Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates


Abstract
1. Autophagy Process
1.1. Initiation of Autophagy
1.2. Autophagosome Formation
1.3. Autophagosome Maturation
1.4. Selective Autophagy
2. Autophagy in the Nervous System and Neurodegenerative Diseases
2.1. Roles of Autophagy in the Nervous System
2.2. Relationship of Autophagy Pathway with Pathogenesis of Neurodegenerative Diseases
2.2.1. Alzheimer’s Disease (AD)
2.2.2. Parkinson’s Disease (PD)
2.2.3. Huntington’s Disease (HD)
2.2.4. Amyotrophic Lateral Sclerosis (ALS)
2.2.5. Hereditary Spastic Paraplegia (HSP)
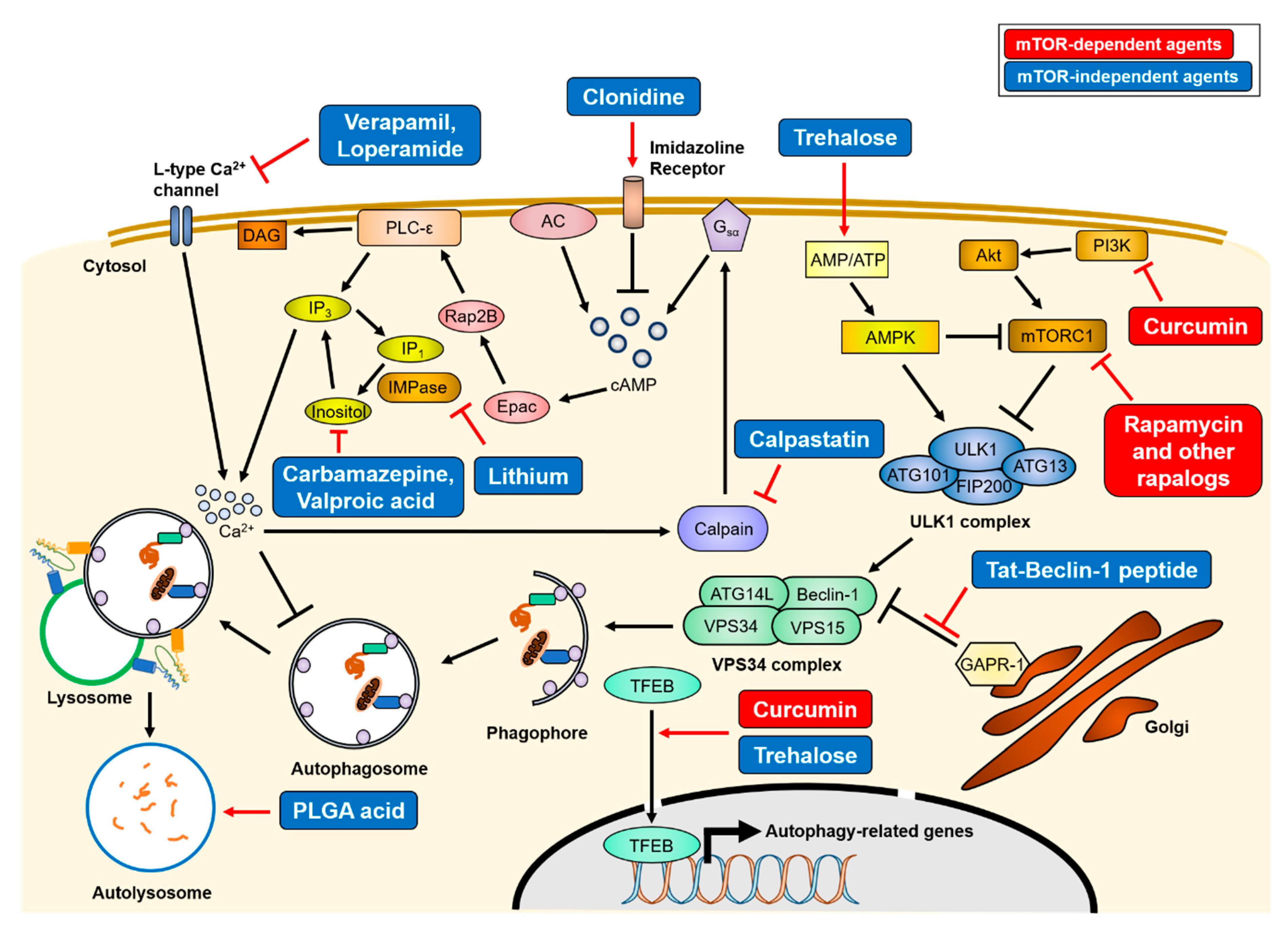
3. Autophagy Upregulation as a Therapeutic Strategy for Neurodegenerative Diseases
3.1. mTOR-Dependent Autophagy Inducing Agents
3.2. mTOR-Independent Autophagy Inducing Agents
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Di Nardo, A.; Wertz, M.H.; Kwiatkowski, E.; Tsai, P.T.; Leech, J.D.; Greene-Colozzi, E.; Goto, J.; Dilsiz, P.; Talos, D.M.; Clish, C.B.; et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum. Mol. Genet. 2014, 23, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.G.; Hurley, J.H. Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 2016, 39, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Turco, E.; Fracchiolla, D.; Martens, S. Recruitment and Activation of the ULK1/Atg1 Kinase Complex in Selective Autophagy. J. Mol. Biol. 2020, 432, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.J.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278. [Google Scholar] [CrossRef]
- Kim, B.W.; Jin, Y.; Kim, J.; Kim, J.H.; Jung, J.; Kang, S.; Kim, I.Y.; Kim, J.; Cheong, H.; Song, H.K. The C-terminal region of ATG101 bridges ULK1 and PtdIns3K complex in autophagy initiation. Autophagy 2018, 14, 2104–2116. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Giordano, F.; Dupont, N.; Grasso, D.; Vaccaro, M.I.; Codogno, P.; Morel, E. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 2017, 36, 2018–2033. [Google Scholar] [CrossRef]
- Molino, D.; Zemirli, N.; Codogno, P.; Morel, E. The Journey of the Autophagosome through Mammalian Cell Organelles and Membranes. J. Mol. Biol. 2017, 429, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell 2018, 45, 114–131.e118. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef]
- Lystad, A.H.; Carlsson, S.R.; de la Ballina, L.R.; Kauffman, K.J.; Nag, S.; Yoshimori, T.; Melia, T.J.; Simonsen, A. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat. Cell Biol. 2019, 21, 372–383. [Google Scholar] [CrossRef]
- Dudley, L.J.; Cabodevilla, A.G.; Makar, A.N.; Sztacho, M.; Michelberger, T.; Marsh, J.A.; Houston, D.R.; Martens, S.; Jiang, X.; Gammoh, N. Intrinsic lipid binding activity of ATG16L1 supports efficient membrane anchoring and autophagy. EMBO J. 2019, 38, e1000554. [Google Scholar] [CrossRef]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef]
- Maruyama, T.; Noda, N.N. Autophagy-regulating protease Atg4: Structure, function, regulation and inhibition. J. Antibiot. 2018, 71, 72–78. [Google Scholar] [CrossRef]
- Sakoh-Nakatogawa, M.; Matoba, K.; Asai, E.; Kirisako, H.; Ishii, J.; Noda, N.N.; Inagaki, F.; Nakatogawa, H.; Ohsumi, Y. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat. Struct. Mol. Biol. 2013, 20, 433–439. [Google Scholar] [CrossRef]
- Martens, S.; Nakamura, S.; Yoshimori, T. Phospholipids in Autophagosome Formation and Fusion. J. Mol. Biol. 2016, S0022-2836, 30455–30457. [Google Scholar] [CrossRef] [PubMed]
- Randall-Demllo, S.; Chieppa, M.; Eri, R. Intestinal epithelium and autophagy: Partners in gut homeostasis. Front. Immunol. 2013, 4, 301. [Google Scholar] [CrossRef] [PubMed]
- Popovic, D.; Dikic, I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep. 2014, 15, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ma, K.; Gao, R.; Mu, C.; Chen, L.; Liu, Q.; Luo, Q.; Feng, D.; Zhu, Y.; Chen, Q. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017, 27, 184–201. [Google Scholar] [CrossRef]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Kochl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell. Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef]
- Bento, C.F.; Puri, C.; Moreau, K.; Rubinsztein, D.C. The role of membrane-trafficking small GTPases in the regulation of autophagy. J. Cell. Sci. 2013, 126, 1059–1069. [Google Scholar] [CrossRef]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell. Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef]
- Imai, K.; Hao, F.; Fujita, N.; Tsuji, Y.; Oe, Y.; Araki, Y.; Hamasaki, M.; Noda, T.; Yoshimori, T. Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J. Cell. Sci. 2016, 129, 3781–3791. [Google Scholar] [CrossRef]
- Feng, Y.; Klionsky, D.J. Autophagic membrane delivery through ATG9. Cell Res. 2017, 27, 161–162. [Google Scholar] [CrossRef]
- Rieter, E.; Vinke, F.; Bakula, D.; Cebollero, E.; Ungermann, C.; Proikas-Cezanne, T.; Reggiori, F. Atg18 function in autophagy is regulated by specific sites within its beta-propeller. J. Cell. Sci. 2013, 126, 593–604. [Google Scholar] [CrossRef]
- Zheng, J.X.; Li, Y.; Ding, Y.H.; Liu, J.J.; Zhang, M.J.; Dong, M.Q.; Wang, H.W.; Yu, L. Architecture of the ATG2B-WDR45 complex and an aromatic Y/HF motif crucial for complex formation. Autophagy 2017, 13, 1870–1883. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Sekito, T.; Niimi, K.; Ohsumi, Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J. Biol. Chem. 2008, 283, 23972–23980. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Otomo, C.; Leitner, A.; Ohashi, K.; Aebersold, R.; Lander, G.C.; Otomo, T. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc. Natl. Acad. Sci. USA 2018, 115, E9792–E9801. [Google Scholar] [CrossRef]
- Bakula, D.; Muller, A.J.; Zuleger, T.; Takacs, Z.; Franz-Wachtel, M.; Thost, A.K.; Brigger, D.; Tschan, M.P.; Frickey, T.; Robenek, H.; et al. WIPI3 and WIPI4 beta-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat. Commun. 2017, 8, 15637. [Google Scholar] [CrossRef] [PubMed]
- Velikkakath, A.K.; Nishimura, T.; Oita, E.; Ishihara, N.; Mizushima, N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 2012, 23, 896–909. [Google Scholar] [CrossRef]
- Osawa, T.; Kotani, T.; Kawaoka, T.; Hirata, E.; Suzuki, K.; Nakatogawa, H.; Ohsumi, Y.; Noda, N.N. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol. 2019, 26, 281–288. [Google Scholar] [CrossRef]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef]
- Lee, J.A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef]
- Zhou, F.; Zou, S.; Chen, Y.; Lipatova, Z.; Sun, D.; Zhu, X.; Li, R.; Wu, Z.; You, W.; Cong, X.; et al. A Rab5 GTPase module is important for autophagosome closure. PLoS Genet. 2017, 13, e1007020. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with Lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell. Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.T.; Zhou, B.; Lin, M.Y.; Cai, Q.; Sheng, Z.H. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J. Cell. Biol. 2015, 209, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Holzbaur, E.L. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 2014, 30, 71–85. [Google Scholar] [CrossRef]
- Nian, F.S.; Li, L.L.; Cheng, C.Y.; Wu, P.C.; Lin, Y.T.; Tang, C.Y.; Ren, B.S.; Tai, C.Y.; Fann, M.J.; Kao, L.S.; et al. Rab18 Collaborates with Rab7 to Modulate Lysosomal and Autophagy Activities in the Nervous System: An Overlapping Mechanism for Warburg Micro Syndrome and Charcot-Marie-Tooth Neuropathy Type 2B. Mol. Neurobiol. 2019, 56, 6095–6105. [Google Scholar] [CrossRef]
- Maruzs, T.; Lorincz, P.; Szatmari, Z.; Szeplaki, S.; Sandor, Z.; Lakatos, Z.; Puska, G.; Juhasz, G.; Sass, M. Retromer Ensures the Degradation of Autophagic Cargo by Maintaining Lysosome Function in Drosophila. Traffic 2015, 16, 1088–1107. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Roney, J.C.; Cheng, X.T.; Li, S.; Cuddy, S.R.; Sheng, Z.H. Neuronal Soma-Derived Degradative Lysosomes Are Continuously Delivered to Distal Axons to Maintain Local Degradation Capacity. Cell Rep. 2019, 28, 51–64.e54. [Google Scholar] [CrossRef]
- Matsui, T.; Jiang, P.; Nakano, S.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell. Biol. 2018, 217, 2633–2645. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Rong, Y.; Liu, M.; Ma, L.; Du, W.; Zhang, H.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.; et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, L. Recent progress in autophagic lysosome reformation. Traffic 2017, 18, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Weller, S.G.; Schroeder, B.; Krueger, E.W.; Chi, S.; Casey, C.A.; McNiven, M.A. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J. Cell. Biol. 2013, 203, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Kraft, C.; Peter, M.; Hofmann, K. Selective autophagy: Ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 2010, 12, 836–841. [Google Scholar] [CrossRef]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet. Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Deng, Z.; Purtell, K.; Lachance, V.; Wold, M.S.; Chen, S.; Yue, Z. Autophagy Receptors and Neurodegenerative Diseases. Trends. Cell Biol. 2017, 27, 491–504. [Google Scholar] [CrossRef]
- Conway, O.; Akpinar, H.A.; Rogov, V.V.; Kirkin, V. Selective Autophagy Receptors in Neuronal Health and Disease. J. Mol. Biol. 2019, S0022-S2836, 2483–2509. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Savova, A.; Danieli, A.; Romanov, J.; Tremel, S.; Ebner, M.; Peterbauer, T.; Sztacho, M.; Trapannone, R.; Tarafder, A.K.; et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 2018, 37, e98308. [Google Scholar] [CrossRef]
- Isogai, S.; Morimoto, D.; Arita, K.; Unzai, S.; Tenno, T.; Hasegawa, J.; Sou, Y.S.; Komatsu, M.; Tanaka, K.; Shirakawa, M.; et al. Crystal structure of the ubiquitin-associated (UBA) domain of p62 and its interaction with ubiquitin. J. Biol. Chem. 2011, 286, 31864–31874. [Google Scholar] [CrossRef]
- Walinda, E.; Morimoto, D.; Sugase, K.; Konuma, T.; Tochio, H.; Shirakawa, M. Solution structure of the ubiquitin-associated (UBA) domain of human autophagy receptor NBR1 and its interaction with ubiquitin and polyubiquitin. J. Biol. Chem. 2014, 289, 13890–13902. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Rohaim, A.; Kato, R.; Dikic, I.; Wakatsuki, S.; Kawasaki, M. A novel mode of ubiquitin recognition by the ubiquitin-binding zinc finger domain of WRNIP1. FEBS J. 2016, 283, 2004–2017. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif—crucial for selective autophagy. J. Cell. Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Lohr, F.; Wakatsuki, S.; Dotsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef]
- Caccamo, A.; Ferreira, E.; Branca, C.; Oddo, S. p62 improves AD-like pathology by increasing autophagy. Mol. Psychiatry 2017, 22, 865–873. [Google Scholar] [CrossRef]
- Ramesh Babu, J.; Lamar Seibenhener, M.; Peng, J.; Strom, A.L.; Kemppainen, R.; Cox, N.; Zhu, H.; Wooten, M.C.; Diaz-Meco, M.T.; Moscat, J.; et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 2008, 106, 107–120. [Google Scholar] [CrossRef]
- Piras, A.; Collin, L.; Gruninger, F.; Graff, C.; Ronnback, A. Autophagic and lysosomal defects in human tauopathies: Analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun. 2016, 4. [Google Scholar] [CrossRef]
- Collin, L.; Bohrmann, B.; Gopfert, U.; Oroszlan-Szovik, K.; Ozmen, L.; Gruninger, F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer’s disease. Brain 2014, 137, 2834–2846. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, S.; Zheng, H. The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy 2019, 15, 583–598. [Google Scholar] [CrossRef]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Xiao, Q.; Gil, S.C.; Yan, P.; Wang, Y.; Han, S.; Gonzales, E.; Perez, R.; Cirrito, J.R.; Lee, J.M. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J. Biol. Chem. 2012, 287, 21279–21289. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Chang, J.C.; Fan, E.Y.; Flajolet, M.; Greengard, P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 17071–17076. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 8, 2813. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Odagiri, S.; Miki, Y.; Maruyama, A.; Nikaido, Y.; Mimura, J.; Mori, F.; Warabi, E.; Yanagawa, T.; Ueno, S.; et al. p62 Deficiency Enhances alpha-Synuclein Pathology in Mice. Brain Pathol. 2015, 25, 552–564. [Google Scholar] [CrossRef]
- Fu, Y.; Wu, P.; Pan, Y.; Sun, X.; Yang, H.; Difiglia, M.; Lu, B. A toxic mutant huntingtin species is resistant to selective autophagy. Nat. Chem. Biol. 2017, 13, 1152–1154. [Google Scholar] [CrossRef]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell. Sci. 2013, 126, 580–592. [Google Scholar] [CrossRef]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef]
- Fox, L.M.; Kim, K.; Johnson, C.W.; Chen, S.; Croce, K.R.; Victor, M.B.; Eenjes, E.; Bosco, J.R.; Randolph, L.K.; Dragatsis, I.; et al. Huntington’s Disease Pathogenesis Is Modified In Vivo by Alfy/Wdfy3 and Selective Macroautophagy. Neuron 2019, 105, 813–821.e816. [Google Scholar] [CrossRef]
- Cykowski, M.D.; Powell, S.Z.; Appel, J.W.; Arumanayagam, A.S.; Rivera, A.L.; Appel, S.H. Phosphorylated TDP-43 (pTDP-43) aggregates in the axial skeletal muscle of patients with sporadic and familial amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2018, 6, 28. [Google Scholar] [CrossRef]
- Brady, O.A.; Meng, P.; Zheng, Y.; Mao, Y.; Hu, F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 2011, 116, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Hiji, M.; Takahashi, T.; Fukuba, H.; Yamashita, H.; Kohriyama, T.; Matsumoto, M. White matter lesions in the brain with frontotemporal lobar degeneration with motor neuron disease: TDP-43-immunopositive inclusions co-localize with p62, but not ubiquitin. Acta Neuropathol. 2008, 116, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Mitsui, S.; Pan, L.; Otomo, A.; Kubo, M.; Sato, K.; Ono, S.; Onodera, W.; Abe, K.; Chen, X.; et al. Functional links between SQSTM1 and ALS2 in the pathogenesis of ALS: Cumulative impact on the protection against mutant SOD1-mediated motor dysfunction in mice. Hum. Mol. Genet. 2016, 25, 3321–3340. [Google Scholar] [CrossRef] [PubMed]
- Gal, J.; Strom, A.L.; Kwinter, D.M.; Kilty, R.; Zhang, J.; Shi, P.; Fu, W.; Wooten, M.W.; Zhu, H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J. Neurochem. 2009, 111, 1062–1073. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, M.; Huang, C.; Liu, X.; Huang, B.; Li, N.; Zhou, H.; Xia, X.G. Pathogenic Ubqln2 gains toxic properties to induce neuron death. Acta Neuropathol. 2015, 129, 417–428. [Google Scholar] [CrossRef]
- Chen, T.; Huang, B.; Shi, X.; Gao, L.; Huang, C. Mutant UBQLN2(P497H) in motor neurons leads to ALS-like phenotypes and defective autophagy in rats. Acta Neuropathol. Commun. 2018, 6, 122. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef]
- Jantrapirom, S.; Lo Piccolo, L.; Yoshida, H.; Yamaguchi, M. Depletion of Ubiquilin induces an augmentation in soluble ubiquitinated Drosophila TDP-43 to drive neurotoxicity in the fly. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864, 3038–3049. [Google Scholar] [CrossRef]
- Yap, C.C.; Winckler, B. Harnessing the power of the endosome to regulate neural development. Neuron 2012, 74, 440–451. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-monitoring and autophagy-deficient mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.-I.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef]
- Nishiyama, J.; Miura, E.; Mizushima, N.; Watanabe, M.; Yuzaki, M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 2007, 3, 591–596. [Google Scholar] [CrossRef]
- Liang, C.C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.L. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 2010, 285, 3499–3509. [Google Scholar] [CrossRef]
- Lattante, S.; de Calbiac, H.; Le Ber, I.; Brice, A.; Ciura, S.; Kabashi, E. Sqstm1 knock-down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum. Mol. Genet. 2015, 24, 1682–1690. [Google Scholar] [CrossRef]
- Wertz, M.H.; Mitchem, M.R.; Pineda, S.S.; Hachigian, L.J.; Lee, H.; Lau, V.; Powers, A.; Kulicke, R.; Madan, G.K.; Colic, M.; et al. Genome-wide In Vivo CNS Screening Identifies Genes that Modify CNS Neuronal Survival and mHTT Toxicity. Neuron 2020, S0896-S6273, 76–89. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.; Gopal, P.P.; Gubas, A.; Tooze, S.A.; Holzbaur, E.L. Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. Elife 2019, 8, e44219. [Google Scholar] [CrossRef] [PubMed]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef]
- Tsvetkov, A.S.; Arrasate, M.; Barmada, S.; Ando, D.M.; Sharma, P.; Shaby, B.A.; Finkbeiner, S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat. Chem. Biol. 2013, 9, 586–592. [Google Scholar] [CrossRef]
- Choy, R.W.; Cheng, Z.; Schekman, R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid beta (Abeta) production in the trans-Golgi network. Proc. Natl. Acad. Sci. USA 2012, 109, E2077–E2082. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, D.; Lee, T.H. Phosphorylation Signaling in APP Processing in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 209. [Google Scholar] [CrossRef]
- Lee, S.; Mankhong, S.; Kang, J.H. Extracellular Vesicle as a Source of Alzheimer’s Biomarkers: Opportunities and Challenges. Int. J. Mol. Sci. 2019, 20, 1728. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.R.; Cheng, K.C.; Chen, Y.R.; Lin, T.Y.; Cheung, C.H.A.; Wu, C.L.; Chiang, H.C. Dysfunction of different cellular degradation pathways contributes to specific beta-amyloid42-induced pathologies. FASEB J. 2018, 32, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Malampati, S.; Zeng, Y.; Durairajan, S.S.K.; Yang, C.B.; Tong, B.C.; Iyaswamy, A.; Shang, W.B.; Sreenivasmurthy, S.G.; Zhu, Z.; et al. A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell 2020, 19, e13069. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.; Song, H.J.; Garza, D.; Neufeld, T.P.; Salvaterra, P.M. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS ONE 2009, 4, e4201. [Google Scholar] [CrossRef]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. ELife 2017, 6, e21776. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Haung, W.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagty in Alzheimer disease: An immuno electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 20009. [Google Scholar] [CrossRef]
- Cho, S.J.; Lim, H.J.; Jo, C.; Park, M.H.; Han, C.; Koh, Y.H. Plasma ATG5 is increased in Alzheimer’s disease. Sci. Rep. 2019, 9, 4741. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef]
- Chong, C.M.; Ke, M.; Tan, Y.; Huang, Z.; Zhang, K.; Ai, N.; Ge, W.; Qin, D.; Lu, J.H.; Su, H. Presenilin 1 deficiency suppresses autophagy in human neural stem cells through reducing gamma-secretase-independent ERK/CREB signaling. Cell Death Dis. 2018, 9, 879. [Google Scholar] [CrossRef]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef]
- Coffey, E.E.; Beckel, J.M.; Laties, A.M.; Mitchell, C.H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014, 263, 111–124. [Google Scholar] [CrossRef]
- Xu, W.; Tan, L.; Yu, J.T. The Role of PICALM in Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 399–413. [Google Scholar] [CrossRef]
- Ando, K.; Brion, J.P.; Stygelbout, V.; Suain, V.; Authelet, M.; Dedecker, R.; Chanut, A.; Lacor, P.; Lavaur, J.; Sazdovitch, V.; et al. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer’s brains. Acta Neuropathol. 2013, 125, 861–878. [Google Scholar] [CrossRef]
- Moreau, K.; Fleming, A.; Imarisio, S.; Lopez Ramirez, A.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.; et al. PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 2014, 5, 4998. [Google Scholar] [CrossRef] [PubMed]
- Lucin, K.M.; O’Brien, C.E.; Bieri, G.; Czirr, E.; Mosher, K.I.; Abbey, R.J.; Mastroeni, D.F.; Rogers, J.; Spencer, B.; Masliah, E.; et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 2013, 79, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Zhu, W.; Plowey, E.D. BECN1/Beclin 1 sorts cell-surface APP/amyloid beta precursor protein for lysosomal degradation. Autophagy 2016, 12, 2404–2419. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Kent, K.; Pierce, A.; Leung, C.; Kang, M.S.; Okada, H.; Honig, L.; Vonsattel, J.P.; Kim, T.W. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann. Neurol. 2005, 58, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.J.; Johnson, G.V. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J. Biol. Chem. 2010, 285, 21978–21987. [Google Scholar] [CrossRef]
- Butzlaff, M.; Hannan, S.B.; Karsten, P.; Lenz, S.; Ng, J.; Vossfeldt, H.; Prussing, K.; Pflanz, R.; Schulz, J.B.; Rasse, T.; et al. Impaired retrograde transport by the Dynein/Dynactin complex contributes to Tau-induced toxicity. Hum. Mol. Genet. 2015, 24, 3623–3637. [Google Scholar] [CrossRef]
- Krüger, U.; Wang, Y.; Kumar, S.; Mandelkow, E.M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 2012, 33, 2291–2305. [Google Scholar] [CrossRef]
- Wang, Y.; Martinez-Vicente, M.; Kruger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Q.; Yuan, Y.H.; Chu, S.F.; Li, G.H.; Chen, N.H. E46K Mutant alpha-Synuclein Is Degraded by Both Proteasome and Macroautophagy Pathway. Molecules 2018, 23, 2839. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell. Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M. Formation of alpha-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef]
- Hoffmann, A.C.; Minakaki, G.; Menges, S.; Salvi, R.; Savitskiy, S.; Kazman, A.; Vicente Miranda, H.; Mielenz, D.; Klucken, J.; Winkler, J.; et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci. Rep. 2019, 9, 544. [Google Scholar] [CrossRef]
- Dinter, E.; Saridaki, T.; Nippold, M.; Plum, S.; Diederichs, L.; Komnig, D.; Fensky, L.; May, C.; Marcus, K.; Voigt, A.; et al. Rab7 induces clearance of alpha-synuclein aggregates. J. Neurochem. 2016, 138, 758–774. [Google Scholar] [CrossRef]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Cao, G.; Wei, G. A30P mutant alpha-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 2019, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Arotcarena, M.L.; Bourdenx, M.; Dutheil, N.; Thiolat, M.L.; Doudnikoff, E.; Dovero, S.; Ballabio, A.; Fernagut, P.O.; Meissner, W.G.; Bezard, E.; et al. Transcription factor EB overexpression prevents neurodegeneration in experimental synucleinopathies. JCI Insight 2019, 4, 129719. [Google Scholar] [CrossRef] [PubMed]
- Torra, A.; Parent, A.; Cuadros, T.; Rodriguez-Galvan, B.; Ruiz-Bronchal, E.; Ballabio, A.; Bortolozzi, A.; Vila, M.; Bove, J. Overexpression of TFEB Drives a Pleiotropic Neurotrophic Effect and Prevents Parkinson’s Disease-Related Neurodegeneration. Mol. Ther. 2018, 26, 1552–1567. [Google Scholar] [CrossRef]
- Garcia-Sanz, P.; Orgaz, L.; Bueno-Gil, G.; Espadas, I.; Rodriguez-Traver, E.; Kulisevsky, J.; Gutierrez, A.; Davila, J.C.; Gonzalez-Polo, R.A.; Fuentes, J.M.; et al. N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov. Disord. 2017, 32, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet. Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef]
- Velayati, A.; Yu, W.H.; Sidransky, E. The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr. Neurol. Neurosci. Rep. 2010, 10, 190–198. [Google Scholar] [CrossRef]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef]
- Manning-Bog, A.B.; Schule, B.; Langston, J.W. Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: A biological link between Gaucher disease and parkinsonism. Neurotoxicology 2009, 30, 1127–1132. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Velayati, A.; Sidransky, E.; Lee, J.C. Membrane-bound α-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol. Genet. Metab. 2013, 108, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Cardoso, S.M. LRRK2 at the Crossroad Between Autophagy and Microtubule Trafficking: Insights into Parkinson’s Disease. Neuroscientist 2017, 23, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J., 3rd; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.W.; Leung, L.K.; Lone, M.; Lizzio, M.A.; Guo, M. Novel ethyl methanesulfonate (EMS)-induced null alleles of the Drosophila homolog of LRRK2 reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis. Model. Mech. 2014, 7, 1351–1363. [Google Scholar] [CrossRef]
- Kett, L.R.; Dauer, W.T. Leucine-rich repeat kinase 2 for beginners: Six key questions. Cold Spring Harb. Perspect. Med. 2012, 2, a009407. [Google Scholar] [CrossRef]
- Alegre-Abarrategui, J.; Christian, H.; Lufino, M.M.; Mutihac, R.; Venda, L.L.; Ansorge, O.; Wade-Martins, R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009, 18, 4022–4034. [Google Scholar] [CrossRef]
- Plowey, E.D.; Cherra, S.J., 3rd; Liu, Y.J.; Chu, C.T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 2008, 105, 1048–1056. [Google Scholar] [CrossRef]
- Ramonet, D.; Daher, J.P.; Lin, B.M.; Stafa, K.; Kim, J.; Banerjee, R.; Westerlund, M.; Pletnikova, O.; Glauser, L.; Yang, L.; et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE 2011, 6, e18568. [Google Scholar] [CrossRef]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Rivero-Rios, P.; Fdez, E.; Blanca Ramirez, M.; Ferrer, I.; Aiastui, A.; Lopez De Munain, A.; Hilfiker, S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 2014, 23, 6779–6796. [Google Scholar] [CrossRef] [PubMed]
- Wallings, R.; Connor-Robson, N.; Wade-Martins, R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum. Mol. Genet. 2019, 28, 2696–2710. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef]
- Bento, C.F.; Ashkenazi, A.; Jimenez-Sanchez, M.; Rubinsztein, D.C. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat. Commun. 2016, 7, 11803. [Google Scholar] [CrossRef]
- Wang, R.; Tan, J.; Chen, T.; Han, H.; Tian, R.; Tan, Y.; Wu, Y.; Cui, J.; Chen, F.; Li, J.; et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J. Cell. Biol. 2019, 218, 267–284. [Google Scholar] [CrossRef]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef]
- Miura, E.; Hasegawa, T.; Konno, M.; Suzuki, M.; Sugeno, N.; Fujikake, N.; Geisler, S.; Tabuchi, M.; Oshima, R.; Kikuchi, A.; et al. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 2014, 71, 1–13. [Google Scholar] [CrossRef]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef]
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef]
- Takeuchi, T.; Nagai, Y. Protein Misfolding and Aggregation as a Therapeutic Target for Polyglutamine Diseases. Brain Sci. 2017, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Maat-Schieman, M.L.; Dorsman, J.C.; Smoor, M.A.; Siesling, S.; Van Duinen, S.G.; Verschuuren, J.J.; den Dunnen, J.T.; Van Ommen, G.J.; Roos, R.A. Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J. Neuropathol. Exp. Neurol. 1999, 58, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Heng, M.Y.; Duong, D.K.; Albin, R.L.; Tallaksen-Greene, S.J.; Hunter, J.M.; Lesort, M.J.; Osmand, A.; Paulson, H.L.; Detloff, P.J. Early autophagic response in a novel knock-in model of Huntington disease. Hum. Mol. Genet. 2010, 19, 3702–3720. [Google Scholar] [CrossRef] [PubMed]
- Erie, C.; Sacino, M.; Houle, L.; Lu, M.L.; Wei, J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur. J. Neurosci. 2015, 42, 1941–1951. [Google Scholar] [CrossRef]
- Nagata, E.; Sawa, A.; Ross, C.A.; Snyder, S.H. Autophagosome-like vacuole formation in Huntington’s disease lymphoblasts. Neuroreport 2004, 15, 1325–1328. [Google Scholar] [CrossRef]
- Rudnicki, D.D.; Pletnikova, O.; Vonsattel, J.P.; Ross, C.A.; Margolis, R.L. A comparison of huntington disease and huntington disease-like 2 neuropathology. J. Neuropathol. Exp. Neurol. 2008, 67, 366–374. [Google Scholar] [CrossRef]
- Wu, J.C.; Qi, L.; Wang, Y.; Kegel, K.B.; Yoder, J.; Difiglia, M.; Qin, Z.H.; Lin, F. The regulation of N-terminal Huntingtin (Htt552) accumulation by Beclin1. Acta Pharmacol. Sin. 2012, 33, 743–751. [Google Scholar] [CrossRef]
- Proenca, C.C.; Stoehr, N.; Bernhard, M.; Seger, S.; Genoud, C.; Roscic, A.; Paganetti, P.; Liu, S.; Murphy, L.O.; Kuhn, R.; et al. Atg4b-dependent autophagic flux alleviates Huntington’s disease progression. PLoS ONE 2013, 8, e68357. [Google Scholar] [CrossRef]
- Jia, K.; Hart, A.C.; Levine, B. Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 2007, 3, 21–25. [Google Scholar] [CrossRef]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Mealer, R.G.; Murray, A.J.; Shahani, N.; Subramaniam, S.; Snyder, S.H. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8. [Google Scholar] [CrossRef]
- Li, H.Y.; Yeh, P.A.; Chiu, H.C.; Tang, C.Y.; Tu, B.P. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS ONE 2011, 6, e23075. [Google Scholar] [CrossRef]
- Williams, K.L.; Warraich, S.T.; Yang, S.; Solski, J.A.; Fernando, R.; Rouleau, G.A.; Nicholson, G.A.; Blair, I.P. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527-e3. [Google Scholar] [CrossRef]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349, 650–655. [Google Scholar] [CrossRef]
- Torres, P.; Ramirez-Nunez, O.; Romero-Guevara, R.; Bares, G.; Granado-Serrano, A.B.; Ayala, V.; Boada, J.; Fontdevila, L.; Povedano, M.; Sanchis, D.; et al. Cryptic exon splicing function of TARDBP interacts with autophagy in nervous tissue. Autophagy 2018, 14, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.L.; Wu, L.S.; Lee, M.; Chang, C.W.; Cheng, W.C.; Fang, Y.S.; Chen, Y.R.; Cheng, P.L.; Shen, C.J. A robust TDP-43 knock-in mouse model of ALS. Acta Neuropathol. Commun. 2020, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed]
- N’Diaye, E.N.; Kajihara, K.K.; Hsieh, I.; Morisaki, H.; Debnath, J.; Brown, E.J. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009, 10, 173–179. [Google Scholar] [CrossRef]
- Rothenberg, C.; Srinivasan, D.; Mah, L.; Kaushik, S.; Peterhoff, C.M.; Ugolino, J.; Fang, S.; Cuervo, A.M.; Nixon, R.A.; Monteiro, M.J. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum. Mol. Genet. 2010, 19, 3219–3232. [Google Scholar] [CrossRef]
- Senturk, M.; Lin, G.; Zuo, Z.; Mao, D.; Watson, E.; Mikos, A.G.; Bellen, H.J. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat. Cell Biol. 2019, 21, 384–396. [Google Scholar] [CrossRef]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef]
- Tumbarello, D.A.; Waxse, B.J.; Arden, S.D.; Bright, N.A.; Kendrick-Jones, J.; Buss, F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol. 2012, 14, 1024–1035. [Google Scholar] [CrossRef]
- Sundaramoorthy, V.; Walker, A.K.; Tan, V.; Fifita, J.A.; McCann, E.P.; Williams, K.L.; Blair, I.P.; Guillemin, G.J.; Farg, M.A.; Atkin, J.D. Defects in optineurin—and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2015, 24, 3830–3846. [Google Scholar] [CrossRef]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006, 67, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Ryu, H.H.; Jun, M.H.; Jang, D.J.; Lee, J.A. The functional analysis of the CHMP2B missense mutation associated with neurodegenerative diseases in the endo-lysosomal pathway. Biochem. Biophys. Res. Commun. 2012, 421, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, C. Hereditary spastic paraplegia. Handb. Clin. Neurol. 2018, 148, 633–652. [Google Scholar] [CrossRef]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef]
- Lee, S.; Park, H.; Zhu, P.P.; Jung, S.Y.; Blackstone, C.; Chang, J. Hereditary spastic paraplegia SPG8 mutations impair CAV1-dependent, integrin-mediated cell adhesion. Sci. Signal. 2020, 13, eaau7500. [Google Scholar] [CrossRef]
- Varga, R.E.; Khundadze, M.; Damme, M.; Nietzsche, S.; Hoffmann, B.; Stauber, T.; Koch, N.; Hennings, J.C.; Franzka, P.; Huebner, A.K.; et al. In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet. 2015, 11, e1005454. [Google Scholar] [CrossRef]
- Chang, J.; Lee, S.; Blackstone, C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Investig. 2014, 124, 5249–5262. [Google Scholar] [CrossRef]
- Vantaggiato, C.; Panzeri, E.; Castelli, M.; Citterio, A.; Arnoldi, A.; Santorelli, F.M.; Liguori, R.; Scarlato, M.; Musumeci, O.; Toscano, A.; et al. ZFYVE26/SPASTIZIN and SPG11/SPATACSIN mutations in hereditary spastic paraplegia types AR-SPG15 and AR-SPG11 have different effects on autophagy and endocytosis. Autophagy 2019, 15, 34–57. [Google Scholar] [CrossRef]
- Vantaggiato, C.; Crimella, C.; Airoldi, G.; Polishchuk, R.; Bonato, S.; Brighina, E.; Scarlato, M.; Musumeci, O.; Toscano, A.; Martinuzzi, A.; et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain 2013, 136, 3119–3139. [Google Scholar] [CrossRef]
- Behne, R.; Teinert, J.; Wimmer, M.; D’Amore, A.; Davies, A.K.; Scarrott, J.M.; Eberhardt, K.; Brechmann, B.; Chen, I.P.; Buttermore, E.D.; et al. Adaptor protein complex 4 deficiency: A paradigm of childhood-onset hereditary spastic paraplegia caused by defective protein trafficking. Hum. Mol. Genet. 2020, 29, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.K.; Itzhak, D.N.; Edgar, J.R.; Archuleta, T.L.; Hirst, J.; Jackson, L.P.; Robinson, M.S.; Borner, G.H.H. AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat. Commun. 2018, 9, 3958. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Borner, G.H.; Edgar, J.; Hein, M.Y.; Mann, M.; Buchholz, F.; Antrobus, R.; Robinson, M.S. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol. Biol. Cell 2013, 24, 2558–2569. [Google Scholar] [CrossRef] [PubMed]
- Khundadze, M.; Ribaudo, F.; Hussain, A.; Rosentreter, J.; Nietzsche, S.; Thelen, M.; Winter, D.; Hoffmann, B.; Afzal, M.A.; Hermann, T.; et al. A mouse model for SPG48 reveals a block of autophagic flux upon disruption of adaptor protein complex five. Neurobiol. Dis. 2019, 127, 419–431. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef]
- Bove, J.; Martinez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Forouzanfar, F.; Read, M.I.; Barreto, G.E.; Sahebkar, A. Neuroprotective effects of curcumin through autophagy modulation. IUBMB Life 2019, 72, 652–664. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, X.; Teng, Z.; Zhang, T.; Li, Y. Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur. J. Pharmacol. 2014, 740, 312–320. [Google Scholar] [CrossRef]
- Liang, J.; Zhou, F.; Xiong, X.; Zhang, X.; Li, S.; Li, X.; Gao, M.; Li, Y. Enhancing the retrograde axonal transport by curcumin promotes autophagic flux in N2a/APP695swe cells. Aging 2019, 11, 7036–7050. [Google Scholar] [CrossRef]
- Jiang, T.F.; Zhang, Y.J.; Zhou, H.Y.; Wang, H.M.; Tian, L.P.; Liu, J.; Ding, J.Q.; Chen, S.D. Curcumin ameliorates the neurodegenerative pathology in A53T alpha-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J. Neuroimmune Pharmacol. 2013, 8, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.J.; Heitmeier, M.R.; Mayer, A.L.; Higgins, C.B.; Crowley, J.R.; Kraft, T.E.; Chi, M.; Newberry, E.P.; Chen, Z.; Finck, B.N.; et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci. Signal. 2016, 9, ra21. [Google Scholar] [CrossRef] [PubMed]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; van Zundert, B.; Hetz, C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Wang, X.; Yu, X.; Duan, W.; Hong, K.; Wang, J.; Han, H.; Li, C. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 2015, 298, 12–25. [Google Scholar] [CrossRef]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell. Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Neurodegenerative Disease | Autophagic Receptor/Adaptor | Target Aggregates | Reference |
|---|---|---|---|
| Alzheimer’s disease | p62 | Aβ | [65] |
| p62 | tau | [66,67,68,69] | |
| NDP52 | tau | [70] | |
| OPTN | tau | [69] | |
| PICALM | Aβ, APP-CTF | [71,72] | |
| Parkinson’s disease | p62 | α-synuclein | [73,74] |
| Huntington’s disease | p62 | mHtt | [75,76] |
| OPTN | mHtt | [77,78] | |
| ALFY | mHtt | [76,79] | |
| Amyotrophic lateral sclerosis | p62 | TDP-43 | [80,81,82] |
| p62 | SOD1 | [83,84] | |
| p62 | UBQLN2 | [85,86] | |
| OPTN | TDP-43 | [78,87] | |
| OPTN | SOD1 | [77,87,88] | |
| UBQLN2 | TDP-43 | [89] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Kang, J.-H.; Lee, S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. Int. J. Mol. Sci. 2020, 21, 3369. https://doi.org/10.3390/ijms21093369
Park H, Kang J-H, Lee S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. International Journal of Molecular Sciences. 2020; 21(9):3369. https://doi.org/10.3390/ijms21093369
Chicago/Turabian StylePark, Hyungsun, Ju-Hee Kang, and Seongju Lee. 2020. "Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates" International Journal of Molecular Sciences 21, no. 9: 3369. https://doi.org/10.3390/ijms21093369
APA StylePark, H., Kang, J.-H., & Lee, S. (2020). Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. International Journal of Molecular Sciences, 21(9), 3369. https://doi.org/10.3390/ijms21093369