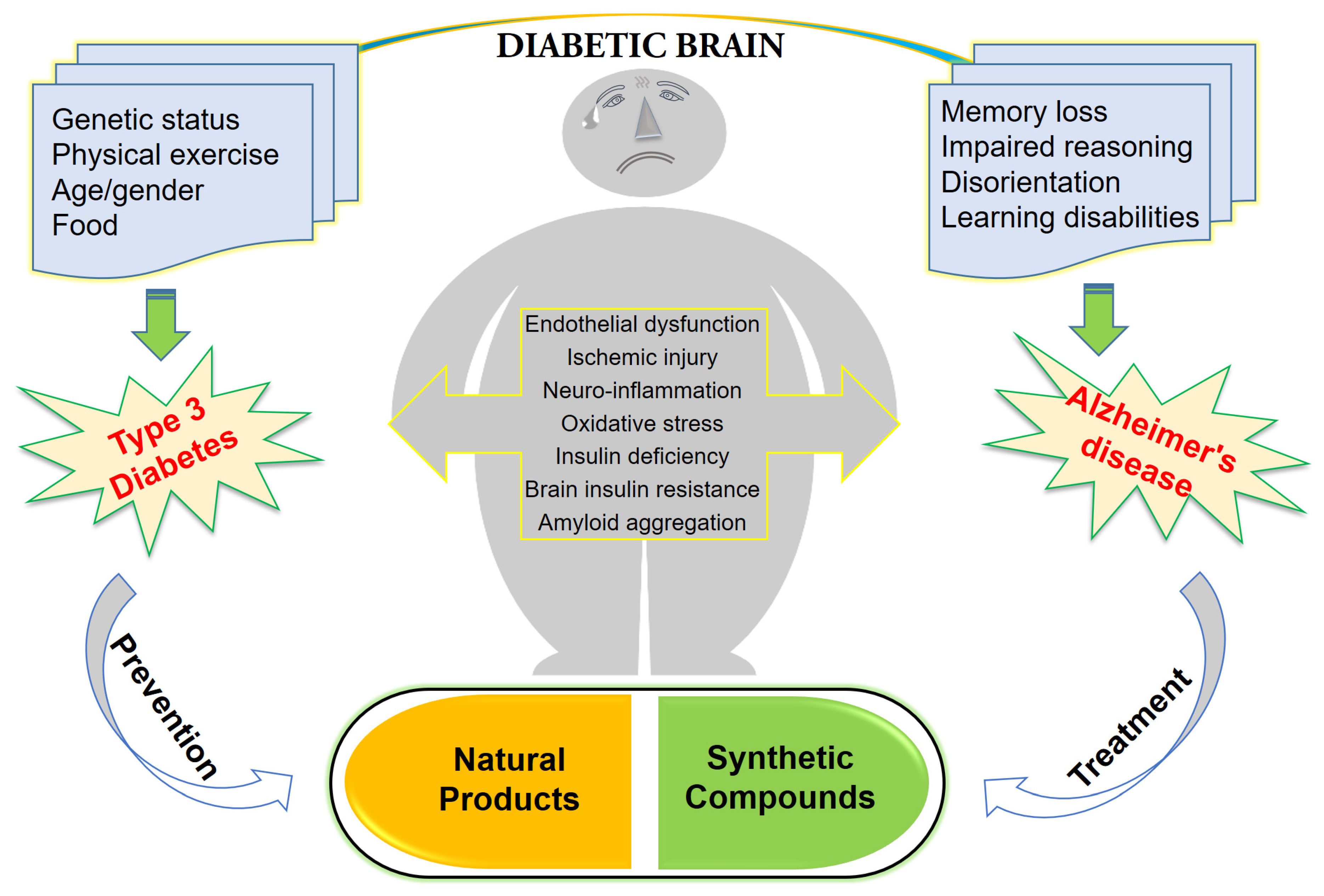
Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease


Abstract
1. Introduction
2. Insulin and Glucagon Signaling in the Central Nervous System (CNS)
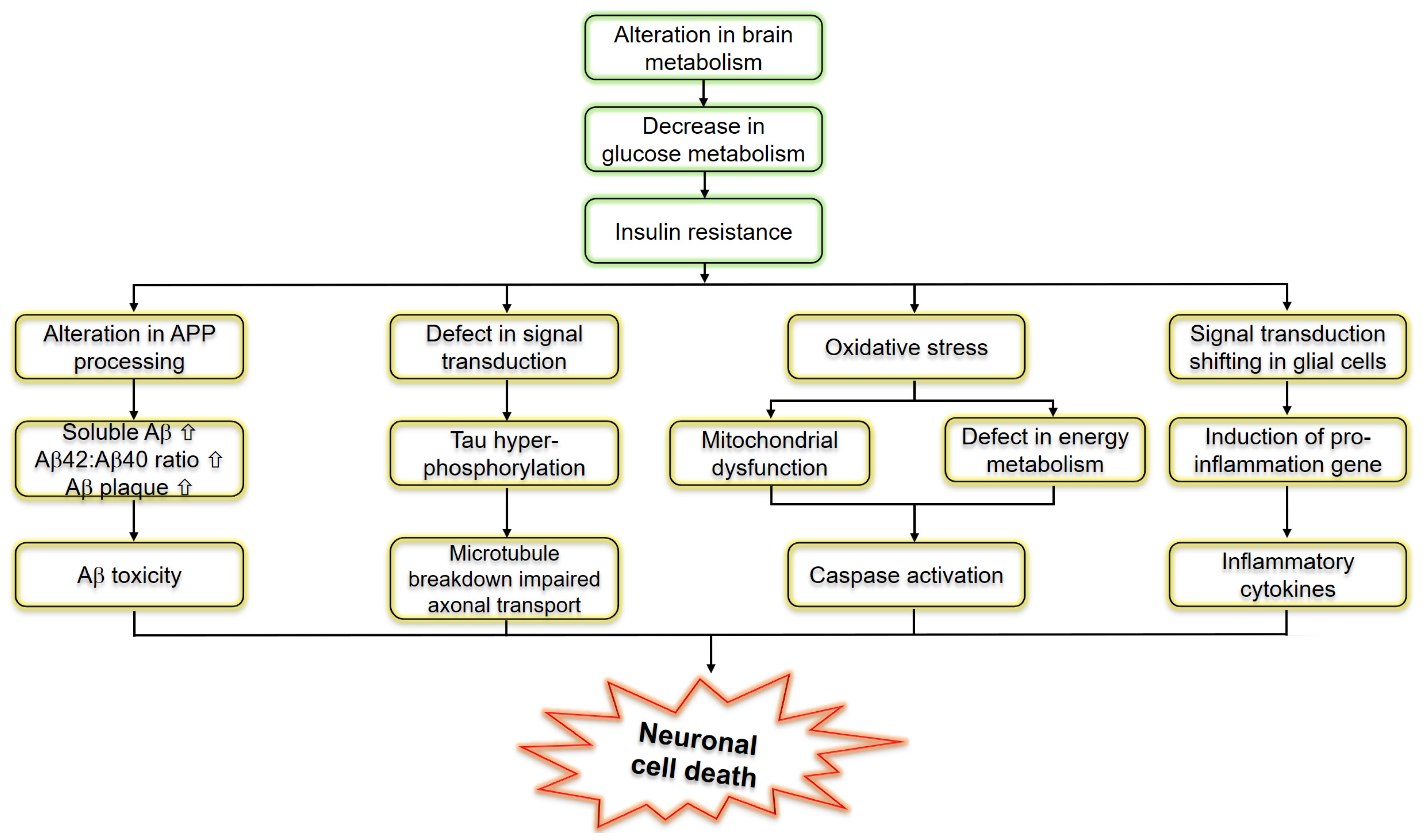
3. The Role of Type 3 Diabetes in Glucose Homeostasis
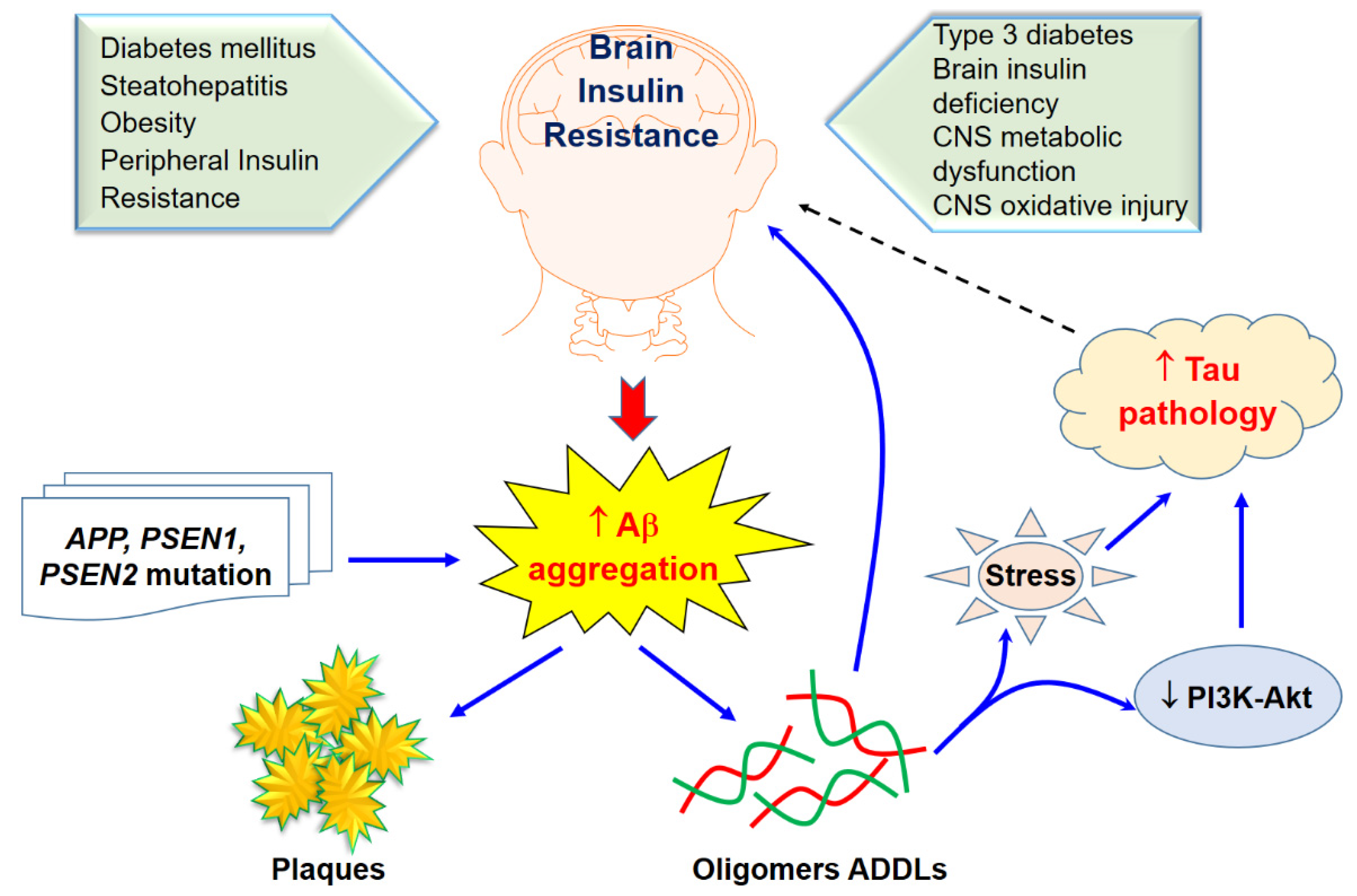
4. Type 3 Diabetes and Aβ Protein Pathology
5. Type 3 Diabetes Regarding Alzheimer’s Disease
6. Therapeutic Approaches to Type 3 Diabetes in Alzheimer’s Disease
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Ngoc, N.B.; Lin, Z.L.; Ahmed, W. Diabetes: What Challenges Lie Ahead for Vietnam? Ann. Glob. Health 2020, 86, 1. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Giau, V.V.; Vo, T.K. Current advances in transdermal delivery of drugs for Alzheimer’s disease. Indian J. Pharmacol. 2017, 49, 145–154. [Google Scholar]
- Bedse, G.; di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef]
- Duarte, J.M.N. Metabolic Alterations Associated to Brain Dysfunction in Diabetes. Aging Dis. 2015, 6, 304–321. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.T.D.; Le, T.T.; Vo, V.G. Role of Insulin Resistance in the Alzheimer’s Disease Progression. Neurochem. Res. 2020. [Google Scholar] [CrossRef]
- Caberlotto, L.; Nguyen, T.P.; Lauria, M.; Priami, C.; Rimondini, R.; Maioli, S.; Cedazo-Minguez, A.; Sita, G.; Morroni, F.; Corsi, M.; et al. Cross-disease analysis of Alzheimer’s disease and type-2 Diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci. Rep. 2019, 9, 3965. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Candeias, E.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Insulin signaling, glucose metabolism and mitochondria: Major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012, 1441, 64–78. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; de Felice, F.G. Insulin Resistance in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 830. [Google Scholar] [CrossRef] [PubMed]
- Rorbach-Dolata, A.; Piwowar, A. Neurometabolic Evidence Supporting the Hypothesis of Increased Incidence of Type 3 Diabetes Mellitus in the 21st Century. Biomed. Res. Int. 2019, 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; Davis-Plourde, K.L.; Conner, S.; Himali, J.J.; Beiser, A.S.; Lee, A.; Rawlings, A.M.; Sedaghat, S.; Ding, J.; Moshier, E.; et al. Association of metformin, sulfonylurea and insulin use with brain structure and function and risk of dementia and Alzheimer’s disease: Pooled analysis from 5 cohorts. PLoS ONE 2019, 14, e0212293. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. The insulin receptor: Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946. [Google Scholar] [CrossRef]
- Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. Embo J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef]
- Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef]
- Lee, C.C.; Huang, C.C.; Hsu, K.S. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology 2011, 61, 867–879. [Google Scholar] [CrossRef]
- Lee, S.-H.; Zabolotny, J.M.; Huang, H.; Lee, H.; Kim, Y.-B. Insulin in the nervous system and the mind: Functions in metabolism, memory, and mood. Mol. Metab. 2016, 5, 589–601. [Google Scholar] [CrossRef]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Han, Y. Insulin inhibits AMPA-induced neuronal damage via stimulation of protein kinase B (Akt). J. Neural Transm. (Vienna, Austria: 1996) 2005, 112, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T. Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn. J. Basic. Med. Sci. 2016, 16, 162–179. [Google Scholar] [CrossRef]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar] [CrossRef] [PubMed]
- Dorn, A.; Bernstein, H.G.; Rinne, A.; Ziegler, M.; Hahn, H.J.; Ansorge, S. Insulin- and glucagonlike peptides in the brain. Anat. Rec. 1983, 207, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Young, W.S., 3rd. Periventricular hypothalamic cells in the rat brain contain insulin mRNA. Neuropeptides 1986, 8, 93–97. [Google Scholar] [CrossRef]
- Gerozissis, K. Brain insulin: Regulation, mechanisms of action and functions. Cell. Mol. Neurobiol. 2003, 23, 1–25. [Google Scholar] [CrossRef]
- Clarke, D.W.; Mudd, L.; Boyd, F.T., Jr.; Fields, M.; Raizada, M.K. Insulin is released from rat brain neuronal cells in culture. J. Neurochem. 1986, 47, 831–836. [Google Scholar] [CrossRef]
- Pomytkin, I.; Costa-Nunes, J.P.; Kasatkin, V.; Veniaminova, E.; Demchenko, A.; Lyundup, A.; Lesch, K.-P.; Ponomarev, E.D.; Strekalova, T. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci. Ther. 2018, 24, 763–774. [Google Scholar] [CrossRef]
- Hancock, M.L.; Meyer, R.C.; Mistry, M.; Khetani, R.S.; Wagschal, A.; Shin, T.; Sui, S.J.H.; Näär, A.M.; Flanagan, J.G. Insulin Receptor Associates with Promoters Genome-wide and Regulates Gene Expression. Cell 2019, 177, 722–736.e22. [Google Scholar] [CrossRef]
- Frolich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Turk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. (Vienna, Austria: 1996) 1998, 105, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Gubellini, P.; Picconi, B.; di Filippo, M.; Calabresi, P. Downstream mechanisms triggered by mitochondrial dysfunction in the basal ganglia: From experimental models to neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 151–161. [Google Scholar] [CrossRef]
- Apelt, J.; Mehlhorn, G.; Schliebs, R. Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J. Neurosci. Res. 1999, 57, 693–705. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Holscher, C. Common pathological processes in Alzheimer disease and type 2 diabetes: A review. Brain Res. Rev. 2007, 56, 384–402. [Google Scholar] [CrossRef]
- Goldstein, B.J. Insulin resistance as the core defect in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 3g–10g. [Google Scholar] [CrossRef]
- Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, Z.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578. [Google Scholar] [CrossRef]
- Hardigan, T.; Ward, R.; Ergul, A. Cerebrovascular complications of diabetes: Focus on cognitive dysfunction. Clin. Sci. (Lond.) 2016, 130, 1807–1822. [Google Scholar] [CrossRef]
- Hoyer, S. The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: An update. J. Neural Transm. (Vienna, Austria: 1996) 2002, 109, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Martins, R.N.; Racchi, M.; Craft, S.; Helmerhorst, E. Amyloid beta antagonizes insulin promoted secretion of the amyloid beta protein precursor. J. Alzheimer’s Dis. Jad. 2002, 4, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.H.; Kar, S.; Quirion, R. Insulin-like growth factor-1-induced phosphorylation of the forkhead family transcription factor FKHRL1 is mediated by Akt kinase in PC12 cells. J. Biol. Chem. 2000, 275, 39152–39158. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of β-Amyloid Precursor Protein Trafficking by Insulin Reduces Intraneuronal β-Amyloid and Requires Mitogen-Activated Protein Kinase Signaling. J. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Delikkaya, B.; Moriel, N.; Tong, M.; Gallucci, G.; de la Monte, S.M. Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-ε4–associated Alzheimer’s disease, Alzheimer’s & Dementia: Diagnosis. Assess. Dis. Monit. 2019, 11, 392–404. [Google Scholar]
- Mittal, K.; Mani, R.J.; Katare, D.P. Type 3 Diabetes: Cross Talk between Differentially Regulated Proteins of Type 2 Diabetes Mellitus and Alzheimer’s Disease. Sci. Rep. 2016, 6, 25589. [Google Scholar] [CrossRef]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. A J. Clin. Ther. 2009, 14, 373–379. [Google Scholar]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- de la Monte, S.M. Type 3 diabetes is sporadic Alzheimer’s disease: Mini-review. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Razay, G.; Vreugdenhil, A.; Wilcock, G. Obesity, abdominal obesity and Alzheimer disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 173–176. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Haring, H.U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef]
- Lillioja, S.; Mott, D.M.; Spraul, M.; Ferraro, R.; Foley, J.E.; Ravussin, E.; Knowler, W.C.; Bennett, P.H.; Bogardus, C. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N. Engl. J. Med. 1993, 329, 1988–1992. [Google Scholar] [CrossRef]
- Li, J.; Bai, L.; Wei, F.; Zhao, J.; Wang, D.; Xiao, Y.; Yan, W.; Wei, J. Therapeutic Mechanisms of Herbal Medicines Against Insulin Resistance: A Review. Front. Pharmacol. 2019, 10, 661. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 589–596. [Google Scholar] [CrossRef]
- Fontaine, J.F.; Barbosa-Silva, A.; Schaefer, M.; Huska, M.R.; Muro, E.M.; Andrade-Navarro, M.A. MedlineRanker: Flexible ranking of biomedical literature. Nucleic Acids Res. 2009, 37, W141–W146. [Google Scholar] [CrossRef]
- Wang, G. Raison d’être of insulin resistance: The adjustable threshold hypothesis. J. R. Soc. Interface 2014, 11, 20140892. [Google Scholar] [CrossRef]
- Nisr, R.B.; Affourtit, C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim. Biophys. Acta 2014, 1837, 270–276. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed]
- Bucht, G.; Adolfsson, R.; Lithner, F.; Winblad, B. Changes in blood glucose and insulin secretion in patients with senile dementia of Alzheimer type. Acta Med. Scand. 1983, 213, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Matioli, M.N.P.S.; Nitrini, R. Mechanisms linking brain insulin resistance to Alzheimer’s disease. Dement. Neuropsychol. 2015, 9, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Tran, A.; Ishimwe, E.; Denner, L.; Dave, N.; Oddo, S.; Dineley, K.T. Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol. Aging 2017, 58, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, H.H.; Chi, T.; Shin, A.C.; Lindtner, C.; Hsieh, W.; Ehrlich, M.; Gandy, S.; Buettner, C. Increased susceptibility to metabolic dysregulation in a mouse model of Alzheimer’s disease is associated with impaired hypothalamic insulin signaling and elevated BCAA levels. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 851–861. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Manning, S.; McClean, P.L.; Coakley, M.F.; O’Halloran, D.J.; Holscher, C.; O’Neill, C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromol. Med. 2013, 15, 102–114. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Clarke, J.R.; Lyra, E.S.N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazao, R.; et al. Alzheimer-associated Abeta oligomers impact the central nervous system to induce peripheral metabolic deregulation. Embo Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Tanokashira, D.; Fukuokaya, W.; Taguchi, A. Involvement of insulin receptor substrates in cognitive impairment and Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1330–1334. [Google Scholar] [PubMed]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stohr, O.; Koch, L.; Kohler, C.; Udelhoven, M.; Leeser, U.; Muller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3315–3324. [Google Scholar]
- Taguchi, A.; Wartschow, L.M.; White, M.F. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science (N. Y.) 2007, 317, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Scales, G.; Leroy, K.; Causevic, M.; Hooper, C.; Irvine, E.E.; Choudhury, A.I.; Drinkwater, L.; Kerr, F.; Al-Qassab, H.; et al. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem. Biophys. Res. Commun. 2009, 386, 257–262. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Pham, Q.T.; Luong, T.N.H.; Le, H.K.; Vo, V.G. Potential Antidiabetic Activity of Extracts and Isolated Compound from Adenosma bracteosum (Bonati). Biomolecules 2020, 10, 201. [Google Scholar] [CrossRef]
- Giau, V.V.; Wu, S.Y.; Jamerlan, A.; An, S.S.A.; Kim, S.; Hulme, J. Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer’s Disease. Nutrients 2018, 10, 1765. [Google Scholar] [CrossRef]
- de Matos, A.M.; de Macedo, M.P.; Rauter, A.P. Bridging Type 2 Diabetes and Alzheimer’s Disease: Assembling the Puzzle Pieces in the Quest for the Molecules With Therapeutic and Preventive Potential. Med. Res. Rev. 2018, 38, 261–324. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Giau, V.V.; Shim, K.; Suk, K.; An, S.S.A.; Kim, S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J. Neurol. Sci. 2017, 376, 242–254. [Google Scholar] [CrossRef]
- van Giau, V.; An, S.S.A.; Hulme, J.P. Mitochondrial therapeutic interventions in Alzheimer’s disease. J. Neurol. Sci. 2018, 395, 62–70. [Google Scholar] [CrossRef]
- Ayaz, M.; Sadiq, A.; Junaid, M.; Ullah, F.; Ovais, M.; Ullah, I.; Ahmed, J.; Shahid, M. Flavonoids as Prospective Neuroprotectants and Their Therapeutic Propensity in Aging Associated Neurological Disorders. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Canhada, S.; Castro, K.; Perry, I.S.; Luft, V.C. Omega-3 fatty acids’ supplementation in Alzheimer’s disease: A systematic review. Nutr. Neurosci. 2018, 21, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Ajith, T.A. A Recent Update on the Effects of Omega-3 Fatty Acids in Alzheimer’s Disease. Curr. Clin. Pharmacol. 2018, 13, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition (Burbank, Los Angeles County, Calif.) 2019, 60, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef]
- Stoykovich, S.; Gibas, K. APOE ε4, the door to insulin-resistant dyslipidemia and brain fog? A case study. Alzheimers Dement. (Amst) 2019, 11, 264–269. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.C.; van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5. [Google Scholar] [CrossRef]
- Frederiksen, K.S.; Gjerum, L.; Waldemar, G.; Hasselbalch, S.G. Effects of Physical Exercise on Alzheimer’s Disease Biomarkers: A Systematic Review of Intervention Studies. J. Alzheimer’s Dis. 2018, 61, 359–372. [Google Scholar] [CrossRef]
- Li, C.; Liu, W.; Li, X.; Zhang, Z.; Qi, H.; Liu, S.; Yan, N.; Xing, Y.; Holscher, C.; Wang, Z. The novel GLP-1/GIP analogue DA5-CH reduces tau phosphorylation and normalizes theta rhythm in the icv. STZ rat model of AD. Brain Behav. 2020, 10, e01505. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, R.; Hou, X.; Wang, C.; Guo, S.; Ning, N.; Sun, C.; Yuan, Y.; Li, L.; Hölscher, C.; et al. DA-JC1 improves learning and memory by antagonizing Aβ31–35-induced circadian rhythm disorder. Mol. Brain 2019, 12, 14. [Google Scholar] [CrossRef]
- Cao, Y.; Holscher, C.; Hu, M.M.; Wang, T.; Zhao, F.; Bai, Y.; Zhang, J.; Wu, M.N.; Qi, J.S. DA5-CH, a novel GLP-1/GIP dual agonist, effectively ameliorates the cognitive impairments and pathology in the APP/PS1 mouse model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 827, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Panagaki, T.; Gengler, S.; Holscher, C. The Novel DA-CH3 Dual Incretin Restores Endoplasmic Reticulum Stress and Autophagy Impairments to Attenuate Alzheimer-Like Pathology and Cognitive Decrements in the APPSWE/PS1DeltaE9 Mouse Model. J. Alzheimer’s Dis. 2018, 66, 195–218. [Google Scholar] [CrossRef] [PubMed]
- de Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Asthana, S.; Cook, D.G.; Baker, L.D.; Cherrier, M.; Purganan, K.; Wait, C.; Petrova, A.; Latendresse, S.; Watson, G.S.; et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: Interactions with apolipoprotein E genotype. Psychoneuroendocrinology 2003, 28, 809–822. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H., 2nd; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimer’s Dis. 2008, 13, 323–331. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Frey, W.H., 2nd; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344. [Google Scholar] [CrossRef]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra, E.S.N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Holscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Holscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 2014, 76 Pt A, 57–67. [Google Scholar] [CrossRef]
- Perry, T.; Haughey, N.J.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J. Pharmacol. Exp. Ther. 2002, 302, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Escribano, L.; Simon, A.M.; Gimeno, E.; Cuadrado-Tejedor, M.; de Maturana, R.L.; Garcia-Osta, A.; Ricobaraza, A.; Perez-Mediavilla, A.; del Rio, J.; Frechilla, D. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2010, 35, 1593–1604. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Upstream Risk Factors | Metabolic Precursors | Pathways | Subclinical Pathology | Disease Outcome | |
|---|---|---|---|---|---|
| Social factors: stress, low socioeconomic status, certain ethnic and racial groups | Obesity Visceral Adiposity | Vascular Processes Blood pressure and hypertension Hyperlipidemia Apolipoprotein E | Cerebral blood flow Atherosclerosis | Amyloid precursor proteins | Alzheimer’s disease |
| Poor diet: high in calories, fat and sugar, low in fiver | Inflammatory/Oxidative processes Inflammation Oxidative stress Endothelial function | Neurofibrillary Tangles Amyloid B deposits | |||
| Physical inactivity Genetics and family history | Hyperglycemia Hyperinsulinemia | Metabolic processes Insulin resistance Insulin-degrading enzyme Peroxisome proliferative-activated receptors | |||
| Early childhood exposures in utero and birth weight | Brain and hippocampal atrophy White matter hyperintensities | ||||
| Compound | Potential Pathway | Study Design | Reference |
|---|---|---|---|
| DA5-CH | Reduces tau phosphorylation and normalizes theta rhythm | Injected intracerebroventricula (ICV), streptozotocin on rat | [89] |
| DA-JC1 | Antagonizing circadian rhythm disorders induced by Aβ31–35 | ICV, amyloid(31–35) AD model | [90] |
| DA5-CH | Improved of hippocampal synaptic plasticity and activation of the PI3K/AKT signaling pathway | APP/PS1 mouse model of AD | [91] |
| DA-CH3 | Reduced ER stress and apoptotic signaling, reduced amyloid plaque load in the brain | APP/PS1 mouse model of AD | [92] |
| Insulin | Prevention of Aβ oligomer induced synapse loss and insulin receptor reduction, amelioration of PKR-mediated ER stress | Rat hippocampal neuronal cultures | [93,94] |
| Insulin | AD patients that are not ε4 carriers have reduced sensitivity to insulin, effecting cognitive performance | AD patients homozygous or not for the ApoE-ε4 allele and normal subjects intravenously injected | [95] |
| Insulin | Improved verbal memory in MCI AD ε4-subjects after acute insulin administration, but not in ε4 carriers | AD patients homozygous or not for the ApoE-ε4 allele, MCI patients and most subjects intranasally administrated | [96,97] |
| Insulin | Chromic intranasal insulin doses enhanced selective attention, retention of new information and functional status of MCI and early AD subjects | AD patients, MCI patients and normal subjects intranasally administrated | [98] |
| Insulin | Only women presented improved working memory after treatment | Healthy men and woman intranasally administrated | [99] |
| Liraglutide | Reduction of tau phosphorylation; protection of insulin reception and synapse loss in a c-AMP dependent manner | Cynomolgus monkeys ICV with Aβ oligomer | [100] |
| Liraglutide | Improvement of memory deficits in novel object recognition test and fear conditioning | Swiss mice injected ICV with Aβ oligomer | [100] |
| Liraglutide | Restored memory deficits in object recognition test and Morris water maze; enhanced LTP; reduced microglial activation; diminished Aβ plaque load | APP/PSEN1 mice | [101,102] |
| Exendin-4 | Decrease in the inhibitory phosphorylation of Ser312IRS1, Ser66IRS1 of INK, while restoring activating Tyr465 IRS1 phosphorylation | Rat hippocampal neural cultures | [69] |
| Exendin-4 | Improvement of spatial memory in the Morris water maze; reduced amyloid plaque LOAD | APP/PS1 mice | [69] |
| Exedin4- Liraglutide | eIF2α phosphorylation reduction | Rat hippocampal neural cultures, APP/PS1 mice, cynomolgus monkeys injected ICV with Aβ oligomer | [94] |
| GLP-1 Exendin-4 | Reduction of neural excitotoxicity | Rat hippocampal neural cultures, rats injected on the basal nucleus with ibotenic acid | [103] |
| Rosiglitazone | Reversal of memory deficits in objects recognition test and the Morris water maze; Aβ levels reduction | AD transgenic mice J20 line | [104] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Van Giau, V. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. https://doi.org/10.3390/ijms21093165
Nguyen TT, Ta QTH, Nguyen TKO, Nguyen TTD, Van Giau V. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(9):3165. https://doi.org/10.3390/ijms21093165
Chicago/Turabian StyleNguyen, Thuy Trang, Qui Thanh Hoai Ta, Thi Kim Oanh Nguyen, Thi Thuy Dung Nguyen, and Vo Van Giau. 2020. "Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 9: 3165. https://doi.org/10.3390/ijms21093165
APA StyleNguyen, T. T., Ta, Q. T. H., Nguyen, T. K. O., Nguyen, T. T. D., & Van Giau, V. (2020). Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. International Journal of Molecular Sciences, 21(9), 3165. https://doi.org/10.3390/ijms21093165