, L. Decaffeinated coffee and nicotine-free tobacco provide neuroprotection in Drosophila models of Parkinson’s disease through an NRF2-dependent mechanism. J. Neurosci. 2010, 30, 5525–5532. [Google Scholar] [CrossRef]




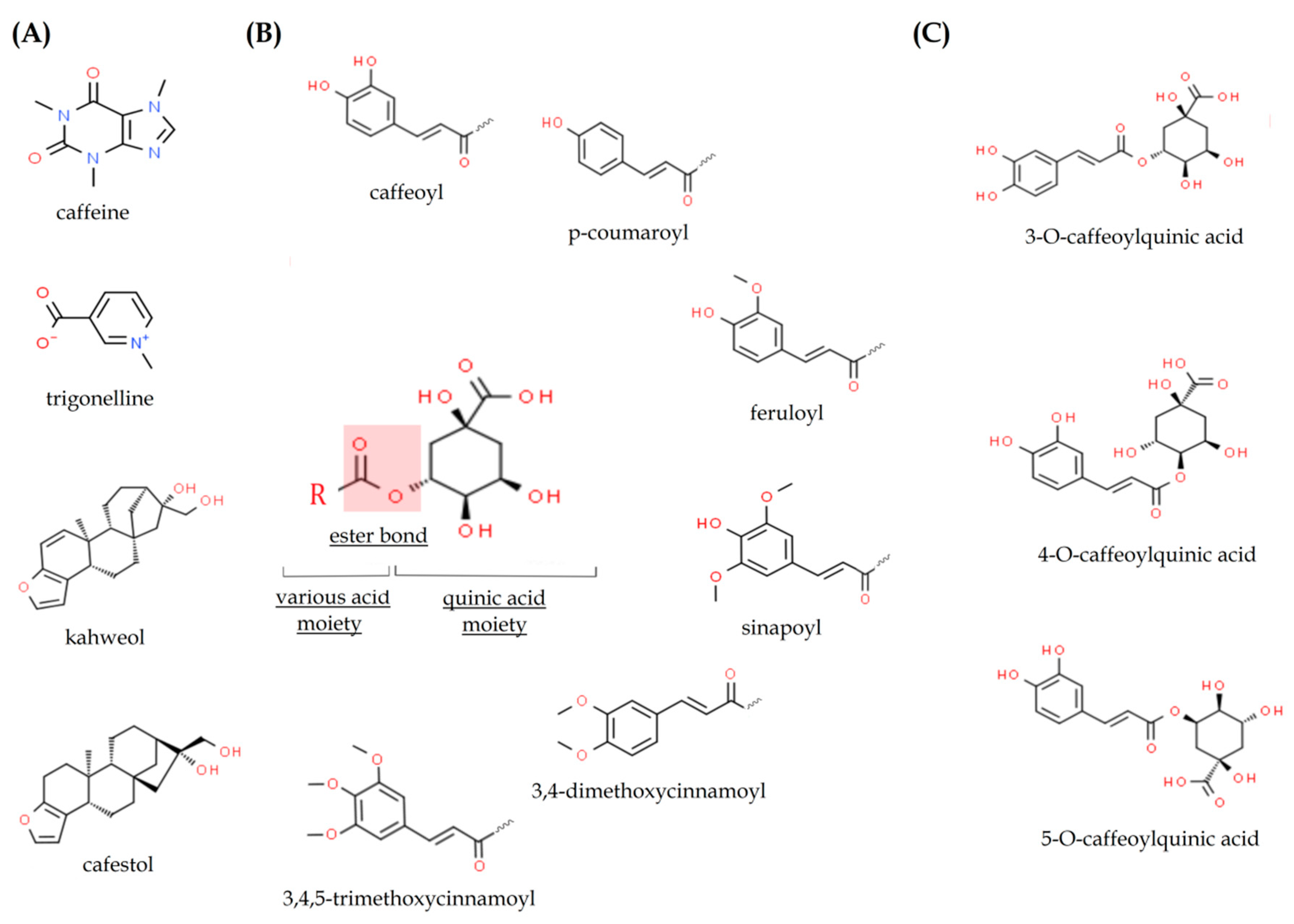{kind=link}
{kind=link}
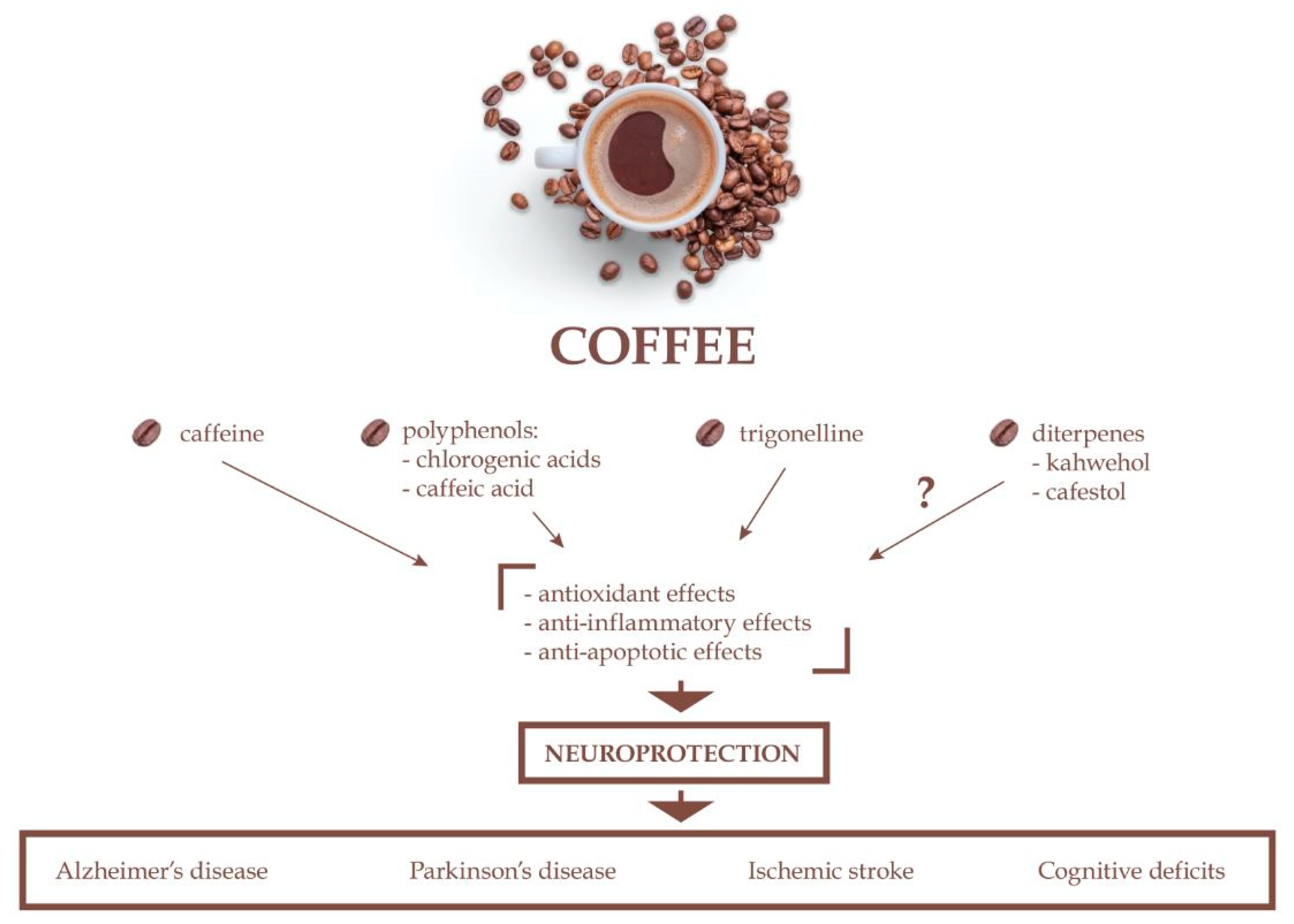{kind=link}
| Compounds | % Content in Dry Weight of Coffee Beans | |
|---|---|---|
| Green Coffee | Roasted Coffee | |
Carbohydrates
| 60 | 43 |
Lipids
| 8–18 | 10–15 |
Proteins
| 9–16 | 7.5–10 |
Other nitrogenous compounds
| 1–6 0.9–3.33 0.88–3.42 2 × 10−6–3 × 10−6 | 1–2 1 0.7–1 0.01–0.04 |
| Melanoidins | – | 25 |
| Minerals | 4 | 3.7–5 |
Organic and inorganic acids and esters
| 6–15 4–14.4 0.7–2.5 2 | 6 1–4 1.4–2.5 <0.3 |
| Compound | Content in Coffee Beverages or Brew Obtained from Blends of Arabica and Robusta Coffee [mg per 100 mL] |
|---|---|
| Water | 94,000–98,500 |
| Aliphatic acids and quinic acid | 692–2140 |
| Polysaccharides (galactomannans and type II arabinogalactans) | 200–700 |
| Lipids | 180–400 |
| Proteins | 120–400 |
| Simple saccharides (arabinose, mannose, galactose, sucrose) | 0–200 |
| Bioactive ingredients: | |
| Melanoidins | 500–1500 |
| Chlorogenic acids | 32–500 |
| Caffeine | 50–380 |
| Trigonelline | 12–50 |
| Diterpenes (cafestol and kahweol) | 0.2–10 |
| N-methylpyridinium | 2.9–8.7 |
| Serotonin | 0–1.4 |
| Polyamines (spermine and spermidine) | 0.4 |
| Phenolic substances | 0.1–0.2 |
| β-carbolins (norharman and harman) | 0.004–0.08 |
| Melatonin | 0.006–0.008 |
| Minerals: | |
| Total ashes | 150–500 |
| Potassium (K) | 115–320 |
| Sodium (Na) | 1–14 |
| Phosphorous (P) | 3–7 |
| Calcium (Ca) | 2–4 |
| Iron (Fe) | 0.02–0.13 |
| Manganese (Mn) | 0.02–0.05 |
| Zinc (Zn) | 0.01–0.05 |
| Vitamins: | |
| B3 | 0.8–10 |
| B9 | 1 |
| C | 0.2 |
| B2 | 0.177 |
| K | 0.1 |
| E | 0.01 |
| B6 | 0.002 |
| B1 | 0.001 |
| Undesirable substances: | |
| Acrylamide | 3.9–840 |
| Furan | 3.8–262 |
| N-alkanoyl-5-hydroxytryptamides | 1.2–34.3 |
| Animals | Treatment | Model | Behavioral Tests | Main Outcomes | Ref. |
|---|---|---|---|---|---|
| APPsw transgenic mice (background C57, B6, SJL and Swiss-Webster mice) | 0.3 mg/mL caffeinated water beginning at 4 months of age for 4 months (daily dose of 1.5 mg caffeine to each mouse) | Genetic model of Alzheimer’s disease | Open-field test, balance beam test, string-suspension, Y-maze test, elevated plus-maze test, Morris water maze test, circular platform test, platform recognition test, radial arm water maze test | (1) Improvement of cognitive task of spatial learning/reference memory, working memory, and recognition/identification, (2) decrease in Aβ production due to reduced expression of presenilin 1 and β-secretase, (3) restored adenosine levels in the brain to normal | [194] |
| APPsw transgenic mice (background C57, B6, SJL and Swiss-Webster mice) | 0.3 mg/mL caffeinated water beginning at 18–19 months of age for 4–5 weeks (daily dose of 1.5 mg caffeine to each mouse) | Genetic model of Alzheimer’s disease | Open-field test, balance beam test, string-suspension, Y-maze test, elevated plus-maze test, Morris water maze test, circular platform test, platform recognition test, radial arm water maze test | (1) Improvement of superior working memory, (2) reduced Aβ deposition in the hippocampus and entorhinal cortex, (3) decrease in brain soluble Aβ levels, (4) aged APPsw mice exhibited memory restoration and reversal of AD pathology, (5) caffeine suppression of β-secretase involves the cRaf-1/NFκB pathway | [195] |
| Albino rats (Morini, Wistar derived strain) | 15, 45, and 80 mg/kg/day (s.c.) for 15 days | – | Staircase test | No effect on memory retention | [192] |
| CF1 mice | 1 mg/mL for 12 months | – | Object recognition test | (1) Aged mice exhibited lower performance in the recognition memory compared with adults, (2) caffeine-treated mice showed similar performance to adult mice in the object recognition test and an improvement compared with their age-matched control mice, (3) caffeine counteracted the age-related increase in BDNF and TrkB immunocontent | [193] |
| CF1 mice | chronic (12 days) treatment with caffeine (1 mg/mL, p.o.); subchronic (4 days) treatment with caffeine (30 mg/kg, i.p.); acute caffeine treatment (30 or 80 mg/kg, i.p.) 30 min treatment before Aβ administration | Aβ25–35-induced neurotoxicity | Inhibitory avoidance test, Y-maze test | (1) Chronic and subchronic treatment with caffeine prevent Aβ-induced cognitive impairment, (2) A2A receptors are engaged in the control of Aβ-induced cognitive dysfunction | [197] |
| Wistar rats | 30 mg/kg (p.o.) daily per 10 days | Aging | Novel object recognition memory test, open field test | (1) Reversed age-related memory deficit, (2) normalized oxygen and NRS levels increased in brains of aged rats, (3) normalized Na+/K+-ATPase activity inhibited in brains of aged rats, (4) A2A receptors affect the impact and formation of free radicals in neuronal preparations | [199] |
| Sprague-Dawley rats | 3 mg/kg/day (i.p.) for 60 days | d-Galactose induced neurodegeneration | Y-maze test | (1) Attenuated memory impairment; (2) reduced oxidative stress via the reduction of 8-oxoguanine; (3) suppressed stress kinases p-JNK; (4) reduced d-galactose-induced neuroinflammation through alleviation of COX-2, NOS-2, TNFα, and IL-1β; (5) reduced cytochrome C, Bax/Bcl2 ratio, caspase-9, caspase-3, and PARP-1 levels; (6) prevented neurodegeneration | [191] |
| THY-Tau22 male mice (C57Bl6/J background) | 0.3 mg/mL caffeinated water beginning at 2 months until 12 months of age (daily dose of 1.5 mg caffeine to each mouse) | Genetic model of Alzheimer’s disease | Morris water maze test | (1) Prevented development of spatial memory impairments, (2) reduced tau phosphorylation and proteolytic fragments, (3) modulated hippocampal neuroinflammatory and oxidative stress markers | [200] |
| Sprague-Dawley rats | 0.3 or 0.6 mg/mL caffeinated water for 3 weeks or just once | – | – | Chronic caffeine treatment (1) induced ventriculomegaly, (2) increased production of CSF, which were associated with the enhancement of the expression of Na+/K+-ATPase and increased CBF | [202] |
| New Zealand white rabbits | 0.5 mg/day or 30 mg/day in the drinking water for 12 weeks | 2% cholesterol-enriched diet | – | (1) Decreased cholesterol-enriched diet-induced increase in Aβ production and accumulation, (2) reduced cholesterol-induced increase in tau phosphorylation, (3) attenuated cholesterol-induced increase in ROS and 8-Iso-PGF2α levels, (4) reduced glutathione depletion, (5) protection against cholesterol-induced endoplasmic reticulum stress, (6) reversed cholesterol-induced decrease in A1 receptor levels | [201] |
| C57BL/6NCrl mice | chronically (twice weekly for 8 weeks) caffeine 5 mg/kg or 20 mg/kg (i.p.), followed 10 min later 10 mg/kg PQ first and 30 mg/kg MB second | Chronic dual-pesticide exposure model of Parkinson’s disease | Horizontal locomotor activity test | Caffeine at 20 mg/kg reduced TH+ neuron loss | [212] |
| Wistar rats | 20 mg/kg (i.p.) 1 h before surgery and twice a day (10 mg/kg, i.p.) for 1 month; apomorphine hydrochloride (0.5 mg/kg, i.p.) 1 week before (baseline) and 4 weeks after the surgery with 1-day interval after the last caffeine injection | 6-OHDA-induced neurotoxicity | Apomorphine-induced rotation tests | Caffeine (1) reduced apomorphine-induced rotations in a 6-OHDA toxicity model, (2) protected the neurons of substantia nigra pars compacta against 6-OHDA toxicity | [211] |
| Wistar rats | 10 and 20 mg/kg (i.p.) daily for 14 days | 6-OHDA-induced neurotoxicity | Apomorphine-induced rotation tests | Caffeine (1) reduced apomorphine-induced rotations in a 6-OHDA toxicity model, (2) reversed decreased noradrenaline and dopamine levels caused by 6-OHDA unilateral intrastriatal injection | [210] |
| Swiss Albino mice | 20 mg/kg (i.p.) for 8 weeks | MPTP-induced neurotoxicity | – | Caffeine (1) partially protected MPTP-induced neurodegenerative changes, (2) modulated MPTP-mediated alterations in the expression and catalytic activity of CYP1A2, expression of adenosine A2A receptor and DAT | [207] |
| Wistar rats | 0.1, 0.3, or 1.0 mg/kg (i.p.) 45 min before the training session | MPTP-induced neurotoxicity | Two-way active avoidance test | Caffeine induced learning and memory improvement, what was independent of the locomotor stimulant effect; observed effects may be realized via dopamine/adenosine-receptor interaction | [206] |
| FVB mice | 10 mg/kg/day (i.p.) for 2 weeks | MPTP-induced neurotoxicity | – | Caffeine (1) protected against loss of dopaminergic neuron in striatum, (2) attenuated gliosis, (3) blocked leakage of the blood–brain barrier in striatum, (3) blocked decreases in levels of striatal tight junction proteins, (4) blocked increases in MMP9 activity | [205] |
| C57BL6 mice | 30 mg/kg (i.p.) for 8 days | MPTP-induced neurotoxicity | Paw grip strength test | Caffeine protected against (1) the reduction of paw grip strength, (2) perturbation in the homeostasis of neurometabolites in the striatum and olfactory bulb | [204] |
| C57BL6 mice | 10, 20, 40 mg/kg (i.p.) | MPTP-induced neurotoxicity | – | Caffeine (1) produced a dose-dependent attenuation of MPTP-induced striatal dopamine loss in both young and retired breeder male, but not female, mice; (2) was less potent or altogether ineffective in female mice as a neuroprotectant after sham surgery compared to ovariectomy or after ovariectomy plus estrogen replacement compared to ovariectomy plus placebo treatment; (3) protection against dopamine loss in young male mice was blocked by estrogen administration | [208] |
| C57BL6 mice | 30 mg/kg (i.p.) | MPTP-induced neurotoxicity | – | Caffeine (1) pre-treatment attenuated MPTP-induced striatal dopamine depletion when it was given 10 min, 30 min, 1 h, or 2 h but not 6 h before MPTP treatment; (2) post-treatment attenuated striatal dopamine loss when it was given 10 min, 30 min, 1 h or 2 h but not 4 h, 8 h or 24 h after MPTP injection; (3) metabolites also provide neuroprotective effect | [209] |
| Sprague–Dawley rats | 1 g/l in drinking water | MPTP-induced neurotoxicity | – | Caffeine treatment (1) initiated simultaneously or during the course of ongoing neurodegeneration reduces loss of nigral dopaminergic neurons, (2) did not modify MPTP-induced decreases in striatal dopamine or tyrosine hydroxylase, (3) attenuated microglia activation in the substantia nigra but not in the striatum of MPTP-treated rats | [213] |
| Wistar rats | 10 or 20 mg/kg/day in the drinking water | 6-OHDA-induced neurotoxicity | Open field test, apomorphine-induced rotation tests | Caffeine treatment (1) blocked partially decreased locomotor activity and a high number of apomorphine-induced rotations, (2) increased dopamine contents and reversed the decrease dopamine level in the striatum, (3) improved the hippocampal neuronal viability, (4) increased TH+ in the striatum, (5) decreased the number of immunopositive cells for histone deacetylase and pro-inflammatory cytokines TNF-α and IL-1β in the 6-OHDA-lesioned group | [214] |
| Mongolian gerbils | 0.1% caffeine drinking solution for 4 weeks | Ischemia model | – | Caffeine treatment (1) reduced the degree of ischemic necrosis of pyramidal cells of the CA1 hippocampal area after 5 min of bilateral carotid occlusion, (2) induced upregulation of A1 adenosine receptors in the CNS, what probably impaired the level of experimentally induced ischemic brain injury | [224] |
| Wistar rat pups | 10 mg/kg (i.p.) immediately following HI induction | HI neonatal model | Water escape test, Morris water maze test | Caffeine treatment (1) attenuated deficits on the Morris water maze test observed in HI animals, (2) might be a potential therapeutic agent in reducing ischemic brain injury | [228] |
| Wistar rat pups | 10 mg/kg/day (i.p.) immediately before HI and at 0, 24, 48 and 72 h post hypoxia | HI neonatal model | – | Caffeine treatment (1) reduced neuronal apoptosis in the developing brain, (2) might be effective in reducing ischemic brain injury | [229] |
| Wistar rat pups | 10 mg/kg (i.p.) immediately after the 120 min of HI and 24 h following the initial injection | HI neonatal model | Rota rod test, silent gap detection, non-spatial water maze test | Caffeine treatment (1) significantly improved some behavioral outcomes in rat with a neonatal HI brain injury induced on postnatal day 6 and (2) partially rescued neuropathology | [230] |
| Sprague-Dawley rat | 10 mg/kg (i.v.) 30 min prior to the induction of ischemia (acute treatment) 20 mg/kg (p.o.) three times daily per dose for the first week and 30 mg/kg (p.o.) three times daily for the second and third weeks; caffeine was withdrawn 24 h prior to ischemia. (chronic treatment) | Reversible forebrain ischemia model | – | Acute caffeine treatment (1) accelerated changes in the magnetic resonance images with increased hippocampal intensity appearing at 24 h post-ischemia, but (2) caused no changes in the extent of neuronal injury in any brain region compared to control-ischemic rats; (3) chronic caffeine treatment caused significantly less neuronal injury | [227] |
| Long-Evans rats | 10 mg/kg of caffeine and 5% or 10% ethanol (0.325 or 0.65 g/kg, respectively) acute or chronic (3 weeks) (p.o.) | Carotid/middle cerebral artery occlusion model of ischemia | – | Caffeine plus ethanol treatment (1) almost entirely eliminated the ischemic injury, (2) initiated at 30-, 60-, 90-, and 120-min post-ischemia significantly reduced the infarct volume; (3) for 3 weeks prior to ischemia eliminates the neuroprotection seen after acute treatment | [273] |
| Long-Evans rats | 2.5 h infusion at doses ranging from 2 to 10 mg/kg for caffeine and from 0.2 to 0.65 g/kg for ethanol | Carotid/middle cerebral artery occlusion model of ischemia | Sensorimotor tests: measurement of forelimb placing and foot-fault asymmetry | Caffeinol (0.2 g/kg of ethanol and 6 mg/kg of caffeine) treatment (1) reduced cortical infarct volume and (2) decreased behavioral dysfunction after transient carotid/middle cerebral artery occlusion | [274] |
| Sprague–Dawley rats | 10 mg/kg caffeine and/or ethanol 0.32 g/kg infusion via the left femoral vein | Carotid/middle cerebral artery occlusion model of ischemia | Sensorimotor tests: measurement of forelimb placing and foot-fault asymmetry, postural reflex | Caffeinol treatment reduced size of excitotoxic lesion and caffeine may augmented the anti-ischemic effect of NMDA receptor blockers | [277] |
| Animals | Treatment | Model | Behavioral Tests | Main Outcomes | Ref. |
|---|---|---|---|---|---|
| Wistar rats | 100 mg/kg (i.p.) for 24 days | Methotrexate-induced cerebellar Purkinje cell damage | – | (1) Reduced Purkinje cell damage and the expression of apoptotic cells, (2) decreased production of MDA and increase in SOD and catalase activity and GSH content in the cerebellum | [310] |
| Wistar rats | 60 mg/kg (p.o.) for 30 days | Cadmium-induced brain damage | – | (1) Restored AChE, SOD, catalase, GSH-Px, and GST activity; (2) restored GSH, vitamins C and E, and lipid peroxidation level; (3) increased membrane-bound ATPase activity; (4) attenuated mitochondrial dysfunction and DNA fragmentation | [311] |
| ICR mice | 3–9 mg/kg (p.o.) 30 min before scopolamine injection | Scopolamine-induced amnesia | Y-maze test, passive avoidance test, Morris water maze test | (1) Attenuation of the scopolamine-induced learning and memory impairment, (2) decreased AChE activity and MDA level in the hippocampus and frontal cortex. | [302] |
| Swiss Albino mice | 1–10 mg/kg (p.o.) for 8 days before scopolamine injection | Scopolamine-induced amnesia | Y-maze test, novel object recognition test | (1) Attenuation of the scopolamine-induced learning and memory impairments, (2) decreased AChE and BChE activities in the cortex and hippocampus, (3) increased free radical scavenging activity | [304] |
| Wistar rats (5 days old pups) | 100 and 200 mg/kg (p.o.) from PD 6 to 28 (with ethanol) | Alcohol-induced brain damage | Morris water maze test | (1) Attenuation of the altered cognitive function in ethanol-exposed pups, decreased AChE and caspase-3 activity, (2) reduced MDA and nitrite levels, (3) increased SOD and catalase activity, (4) decreased TNF-α and IL-1β levels, as well as decreased level of p65 of NF-κB in the cerebral cortex and hippocampus | [312] |
| C57BL/6 mice | 100 mg/kg (i.p.) for 5 days | 3-Nitropropionic acid induced neurotoxicity | – | Reduction of the 3-nitropropionic acid induced toxicity and genotoxicity | [313] |
| Wistar rats | 15–60 mg/kg (p.o.) for 7 days before ischemia induction | Focal cerebral ischemia/reperfusion injury | Neurological deficit scoring | (1) Reduced mortality and improved neurological deficit scores, (2) decreased cerebral infarction area, (3) reduced ICAM-1 and VCAM-1 levels, (4) increased erythropoietin and HIF-1α levels, and (5) increased expression of NGF in the brain | [314] |
| Sprague-Dawley rats | 20–500 mg/kg (p.o) for 7 days before ischemia induction | Cerebral ischemia/reperfusion injury | Neurological deficit scoring, step-down test, Y maze test | (1) Attenuation of the learning and memory impairments; (2) improved neurological deficit scores; (3) decreased cerebral infarction volume, cerebral water content and cerebral index; (4) promoted BDNF and NGF expression; (5) increased SOD activity and GSH levels; (6) decreased production of ROS, LDH, and MDA; (7) inhibited expression of caspase 3 and 9; and (8) promoted Nrf2, NQO-1, and HO-1 expression | [315] |
| Sprague-Dawley rats | 3–30 mg/kg (i.p.) twice at 0 h and 2 h after ischemia induction | Focal cerebral ischemia/reperfusion injury | Balance-beam test | (1) Reduced sensory-motor functional deficits, infarct volume, BBB damage, and brain edema and (2) decreased lipid peroxidation and the expressions of matrix metalloproteinases | [316] |
| Charles foster albino rats | 10 mg/kg (i.n.) after 2 h of occlusion | Global cerebral ischemia/reperfusion injury | – | (1) Reduced cerebral infarction volume and BBB damage; (2) restored the brain water content; (3) reduced calcium, nitrate, and glutamate levels in the cortex, hippocampus, cerebellum, and cerebrospinal fluid, and (4) decreased expression of TNF-α, iNOS, and caspase-3 | [317] |
| Mongolian gerbils | 100 µg/kg (i.p.) 60 min before injection of PEP-1-rpS3 | Transient cerebral ischemia/reperfusion injury | – | Enhanced neuroprotective activity of PEP-1-rpS3 against the ischemia-induced hippocampal damage | [318] |
| Wistar rats | 15–60 mg/kg (i.p.) 30 min after ischemia induction | Transient global ischemia/reperfusion injury | Morris water maze test | (1) Attenuation of the spatial memory impairment; (2) decreased CA1 pyramidal cell loss; (3) increased Bcl-2, SOD2, and CD31 expressions; and (4) decreased endothelin-1 expression | [319] |
| Mongolian gerbils | 7.5–30 mg/kg (i.p.) for 5 days before ischemia induction | Transient global cerebral ischemia injury | 8 Arm radial maze test, passive avoidance task | (1) Attenuation of cognitive impairment; (2) decreased CA1 pyramidal cell loss; (3) increased SOD2 expression; (4) reduced production of ROS, TNF-α, and IL-2 and elevated expression of IL-4 and IL-13 | [320] |
| Sprague-Dawley rats | 20–60 mg/kg (i.p.) 60 min before 6-OHDA injection, for 7 days | 6-OHDA-induced neurotoxicity | Rotarod test, apomorphine-induced rotational test | (1) Reversed motor deficits, (2) attenuated decrease in striatal dopamine concentration, (3) reduced α-synuclein accumulation, (4) increased SOD and GSH-Px activities, and (5) restored Bcl-2/Bax expression in the striatum | [305] |
| C57BL/6J mice | 50 mg/kg (p.o.) for 1 week before rotenone exposure, and then 5 days/week during the 4 weeks of rotenone treatment | Rotenone-induced neurotoxicity | – | (1) Prevented degeneration of dopaminergic neurons in the substantia nigra, (2) upregulated metallothionein-1 and 2 in striatal astrocytes | [321] |
| Swiss Albino mice | 50 mg/kg (p.o.) for 24 days | MPTP-induced neurotoxicity | Rotarod test, pole test, traction test, catalepsy test | (1) Improved motor coordination and neurobehavioral activity; (2) improved mitochondria function; (3) reduced ROS generation; (4) increased SOD and mitochondrial GSH activity; (5) inhibited activation of proapoptotic proteins (Bax and caspase-3); (6) elevated expression of Bcl-2; (7) improved phosphorylation state of Akt, ERK1/2, and GSK3β | [322] |
| C57BL/6J mice | 100 mg/kg (i.p.) for 7 days before LPS injection | LPS-induced neurotoxicity | – | Attenuation of the LPS-induced IL-1β and TNF-α release in the substantia nigra | [323] |
| Kunming mice | 1 ml (p.o.) twice daily for 35 days | Kainic acid-induced neurotoxicity | Y maze test | (1) Attenuation of learning and memory impairment, (2) increased number of nNOS-positive neurons in the hippocampal CA1–4 regions | [324] |
| Swiss Albino mice | 5 mg/kg (p.o.) for 15 days, last injection 30 min before pilocarpine | Pilocarpine-induced seizures | Seizure assessment (duration of clonic and tonic seizure) | (1) Anticonvulsant-like effect; (2) attenuated neuronal loss in the hippocampal CA1 region; (3) restored glutamate and GABA levels; (4) decreased NMDA, mGluR1, and mGluR5 receptor expression; (5) decreased lipid peroxidation and nitrite content; (6) increased SOD, catalase, and GSH activity; (7) restored AChE and monoamine oxidase activity | [325] |
| APP/PS2 transgenic mice | 40 mg/kg (p.o.) for 180 days | Genetic model of Alzheimer’s disease | Morris water maze test | (1) Improved spatial memory, (2) decreased neuronal damage in the hippocampus, (3) inhibited autophagy, and (4) activation of the mTOR/TFEB signaling pathway | [299] |
| Animals | Treatment | Model | Behavioral Tests | Main Outcomes | Ref. |
|---|---|---|---|---|---|
| Mice (KM strain) | 10 and 30 mg/kg (p.o.) 30 min before aluminum injection and then for 10 consecutive days | Aluminum-induced neurotoxicity | Passive avoidance task, water maze test | (1) Attenuation of the aluminum-induced impairment of learning and memory, (2) decreased MDA level, (3) increased choline acetyltransferase expression, (4) decreased expression of amyloid precursor protein of Aβ, and 5-LOX | [349] |
| Male Wistar rats | 100 mg/kg (p.o.) for 11 days | Aluminum-induced neurotoxicity | Morris water maze test | (1) Improved memory; (2) reduced AChE, catalase, and GST activity; (3) reduced GSH and nitrite levels | [350] |
| Wistar rats | 10–40 mg/kg (p.o.) for 21 days | Streptozotocin- induced dementia | Object recognition test, Morris water maze test, locomotor activity test | (1) Attenuation of the streptozotocin -induced learning and memory impairments; (2) increase in AChE activity; (3) increase in MDA, nitrite, and protein carbonyl levels; and (4) decrease in GSH level | [351] |
| Sprague–Dawley rats | 100 mg/kg (i.p.) for 2 weeks | Aβ1–40-induced neurotoxicity | Morris water maze test | (1) Improved cognitive deficits, (2) decreased AChE activity and nitrite generation, (3) increased activity of catalase and GSH, (4) reduced IL-6 and TNF-α levels, (5) decreased NF-κB-p65 protein expression and caspase-3 activity, and (6) decreased p53 and p-p38 MAPK protein expression | [352] |
| Wistar rats | 10–100 mg/kg (p.o.) for 30 days | – | Step-down inhibitory avoidance test, open field test | (1) Improved learning and memory; (2) decreased AChE activity in the cerebral cortex and striatum; and (3) increased AChE activity in the cerebellum, hippocampus, hypothalamus, and pons | [353] |
| Wistar rats | 4 mg/kg (i.p.) 30 min before pilocarpine injection | Pilocarpine-induced seizures | Seizure assessment (latency to the first seizure, % seizures) | (1) Anticonvulsant-like effect, (2) decreased lipid peroxidation level and nitrite content, (3) increased SOD and catalase activity | [355] |
| Wistar rats | 20 mg/kg (i.p.) for 5 days before quinolinic acid administration | Quinolinic acid-induced neurotoxicity | Circling behavior test, cylinder test | Attenuation of the quinolinic acid-induced behavioral alterations | [344] |
| Male Wistar rats | 5 and 10 mg/kg (p.o.) for 21 days | Quinolinic acid-induced neurotoxicity | Locomotor activity test, rotarod test | (1) Improvement of locomotor activity and motor coordination, (2) restored redox status in striatum | [356] |
| Fisher rats | 50 mg/kg (i.p.) 4 injections | Kainic acid-induced neurotoxicity | Seizure assessment (latency to seizures, seizure severity) | (1) Prolonged latency to seizures, (2) reduced neuronal loss in the CA3 hippocampal field | [357] |
| CF1 mice | 4 and 8 mg/kg (i.p.) 30 min before seizure induction | Pilocarpine- and pentylenetetrazole-induced seizures | Seizure assessment (latency to the first seizure, % seizures) | (1) No anticonvulsant-like effect, (2) protection against pilocarpine-induced genotoxic damage in the hippocampus | [358] |
| CF1 mice | 1–8 mg/kg (i.p.) 30 min before pentylenetetrazole injection, once every three day, for a total of 6 injections | Pentylenetetrazole -induced kindling | Seizure assessment (latency to the first seizure and the occurrence of clonic forelimb seizures) | (1) No antiepileptogenic-like effect, (2) protection against kindling-induced genotoxic damage in cerebral cortex, (3) decreased ROS production | [359] |
| Mongolian gerbils | 10 and 20 mg/kg (p.o.) for 3 days before ischemia induction | Transient cerebral ischemia injury | (1) Decreased cell damage in the ischemic hippocampal CA1 region, (2) inhibition of microglia activation | [360] | |
| Swiss mice | 2–60 mg/kg (i.p.) for 5 days | Focal cerebral ischemia injury | Neurological deficit scoring, passive avoidance test, Y-maze test, water maze test, open field test | (1) Reduced infarcted area and improved neurological deficit scores, (2) improvement of working, spatial, and long-term aversive memory deficits, (3) attenuation of the ischemia-induced reduction in synaptophysin expression, and (4) increase in caspase 3 expression | [361] |
| Sprague–Dawley rats | 50 mg/kg (i.p.) immediately after ischemia induction and then repeatedly for 12 h | Cerebral ischemia/reperfusion injury | Neurological deficit scoring | (1) Improved neurological deficit scores, (2) reduced infraction volume, (3) decreased 5-LOX expression | [362] |
| Sprague-Dawley rats | 50 mg/kg (i.p.) 30 min before ischemia induction and 0, 1, 2 h after reperfusion in 1st day, and twice daily in the 2nd to 5th day | Focal cerebral ischemia/reperfusion injury | Neurological deficit scoring, inclined board test | (1) Reduction of neurological deficits, (2) decreased neuron loss, infarct volume, brain atrophy, and astrocyte proliferation, (3) inhibition of leukotriene production | [363] |
| Sprague–Dawley rats | 10–50 mg/kg (i.p.) 30 min before ischemia induction | Global cerebral ischemia-reperfusion injury | Morris water maze test | (1) Attenuation of the ischemia-induced spatial learning and memory deficits, (2) reduced hippocampal neurons injury, (3) decreased MDA level, (4) increased SOD activity, and (5) suppressed 5-LOX overexpression | [364] |
| ICR mice | 10 and 50 mg/kg (i.p.) 30 min, 2 and 6 h after cryoinjury on the 1st day and twice daily on days 2 to 7 | Brain cryoinjury | – | (1) Reduced astrocyte proliferation and glial scar wall formation, (2) decreased expression of GFAP protein, (3) decreased SOD activity and (4) increased MDA level | [365] |
| Sprague-Dawley rats | 50 mg/kg (p.o.) 10.5, 5.5, and 0.5 h before LPS injection | LPS-induced neurotoxicity | – | Attenuation of the LPS-induced loss of dopaminergic neurons and microglial activation in the substantia nigra | [367] |
| C57BL/6 mice | 0.5–2% in diet, for 4 weeks | MPTP-induced neurotoxicity | _ | (1) Decreased inflammatory cytokines levels; (2) suppressed NO, prostaglandin E2, and GFAP production; (3) reserved BDNF, GDNF, and tyrosine hydroxylase levels; (4) improved synthesis of dopamine | [370] |
| C57BL/6J mice | 50 mg/kg (p.o.) for 1 week before rotenone exposure, and then 5 days/week during the 4 weeks of rotenone treatment | Rotenone-induced neurotoxicity | – | (1) Prevented degeneration of dopaminergic neurons in the substantia nigra, (2) upregulated metallothionein-1 and 2 in striatal astrocytes | [321] |
| Animals | Treatment | Model | Behavioral Tests | Main Outcomes | Ref. |
|---|---|---|---|---|---|
| ddY mice | 500 mg/kg (p.o.) for 15 days | Aβ25–35-induced memory impairment | Morris water maze test | Attenuated memory impairment | [393] |
| Wistar rats | 100 mg/kg (p.o.) for 3 days | Aβ25–35 induced neurotoxicity | Y maze test, novel object recognition task | (1) Attenuated learning and memory impairment; (2) alleviated hippocampal neuronal loss; (3) improved mitochondrial membrane potential; (4) restored MDA, protein carbonyl, and GSH levels; (5) reduced SOD and LDH activity; (6) reduced GFAP, S100b, COX-2, TNF-α, and IL-6 level in the hippocampus | [394] |
| Swiss Albino mice | 50 and 100 mg/kg (p.o.) for 28 days | LPS-induced neurotoxicity | Morris water maze test, Y maze test | (1) Attenuated learning and memory disturbances, (2) decreased AChE activity, (3) restored SOD activity, (4) restored GSH and lipid peroxidation levels, (5) decreased TNF-α and IL-6 levels, and (6) increased BDNF level | [395] |
| Wistar rats | 20–80 mg/kg (p.o.) for 7 days | LPS-induced neurotoxicity | Y maze test, Novel object discrimination test, passive avoidance test | (1) Attenuated learning and memory disturbances; (2) decreased MDA level and AChE activity; (3) increased SOD and catalase activity; (4) reduced GSH level; and (5) decreased NF-κB, TLR4, and TNF-α levels | [396] |
| Swiss Albino mice | 20–80 mg/kg (p.o.) for 6 weeks | d-Galactose induced cognitive impairment | Morris water maze test, Y maze test | (1) Attenuated learning and memory disturbances, (2) decreased AChE activity, (3) decreased AGEs and MDA levels, (4) increased SOD activity and GSH level | [397] |
| Wistar rats | 50 and 100 mg/kg (i.p.) for 3 days | 6-OHDA-induced neurotoxicity | Apomorphine-induced rotation test | (1) Reduced rotational behavior, (2) increased viability of neurons in substantia nigra, (3) prevented apoptosis, (4) reduced MDA and nitrite levels, and (5) increased GSH level | [398] |
| Wistar rats | Trigonella foenum-graecum extract (82% trigonelline) 30–100 mg/kg (p.o.), 2 weeks after 6-OHDA injection | 6-OHDA-induced neurotoxicity | Apomorphine-induced rotation test | Increased number of ipsilateral rotations | [399] |
| C57BL/6 mice | Trigonella foenum-graecum extract (82% trigonelline) 30 mg/kg (p.o.), 60 min before or after MPTP | MPTP-induced neurotoxicity | Open field test | Improved spontaneous locomotor activity in the pre-treatment schedule | [399] |
| Sprague–Dawley rats | 25–100 mg/kg (i.p.) twice (30 min before and immediately after ischemia induction) | Cerebral ischemia/reperfusion injury | Neurological deficit scoring, rotarod test | (1) Improved motor coordination and neurodeficit scores, (2) decreased cerebral infarction volume, (3) reduced nitrite and MDA levels, (4) increased GSH level, and (5) decreased expression of myeloperoxidase | [400] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Socała, K.; Szopa, A.; Serefko, A.; Poleszak, E.; Wlaź, P. Neuroprotective Effects of Coffee Bioactive Compounds: A Review. Int. J. Mol. Sci. 2021, 22, 107. https://doi.org/10.3390/ijms22010107
Socała K, Szopa A, Serefko A, Poleszak E, Wlaź P. Neuroprotective Effects of Coffee Bioactive Compounds: A Review. International Journal of Molecular Sciences. 2021; 22(1):107. https://doi.org/10.3390/ijms22010107
Chicago/Turabian StyleSocała, Katarzyna, Aleksandra Szopa, Anna Serefko, Ewa Poleszak, and Piotr Wlaź. 2021. "Neuroprotective Effects of Coffee Bioactive Compounds: A Review" International Journal of Molecular Sciences 22, no. 1: 107. https://doi.org/10.3390/ijms22010107
APA StyleSocała, K., Szopa, A., Serefko, A., Poleszak, E., & Wlaź, P. (2021). Neuroprotective Effects of Coffee Bioactive Compounds: A Review. International Journal of Molecular Sciences, 22(1), 107. https://doi.org/10.3390/ijms22010107