Orphan Nuclear Receptor RORα Regulates Enzymatic Metabolism of Cerebral 24S-Hydroxycholesterol through CYP39A1 Intronic Response Element Activation

 ,
,  ,
,
Abstract
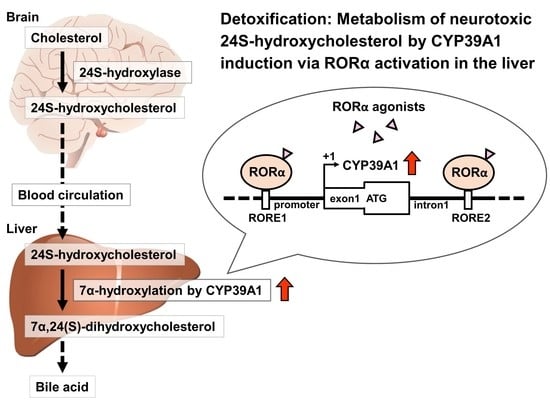
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. RORα Bound to ROREs of CYP39A1 Promoter and Intronic Regions
2.2. RORα and RORE Responses in CYP39A1 Promoter and Intronic Regions
2.3. CYP39A1 Expression Increased in Cells Overexpressing RORα
2.4. CYP39A1 Expression Decreased Following RORα Knockdown
2.5. CYP39A1 Expression Increased upon RORα Ligand Administration
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Electrophoretic Mobility Shift Assay (EMSA)
4.3. Chromatin Immunoprecipitation (ChIP)
4.4. Luciferase Reporter Assays
4.5. Quantitative Reverse Transcription-PCR (qRT-PCR)
4.6. Western Blotting
4.7. Overexpression Analysis
4.8. siRNA Knockdown Analysis
4.9. RORα Agonist Treatments
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 24S-OHC | 24S-hydroxycholesterol |
| AD | Alzheimer’s disease |
| Aβ | amyloid-β peptide |
| BBB | blood brain barrier |
| ChIP | chromatin immunoprecipitation |
| DMEM | Dulbecco’s modified Eagle’s medium |
| EMSA | electrophoretic mobility shift assay |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-coenzyme A |
| IκB | inhibitor of nuclear factor-κB |
| LDH | lactate dehydrogenase |
| LXR | liver x receptor |
| mt | mutant-type |
| NASH | nonalcoholic steatohepatitis |
| REV-ERB | reverse orientation the c-erbA-1 gene |
| RORα | retinoic acid receptor-related orphan receptor α |
| RORE | RORα response element |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| qRT-PCR | quantitative reverse transcription-PCR |
| siRNA | small interfering RNA |
| TSS | transcription start site |
| wt | wild-type |
References
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef]
- Iuliano, L.; Crick, P.J.; Zerbinati, C.; Tritapepe, L.; Abdel-Khalik, J.; Poirot, M.; Wang, Y.; Griffiths, W.J. Cholesterol metabolites exported from human brain. Steroids 2015, 99, 189–193. [Google Scholar] [CrossRef]
- Iuliano, L. Pathways of cholesterol oxidation via non-enzymatic mechanisms. Chem. Phys. Lipids 2011, 164, 457–468. [Google Scholar] [CrossRef]
- Björkhem, I.; Lütjohann, D.; Diczfalusy, U.; Ståhle, L.; Ahlborg, G.; Wahren, J. Cholesterol homeostasis in human brain: Turnover of 24S- hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 1998, 39, 1594–1600. [Google Scholar]
- Li-Hawkins, J.; Lund, E.G.; Bronson, A.D.; Russell, D.W. Expression cloning of an oxysterol 7α-hydroxylase selective for 24- hydroxycholesterol. J. Biol. Chem. 2000, 275, 16543–16549. [Google Scholar] [CrossRef] [PubMed]
- Heverin, M.; Meaney, S.; Lütjohann, D.; Diczfalusy, U.; Wahren, J.; Björkhem, I. Crossing the barrier: Net flux of 27-hydroxycholesterol into the human brain. J. Lipid Res. 2005, 46, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Heverin, M.; Bogdanovic, N.; Lütjohann, D.; Bayer, T.; Pikuleva, I.; Bretillon, L.; Diczfalusy, U.; Winblad, B.; Björkhem, I. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J. Lipid Res. 2004, 45, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Gamba, P.; Testa, G.; Gargiulo, S.; Staurenghi, E.; Poli, G.; Leonarduzzi, G. Oxidized cholesterol as the driving force behind the development of Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Ghribi, O. Does the oxysterol 27-hydroxycholesterol underlie Alzheimer’s disease-Parkinson’s disease overlap? Exp. Gerontol. 2015, 68, 13–18. [Google Scholar] [CrossRef]
- Gongol, B.; Marin, T.L.; Jeppson, J.D.; Mayagoitia, K.; Shin, S.; Sanchez, N.; Kirsch, W.M.; Vinters, H.V.; Wilson, C.G.; Ghribi, O.; et al. Cellular hormetic response to 27-hydroxycholesterol promotes neuroprotection through AICD induction of MAST4 abundance and kinase activity. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Bogdanovic, N.; Bretillon, L.; Lund, E.G.; Diczfalusy, U.; Lannfelt, L.; Winblad, B.; Russell, D.W.; Björkhem, I. On the turnover of brain cholesterol in patients with Alzheimer’s disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neurosci. Lett. 2001, 314, 45–48. [Google Scholar] [PubMed]
- Björkhem, I.; Cedazo-Minguez, A.; Leoni, V.; Meaney, S. Oxysterols and neurodegenerative diseases. Mol. Asp. Med. 2009, 30, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Tanzi, R.E.; Kovacs, D.M. Alzheimer’s disease: The cholesterol connection. Nat. Neurosci. 2003, 6, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Lütjohann, D.; Papassotiropoulos, A.; Björkhem, I.; Locatelli, S.; Bagli, M.; Oehring, R.D.; Schlegel, U.; Jessen, F.; Rao, M.L.; Von Bergmann, K.; et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J. Lipid Res. 2000, 41, 195–198. [Google Scholar]
- Yamanaka, K.; Saito, Y.; Yamamori, T.; Urano, Y.; Noguchi, N. 24(S)-hydroxycholesterol induces neuronal cell death through necroptosis, a form of programmed necrosis. J. Biol. Chem. 2011, 286, 24666–24673. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. 24S-hydroxycholesterol in plasma: A marker of cholesterol turnover in neurodegenerative diseases. Biochimie 2013, 95, 595–612. [Google Scholar] [CrossRef]
- Guissart, C.; Latypova, X.; Rollier, P.; Khan, T.N.; Stamberger, H.; McWalter, K.; Cho, M.T.; Kjaergaard, S.; Weckhuysen, S.; Lesca, G.; et al. Dual Molecular Effects of Dominant RORA Mutations Cause Two Variants of Syndromic Intellectual Disability with Either Autism or Cerebellar Ataxia. Am. J. Hum. Genet. 2018, 102, 744–759. [Google Scholar] [CrossRef]
- Acquaah-Mensah, G.K.; Agu, N.; Khan, T.; Gardner, A. A regulatory role for the insulin- and BDNF-Linked RORA in the hippocampus: Implications for Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 827–838. [Google Scholar] [CrossRef]
- Jetten, A.M.; Kang, H.S.; Takeda, Y. Retinoic acid-related orphan receptors α and γ: Key regulators of lipid/glucose metabolism, inflammation, and insulin sensitivity. Front. Endocrinol. 2013, 4, 1–8. [Google Scholar] [CrossRef]
- Hamilton, B.A.; Frankel, W.N.; Kerrebrock, A.W.; Hawkins, T.L.; FitzHugh, W.; Kusumi, K.; Russell, L.B.; Mueller, K.L.; Van Berkel, V.; Birren, B.W.; et al. Disruption of the nuclear hormone receptor RORα in staggerer mice. Nature 1996, 379, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Boukhtouche, F.; Vodjdani, G.; Jarvis, C.I.; Bakouche, J.; Staels, B.; Mallet, J.; Mariani, J.; Lemaigre-Dubreuil, Y.; Brugg, B. Human retinoic acid receptor-related orphan receptor α1 overexpression protects neurones against oxidative stress-induced apoptosis. J. Neurochem. 2006, 96, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Mutemberezi, V.; Guillemot-Legris, O.; Muccioli, G.G. Oxysterols: From cholesterol metabolites to key mediators. Prog. Lipid Res. 2016, 64, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kumar, N.; Crumbley, C.; Griffin, P.R.; Burris, T.P. A second class of nuclear receptors for oxysterols: Regulation of RORα and RORγ activity by 24S-hydroxycholesterol (cerebrosterol). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 917–923. [Google Scholar] [CrossRef]
- Wada, T.; Hong, S.K.; Angers, M.; Gong, H.; Bhatia, S.; Khadem, S.; Ren, S.; Ellis, E.; Strom, S.C.; Jetten, A.M.; et al. Identification of oxysterol 7α-hydroxylase (Cyp7b1) as a novel retinoid-related orphan receptor α (RORα) (NR1F1) target gene and a functional cross-talk between RORα and liver X receptor (NR1H3). Mol. Pharmacol. 2008, 73, 891–899. [Google Scholar] [CrossRef]
- Pathak, P.; Li, T.; Chiang, J.Y.L. Retinoic acid-related orphan receptor α regulates diurnal rhythm and fasting induction of sterol 12α-hydroxylase in bile acid synthesis. J. Biol. Chem. 2013, 288, 37154–37165. [Google Scholar] [CrossRef]
- Kojetin, D.J.; Burris, T.P. REV-ERB and ROR nuclear receptors as drug targets. Nat. Rev. Drug Discov. 2014, 13, 197–216. [Google Scholar] [CrossRef]
- Jelinek, D.F.; Andersson, S.; Slaughter, C.A.; Russell, D.W. Cloning and regulation of cholesterol 7α-hydroxylase, the rate-limiting enzyme in bile acid biosynthesis. J. Biol. Chem. 1990, 265, 8190–8197. [Google Scholar]
- Reed, B.D.; Charos, A.E.; Szekely, A.M.; Weissman, S.M.; Snyder, M. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet. 2008, 4, e1000133. [Google Scholar] [CrossRef]
- Björkhem, I.; Andersson, U.; Ellis, E.; Alvelius, G.; Ellegård, L.; Diczfalusy, U.; Sjövall, J.; Einarsson, C. From brain to bile: Evidence that conjugation and ω-hydroxylation are important for elimination of 24S-hydroxycholesterol (cerebrosterol) in humans. J. Biol. Chem. 2001, 276, 37004–37010. [Google Scholar] [CrossRef]
- Wang, Y.; Kumar, N.; Nuhant, P.; Cameron, M.D.; Istrate, M.A.; Roush, W.R.; Griffin, P.R.; Burris, T.P. Identification of SR1078, a synthetic agonist for the orphan nuclear receptors RORα and RORγ. ACS Chem. Biol. 2010, 5, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Shaul, O. How introns enhance gene expression. Int. J. Biochem. Cell Biol. 2017, 91, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.K.; Angers, M.; Ju, Y.B.; Wu, X.; Gimble, J.M.; Wada, T.; Xie, W.; Collins, J.B.; Grissom, S.F.; Jetten, A.M. Gene expression profiling reveals a regulatory role for RORα and RORγ in phase I and phase II metabolism. Physiol. Genom. 2007. 31, 281–294.
- Takeda, Y.; Kang, H.S.; Lih, F.B.; Jiang, H.; Blaner, W.S.; Jetten, A.M. Retinoid acid-related orphan receptor γ, RORγ, participates in diurnal transcriptional regulation of lipid metabolic genes. Nucleic Acids Res. 2014, 42, 10448–10459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Papazyan, R.; Damle, M.; Fang, B.; Jager, J.; Feng, D.; Peed, L.C.; Guan, D.; Sun, Z.; Lazar, M.A. The hepatic circadian clock fine-tunes the lipogenic response to feeding through RORα/γ. Genes Dev. 2017, 31, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.A.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXRα. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]
- Wada, T.; Kang, H.S.; Jetten, A.M.; Xie, W. The emerging role of nuclear receptor RORα and its crosstalk with LXR in xeno- and endobiotic gene regulation. Exp. Biol. Med. 2008, 233, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Billon, C.; Sitaula, S.; Burris, T.P. Inhibition of RORα/γ suppresses atherosclerosis via inhibition of both cholesterol absorption and inflammation. Mol. Metab. 2016, 5, 997–1005. [Google Scholar] [CrossRef]
- Matsuoka, H.; Tokunaga, R.; Katayama, M.; Hosoda, Y.; Miya, K.; Sumi, K.; Ohishi, A.; Kamishikiryo, J.; Shima, A.; Michihara, A. Retinoic acid receptor-related orphan receptor α reduces lipid droplets by upregulating neutral cholesterol ester hydrolase 1 in macrophages. BMC Mol. Cell Biol. 2020, 21, 32. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, H.J.; Na, H.; Nam, M.W.; Kim, J.Y.; Kim, J.S.; Koo, S.H.; Lee, M.O. RORα Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Rep. 2017, 20, 124–135. [Google Scholar] [CrossRef]
- Wang, Y.; Billon, C.; Walker, J.K.; Burris, T.P. Therapeutic Effect of a Synthetic RORα/γ Agonist in an Animal Model of Autism. ACS Chem. Neurosci. 2016, 7, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Shafaati, M.; Olin, M.; Båvner, A.; Pettersson, H.; Rozell, B.; Meaney, S.; Parini, P.; Björkhem, I. Enhanced production of 24S-hydroxycholesterol is not sufficient to drive liver X receptor target genes in vivo. J. Intern. Med. 2011, 270, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPARγ Agonists as Therapeutics for the Treatment of Alzheimer’s Disease. Neurotherapeutics 2008, 5, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Boo, K.; Yu, Y.S.; Oh, S.K.; Kim, H.; Jeon, Y.; Bhin, J.; Hwang, D.; Kim, I.K.; Lee, J.S.; et al. RORα controls hepatic lipid homeostasis via negative regulation of PPARγ transcriptional network. Nat. Commun. 2017, 8, 162. [Google Scholar] [CrossRef]
- Matsuoka, H.; Shima, A.; Kuramoto, D.; Kikumoto, D.; Matsui, T.; Michihara, A. Phosphoenolpyruvate carboxykinase, a key enzyme that controls blood glucose, is a target of retinoic acid receptor-related orphan receptor α. PLoS ONE 2015, 10, e0137955. [Google Scholar] [CrossRef]
- Delerive, P.; Monté, D.; Dubois, G.; Trottein, F.; Fruchart-Najib, J.; Mariani, J.; Fruchart, J.C.; Staels, B. The orphan nuclear receptor RORα is a negative regulator of the inflammatory response. EMBO Rep. 2001, 2, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, H.; Tamura, A.; Kinehara, M.; Shima, A.; Uda, A.; Tahara, H.; Michihara, A. Levels of tight junction protein CLDND1 are regulated by microRNA-124 in the cerebellum of stroke-prone spontaneously hypertensive rats. Biochem. Biophys. Res. Commun. 2018, 498, 817–823. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuoka, H.; Katayama, M.; Ohishi, A.; Miya, K.; Tokunaga, R.; Kobayashi, S.; Nishimoto, Y.; Hirooka, K.; Shima, A.; Michihara, A. Orphan Nuclear Receptor RORα Regulates Enzymatic Metabolism of Cerebral 24S-Hydroxycholesterol through CYP39A1 Intronic Response Element Activation. Int. J. Mol. Sci. 2020, 21, 3309. https://doi.org/10.3390/ijms21093309
Matsuoka H, Katayama M, Ohishi A, Miya K, Tokunaga R, Kobayashi S, Nishimoto Y, Hirooka K, Shima A, Michihara A. Orphan Nuclear Receptor RORα Regulates Enzymatic Metabolism of Cerebral 24S-Hydroxycholesterol through CYP39A1 Intronic Response Element Activation. International Journal of Molecular Sciences. 2020; 21(9):3309. https://doi.org/10.3390/ijms21093309
Chicago/Turabian StyleMatsuoka, Hiroshi, Miyu Katayama, Ami Ohishi, Kaoruko Miya, Riki Tokunaga, Sou Kobayashi, Yuya Nishimoto, Kazutake Hirooka, Akiho Shima, and Akihiro Michihara. 2020. "Orphan Nuclear Receptor RORα Regulates Enzymatic Metabolism of Cerebral 24S-Hydroxycholesterol through CYP39A1 Intronic Response Element Activation" International Journal of Molecular Sciences 21, no. 9: 3309. https://doi.org/10.3390/ijms21093309
APA StyleMatsuoka, H., Katayama, M., Ohishi, A., Miya, K., Tokunaga, R., Kobayashi, S., Nishimoto, Y., Hirooka, K., Shima, A., & Michihara, A. (2020). Orphan Nuclear Receptor RORα Regulates Enzymatic Metabolism of Cerebral 24S-Hydroxycholesterol through CYP39A1 Intronic Response Element Activation. International Journal of Molecular Sciences, 21(9), 3309. https://doi.org/10.3390/ijms21093309