Leukamenin E Induces K8/18 Phosphorylation and Blocks the Assembly of Keratin Filament Networks Through ERK Activation

Abstract
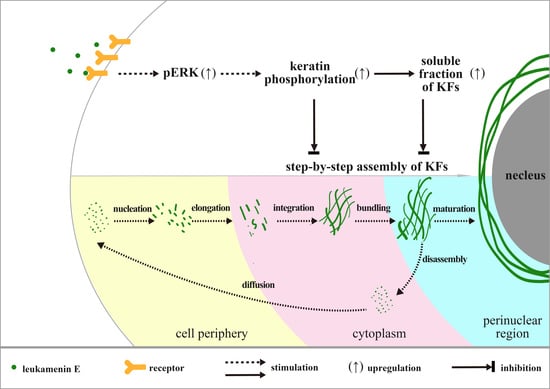
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
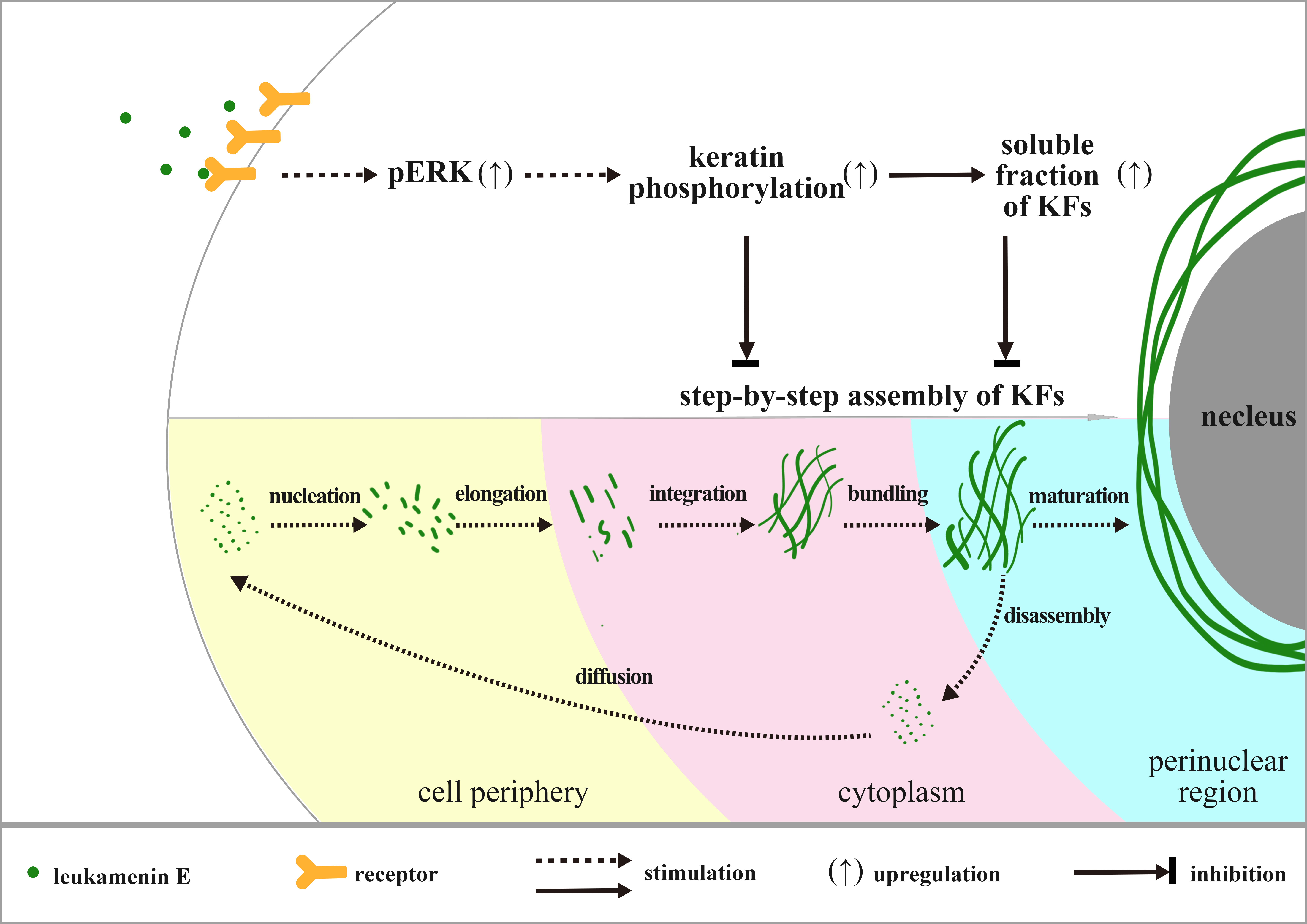
2.1. Effects of Leukamenin E on Cell Viability
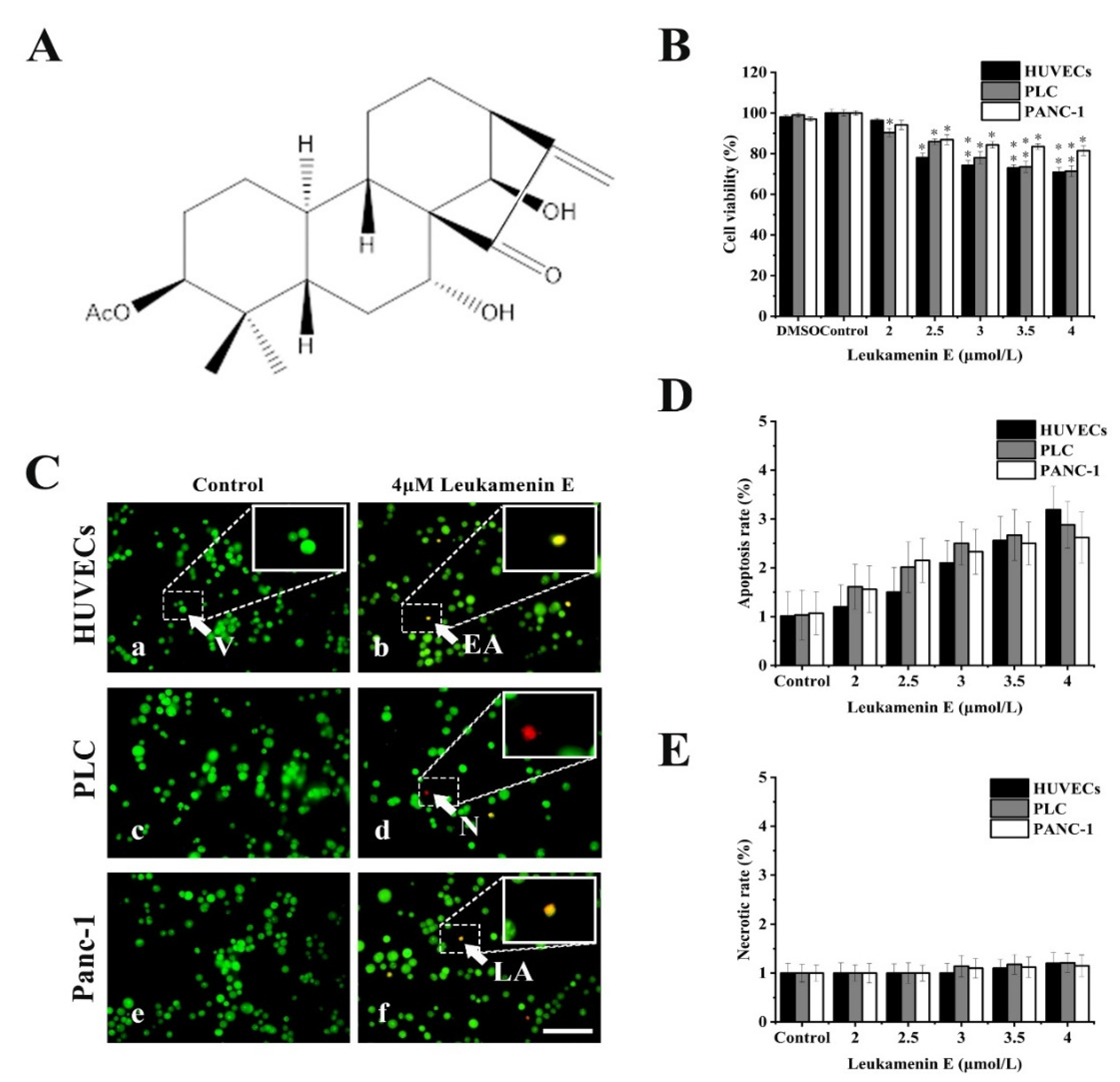
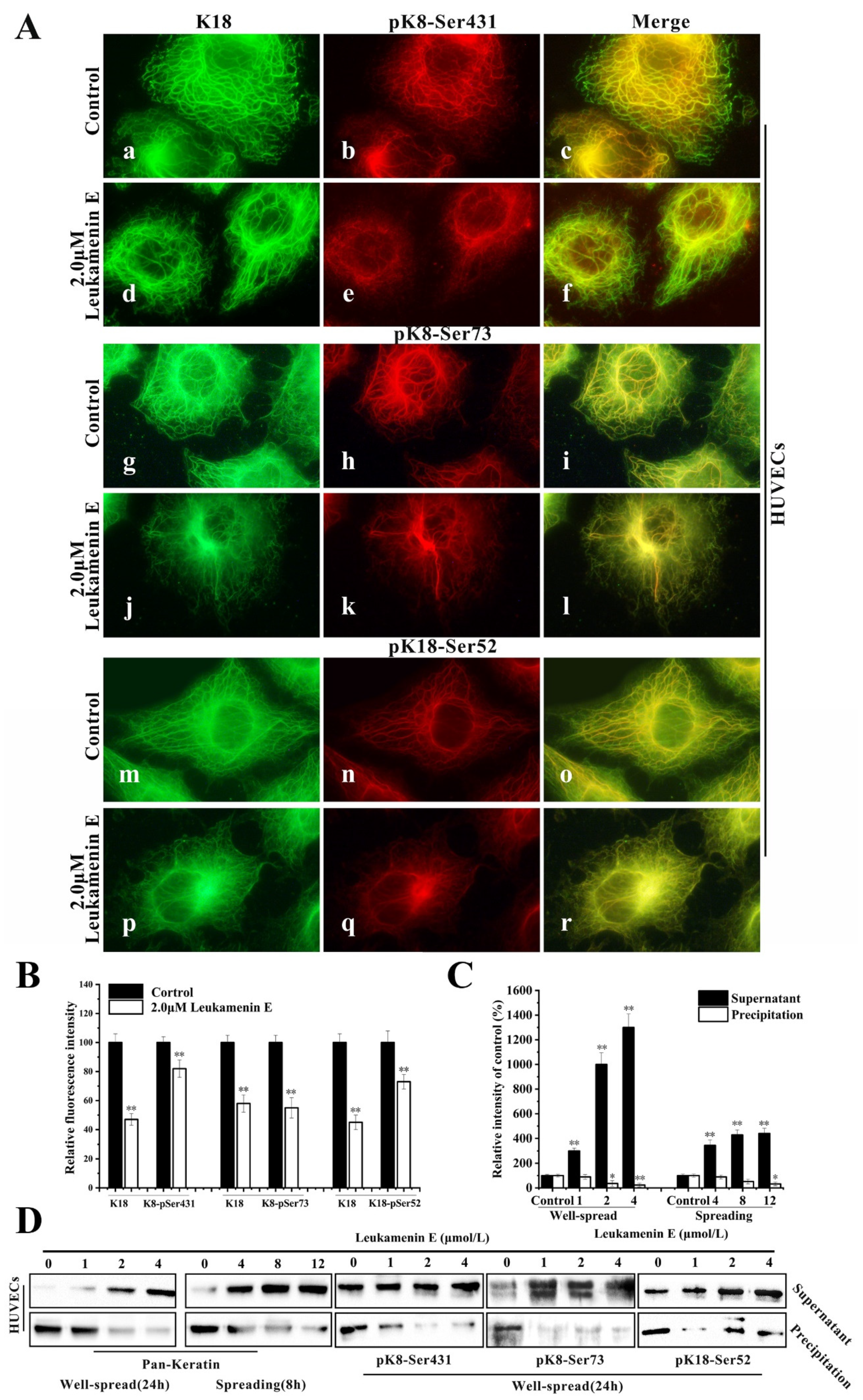
2.2. Leukamenin E Results in Inhibition of Keratin Assembly Independently of Other Major Cytoskeletal Components in PLC and HUVECs
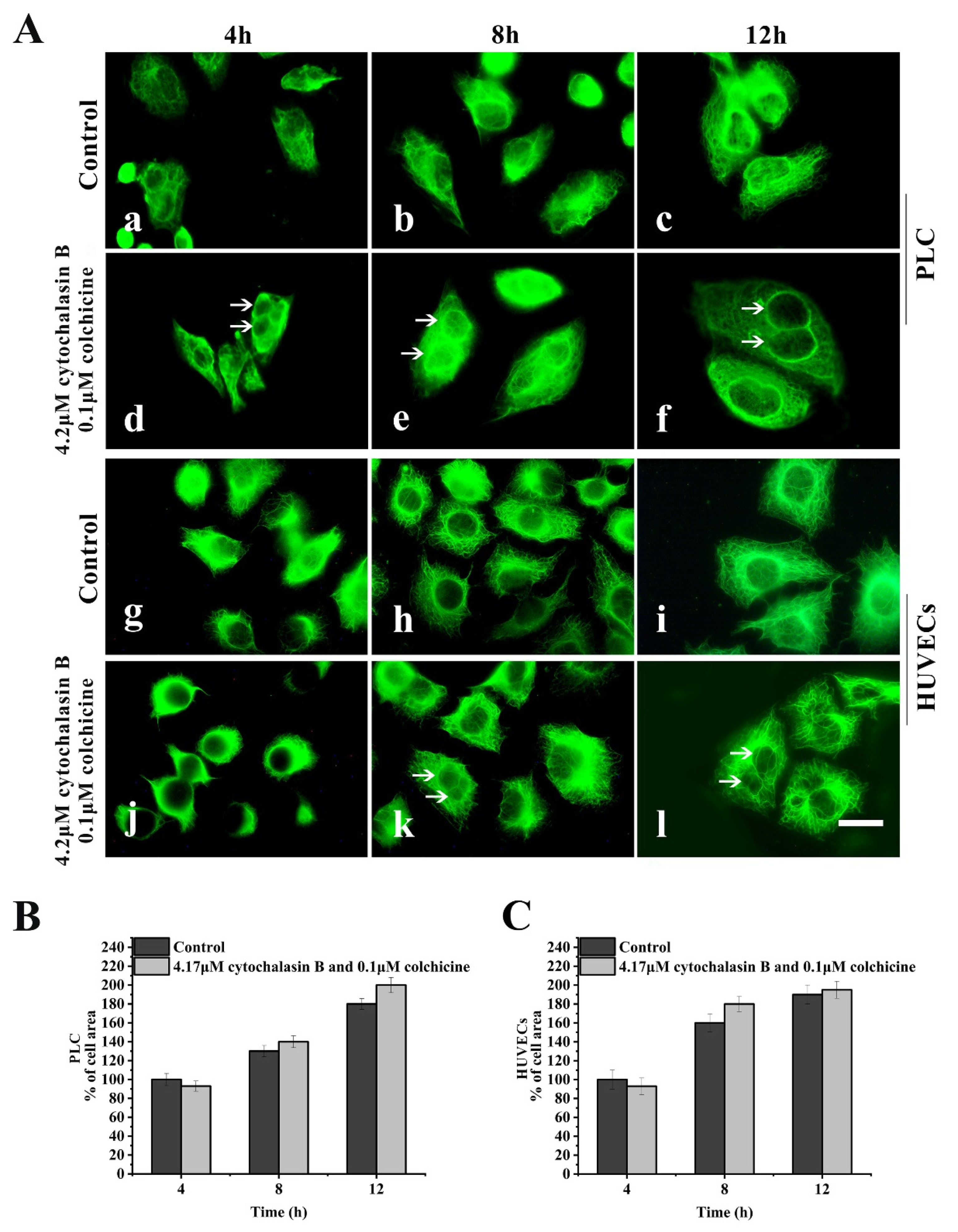
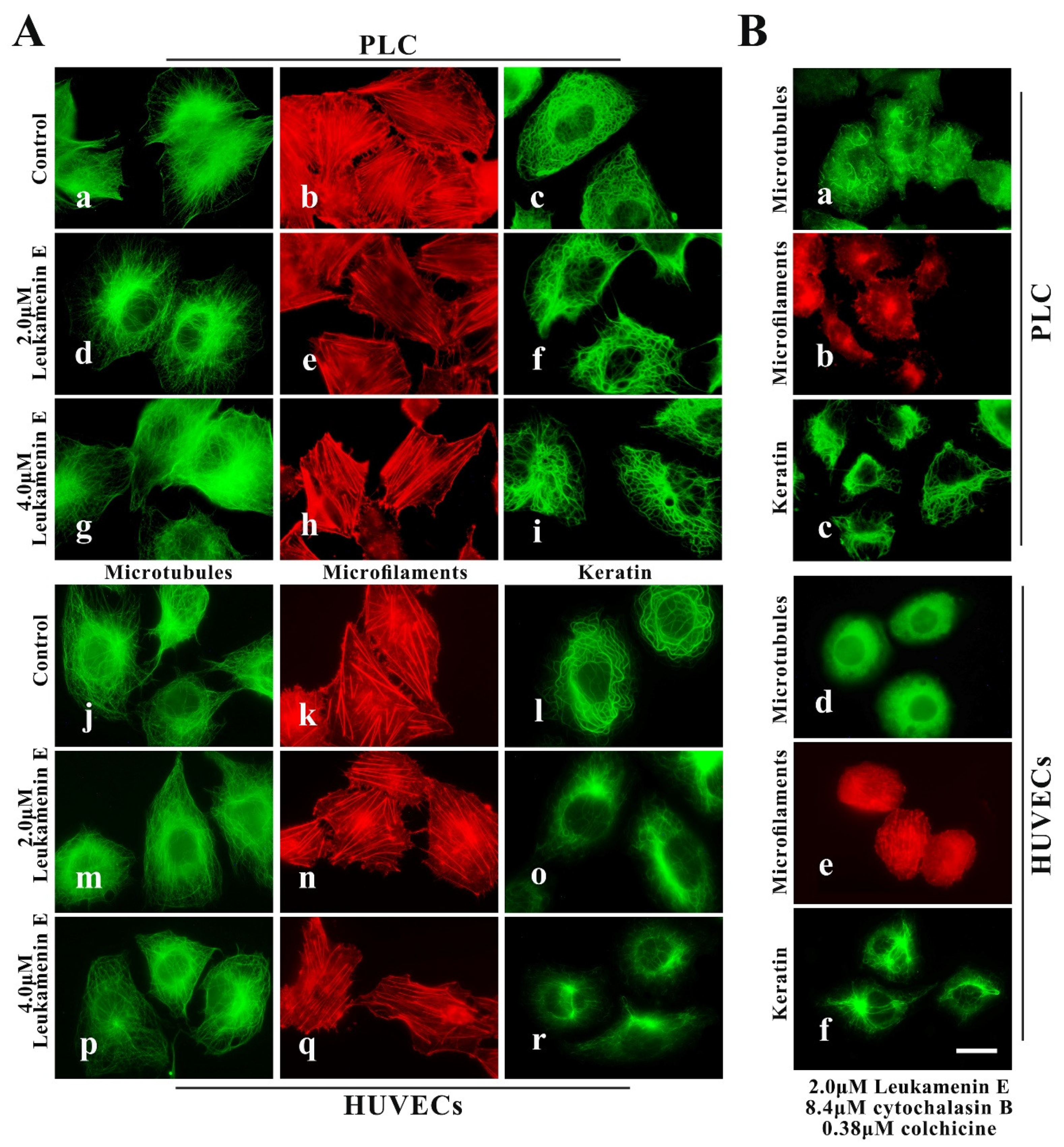
2.2.1. PLC and HUVEC Have a KFs Network Independent of Other Cytoplasmic Skeletons
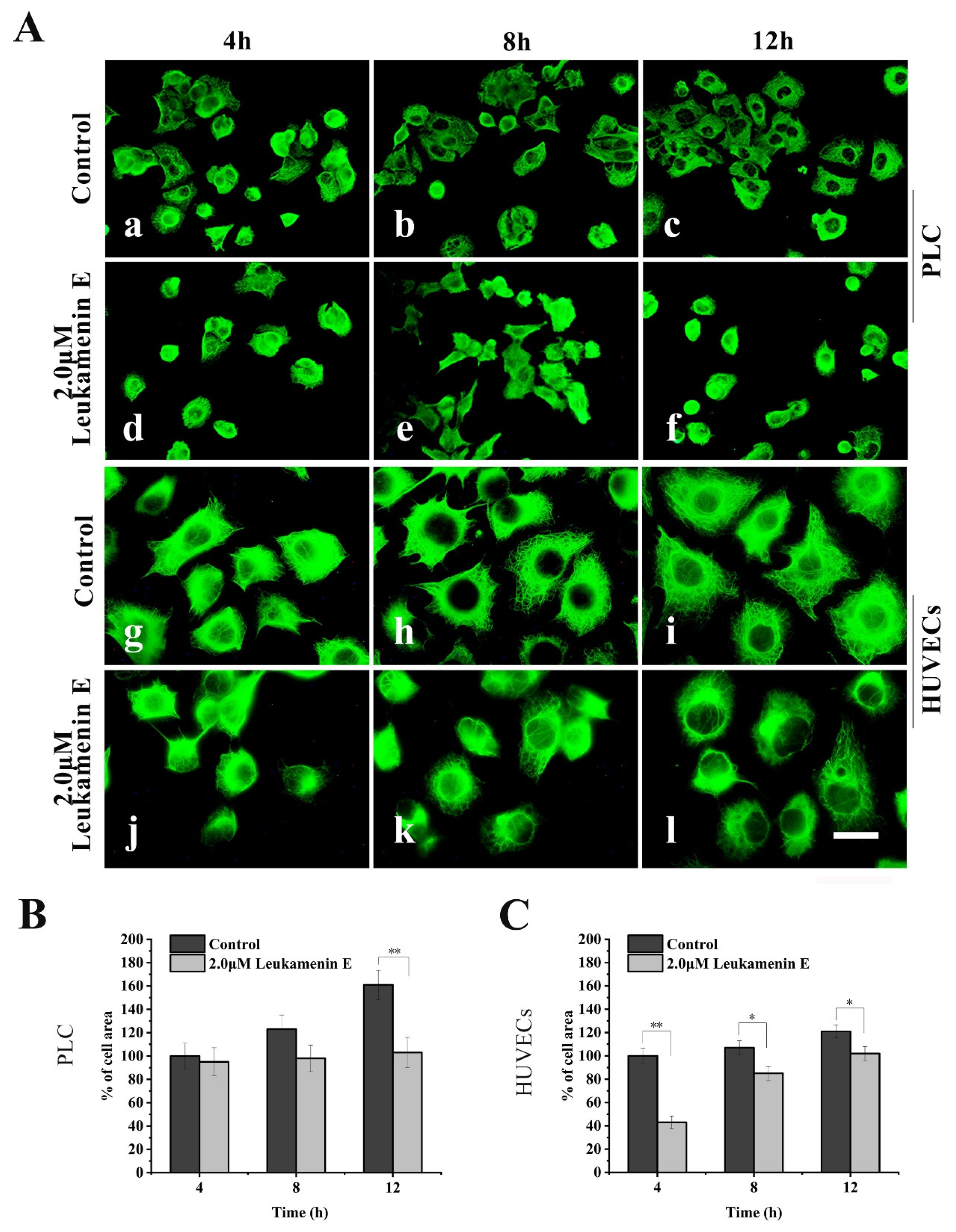
2.2.2. Leukamenin E Destroys the Keratin Network in Well-Spread Cells and Inhibits the Assembly of KFs in Spreading Cells
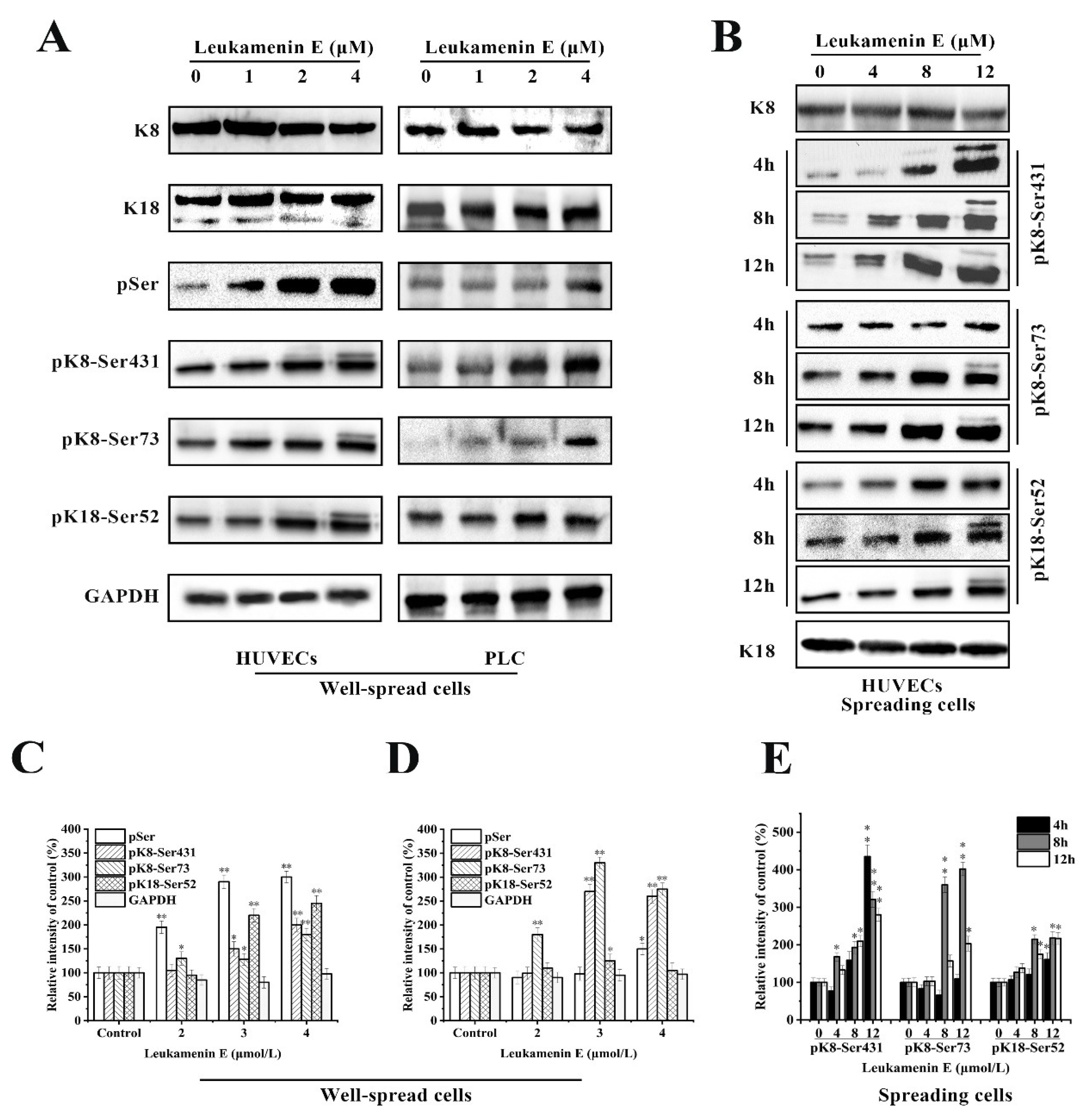
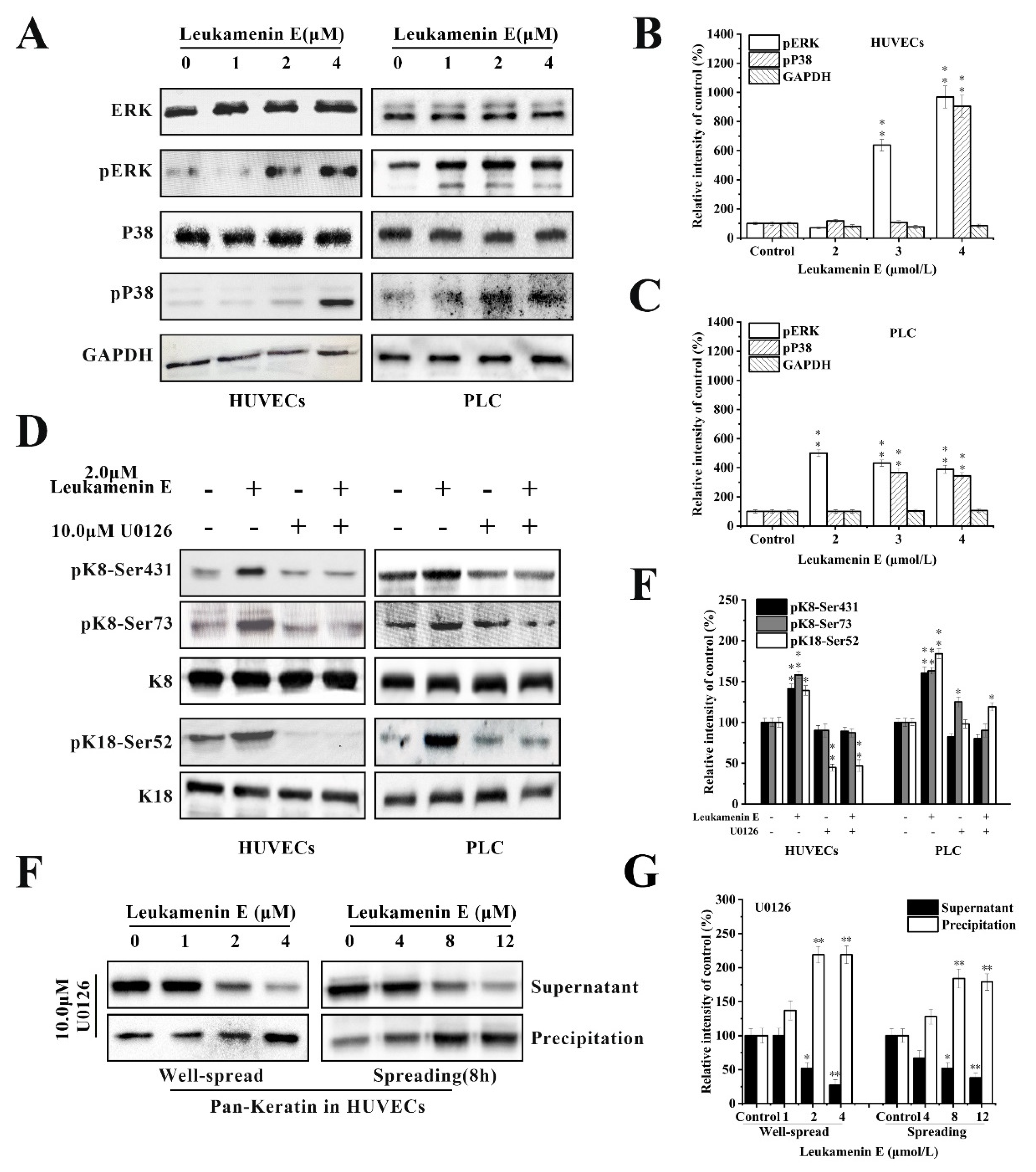
2.3. Leukamenin E Increases Keratin Solubility by Phosphorylation of K8 and K18 and Inhibits the Assembly of KFs
2.3.1. Involvement of ERK in Leukamenin E-Induced KFs Phosphorylation of PLC and HUVECs
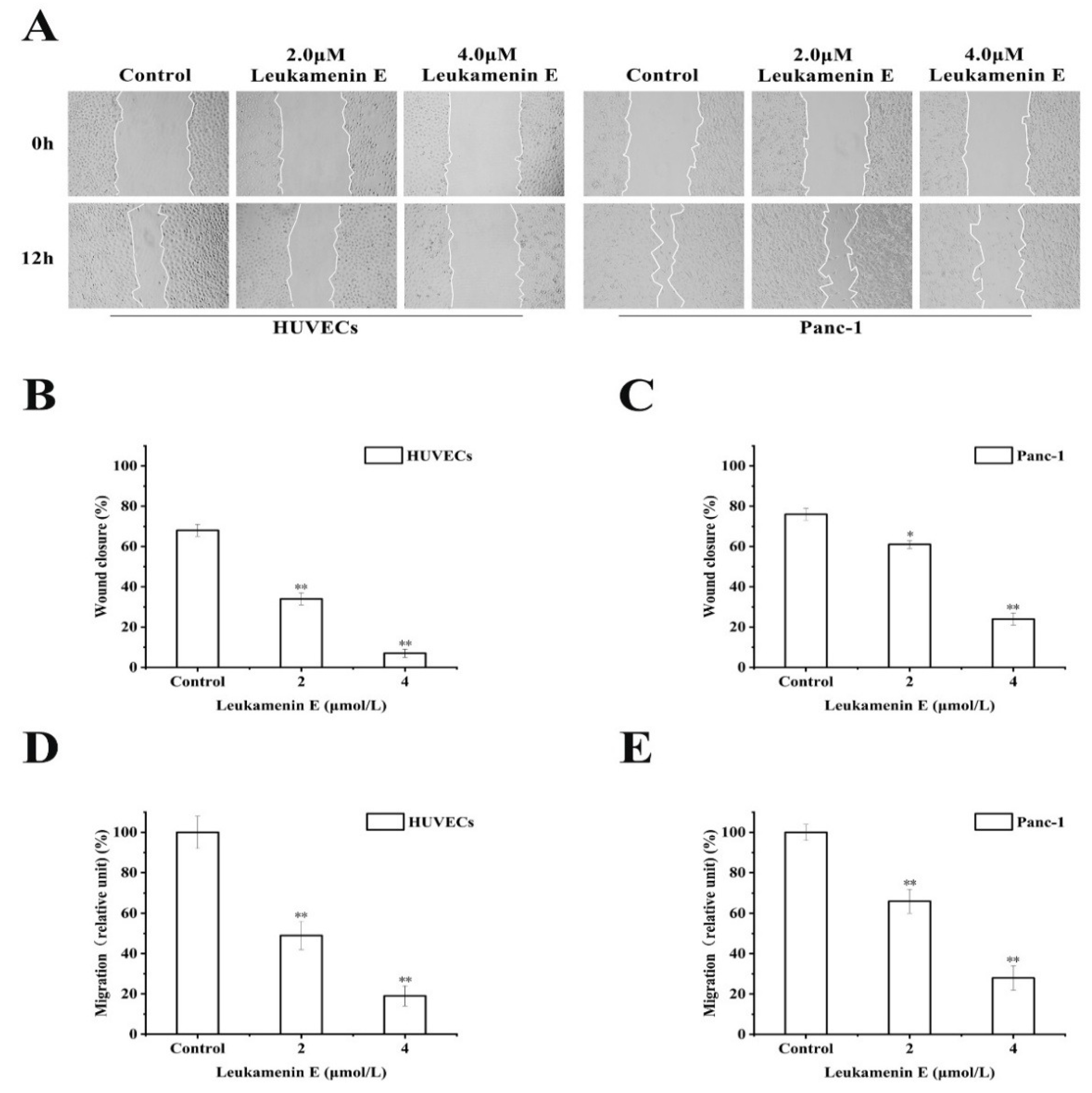
2.3.2. Transwell Migration and Wound Healing Assay
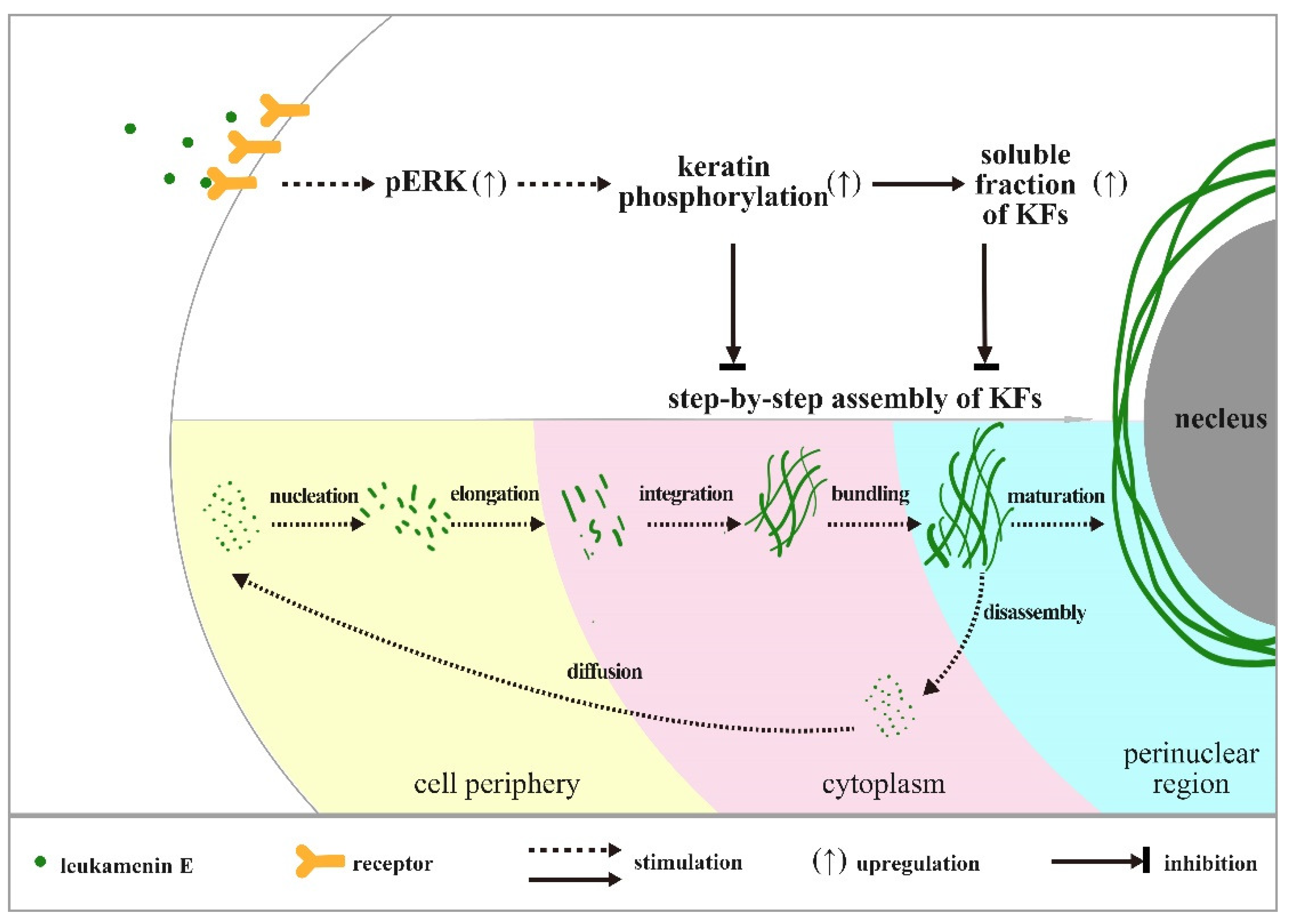
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability and Apoptosis Assay
4.3. Cell Spreading and Well-Spread Treatment
4.4. Immunofluorescence Assay
4.4.1. KFs Staining
4.4.2. MTs Staining
4.4.3. MFs Staining
4.5. Western Blot
4.6. Transwell Migration and Wound Healing Assay
4.7. Materials
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Windoffer, R.; Beil, M.; Magin, T.M.; Leube, R.E. Cytoskeleton in motion: The dynamics of keratin intermediate filaments in epithelia. J. Cell Biol. 2011, 194, 669–678. [Google Scholar] [CrossRef]
- Toivola, D.M.; Strnad, P.; Habtezion, A.; Omary, M. Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 2010, 20, 79–91. [Google Scholar] [CrossRef]
- Hesse, M.; Magin, T.M.; Weber, K. Genes for intermediate filament proteins and the draft sequence of the human genome: Novel keratin genes and a surprisingly high number of pseudogenes related to keratin genes 8 and 18. Cell Sci. 2001, 114, 2569–2575. [Google Scholar]
- Coulombe, P.A.; Wong, P. Cytoplasmic intermediate filaments revealed as dynamic and multipurpose scaffolds. Nat. Cell Biol. 2004, 6, 699–706. [Google Scholar] [CrossRef]
- Peterson, J.R.; Mitchison, T.J. Small molecules, big impact: A history of chemical inhibitors and the cytoskeleton. Chem. Biol. 2002, 9, 1275–1285. [Google Scholar] [CrossRef]
- Sawant, M.S.; Leube, R.E. Consequences of keratin phosphorylation for cytoskeletal organization and epithelial functions. Int. Rev. Cell Mol. Biol. 2016, 330, 171–225. [Google Scholar]
- Chou, C.F.; Omary, M.B. Mitotic arrest-associated enhancement of O-linked glycosylation and phosphorylation of human keratins 8 and 18. J. Biol. Chem. 1993, 268, 4465–4472. [Google Scholar]
- Herrmann, H.; Strelkov, S.V.; Burkhard, P.; Aebi, U. Intermediate filaments: Primary determinants of cell architecture and plasticity. J. Clin. Investig. 2009, 119, 1772–1783. [Google Scholar] [CrossRef]
- Park, M.K.; Lee, H.J.; Shin, J.; Noh, M.; Kim, S.Y.; Lee, C.H. Novel participation of transglutaminase-2 through c-Jun N-terminal kinase activation in sphingosylphosphorylcholine-induced keratin reorganization of PANC-1 cells. Biochim. Biophys. Acta 2011, 1811, 1021–1029. [Google Scholar] [CrossRef]
- Busch, T.; Armacki, M.; Eiseler, T.; Joodi, G.; Temme, C.; Jansen, J.; Wichert, G.; Omary, M.B.; Spatz, J.; Seufferlein, T. Keratin 8 phosphorylation regulates keratin reorganization and migration of epithelial tumor cells. J. Cell Sci. 2012, 125, 2148–2159. [Google Scholar] [CrossRef]
- Lee, E.J.; Park, M.K.; Kim, H.J.; Kang, J.H.; Kim, Y.R.; Kang, G.J.; Byun, H.J.; Lee, C.H. 12-O-Tetradecanoylphorbol-13-Acetate induces keratin 8 phosphorylation and reorganization via expression of transglutaminase-2. Biomol. Ther. 2014, 22, 122–128. [Google Scholar] [CrossRef]
- Zatloukal, K.; French, S.M.; Stumptner, C.; Strnad, P.; Harada, M.; Toivola, D.M.; Cadrin, M.; Omary, M.B. From Mallory to Mallory-Denk bodies: What, how and why? Exp. Cell Res. 2007, 313, 2033–2049. [Google Scholar] [CrossRef]
- Strnad, P.; Paschke, S.; Jang, K.H.; Ku, N.O. Keratins: Markers and modulators of liver disease. Curr. Opin. Gastroenterol. 2012, 28, 209–216. [Google Scholar] [CrossRef]
- Omary, M.B.; Ku, N.O.; Tao, G.Z.; Toivola, D.M.; Liao, J. “Heads and tails” of intermediate filament phosphorylation: Multiple sites and functional insights. Trends Biochem. Sci. 2006, 31, 383–394. [Google Scholar] [CrossRef]
- Shi, Y.; Sun, S.H.; Liu, Y.L.; Li, J.F.; Zhang, T.; Wu, H.; Chen, X.Y.; Chen, D.X.; Zhou, Y.S. Keratin 18 phosphorylation as a progression marker of chronic hepatitis B. Virol. J. 2010, 7, 70. [Google Scholar] [CrossRef]
- Park, M.K.; Park, Y.; Shim, J.; Lee, H.J.; Kim, S.; Lee, C.H. Novel involvement of leukotriene B4 receptor 2 through ERK activation by PP2A down-regulation in leukotriene B4-induced keratin phosphorylation and reorganization of pancreatic cancer cells. Biochim. Biophys. Acta 2012, 1823, 2120–2129. [Google Scholar] [CrossRef]
- Park, M.K.; Park, S.; Kim, H.J.; Kim, H.J.; Kim, E.J.; Kim, S.Y.; Kang, G.J.; Byun, H.J.; Kim, S.H.; Lee, H.; et al. Novel effects of FTY720 on perinuclear reorganization of keratin network induced by sphingosylphosphorylcholine: Involvement of protein phosphatase 2A and G-protein-coupled receptor-12. Eur. J. Pharmacol. 2016, 775, 86–95. [Google Scholar] [CrossRef]
- Beil, M.; Micoulet, A.; Wichert, G.; Paschke, S.; Walther, P.; Omary, M.B.; Veldhoven, P.P.; Gern, U.; Wolff-Hieber, E.; Eggermann, J.; et al. Sphingosylphosphorylcholine regulates keratin network architecture and visco-elastic properties of human cancer cells. Nat. Cell Biol. 2003, 5, 803–811. [Google Scholar] [CrossRef]
- Alam, H.; Gangadaran, P.; Bhate, A.V.; Chaukar, D.A.; Sawant, S.S.; Tiwari, R.; Bobade, J.; Kannan, S.; D’cruz, A.K.; Kane, S.; et al. Loss of keratin 8 phosphorylation leads to increased tumor progression and correlates with clinico-pathological parameters of OSCC patients. PLoS ONE 2011, 6, 1–12. [Google Scholar] [CrossRef]
- Mizuuchi, E.; Semba, S.; Kodama, Y.; Yokozaki, H. Down-modulation of keratin 8 phosphorylation levels by PRL-3 contributes to colorectal carcinoma progression. Int. J. Cancer 2009, 124, 1802–1810. [Google Scholar] [CrossRef]
- Khapare, N.; Kundu, S.; Sehgal, L.; Sawant, M.; Priya, R.; Gosavi, P.; Gupta, N.; Alam, H.; Karkhanis, M.; Naik, N.; et al. Plakophilin3 loss leads to an increase in PRL3 levels promoting K8 dephosphorylation, which is required for transformation and metastasis. PLoS ONE 2012, 7, 1–14. [Google Scholar] [CrossRef]
- Song, D.G.; Kim, Y.S.; Jung, B.C.; Rhee, K.J.; Pan, C.H. Parkin induces upregulation of 40S ribosomal protein SA and posttranslational modification of cytokeratins 8 and 18 in human cervical cancer cells. Appl. Biochem. Biotechnol. 2013, 171, 1630–1638. [Google Scholar] [CrossRef]
- Sun, H.D.; Xu, Y.; Jiang, B. Diterpenoids from Isodon Species; Science Press: Beijing, China, 2001. [Google Scholar]
- Sun, H.D.; Huang, S.X.; Han, Q.B. Diterpenoids from Isodon species and their biological activities. Nat. Prod. Rep. 2006, 23, 673–698. [Google Scholar] [CrossRef]
- Sarwar, M.S.; Xia, Y.X.; Liang, Z.M.; Tsang, S.W.; Zhang, H.J. Mechanistic pathways and molecular targets of plant-derived anticancer ent-kaurane diterpenes. Biomolecules 2020, 10, 144. [Google Scholar] [CrossRef]
- Zhu, J.H.; Chen, Y.P.; Chen, Z.R.; Wei, J.X.; Zhang, H.; Ding, L. Leukamenin E, an ent-kaurane diterpenoid, is a novel and potential keratin intermediate filament inhibitor. Eur. J. Pharmacol. 2019, 846, 86–99. [Google Scholar] [CrossRef]
- Ding, L.; Hou, Q.; Zhou, Q.Y.; Zhang, Q.; Hou, T.D.; Liu, G.A. Structure–activity relationships of eight ent-kaurene diterpenoids from three Isodon plants. Res. Chem. Intermed. 2010, 36, 443–452. [Google Scholar] [CrossRef]
- Zhen, T.; Wu, C.F.; Liu, P.; Wu, H.Y.; Zhou, G.B.; Lu, Y.; Liu, J.X.; Liang, Y.; Li, K.K.; Wang, Y.Y.; et al. Targeting of AML1-ETO in t (8;21) leukemia by oridonin generates a tumor suppressor–like protein. Sci. Transl. Med. 2012, 127, 127–138. [Google Scholar] [CrossRef]
- Liu, C.X.; Yin, Q.Q.; Zhou, H.C.; Wu, Y.L.; Pu, J.X.; Xia, L.; Liu, W.; Huang, X.; Jiang, T.; Wu, M.X.; et al. Adenanthin targets peroxiredoxin I and II to induce differentiation of leukemic cells. Nat. Chem. Biol. 2012, 8, 486–493. [Google Scholar] [CrossRef]
- Ding, L.; Chen, Z.R.; Chen, Y.P.; Li, P.W.; Liu, G.A. NADPH oxidase-derived ROS mediated the differentiation and reorganization of cytoskeletalon induced by epinodosin in HL-60 cells. Chin. J. Cell Biol. 2016, 38, 273–284. [Google Scholar]
- Wei, J.X.; Ding, L.; Liu, Y.; Li, P.W.; Liu, G.A. Induction effects of rabdosin B on differentiation of human promyelocytic leukemia HL-60 cells. Chin. Tradit. Herbal Drugs 2018, 49, 2591–2600. [Google Scholar]
- Caulín, C.; Salvesen, G.; Oshima, R. Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J. Cell Biol. 1997, 138, 1379–1394. [Google Scholar] [CrossRef]
- Bargagna-Mohan, P.; Hamza, A.; Kim, Y.E.; Khuan, Y.; Mor-Vaknin, N.; Wendschlag, N.; Liu, J.; Evans, R.M.; Markovitz, D.M.; Zhan, C.G.; et al. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem. Biol. 2007, 14, 623–634. [Google Scholar] [CrossRef]
- Yoshida, S.; Handa, Y.; Suzuki, T.; Ogawa, M.; Suzuki, M.; Tamai, A.; Abe, A.; Katayama, E.; Sasakawa, C. Microtubule-severing activity of Shigella is pivotal for intercellular spreading. Science 2006, 314, 985–989. [Google Scholar] [CrossRef]
- Herrmann, H.; Aebi, U. Intermediate filaments and their associates: Multi-talented structural elements specifying cytoarchitecture and cytodynamics. Curr. Opin. Cell Biol. 2000, 12, 79–90. [Google Scholar] [CrossRef]
- Fuchs, E.; Weber, K. Intermediate filaments: Structure, dynamics, function, and disease. Annu. Rev. Biochem. 1994, 63, 345–382. [Google Scholar] [CrossRef]
- Ku, N.O.; Omary, M.B. Phosphorylation of human keratin 8 in vivo at conserved head domain serine 23 and at epidermal growth factor-stimulated tail domain serine 431. J. Biol. Chem. 1997, 272, 7556–7564. [Google Scholar] [CrossRef]
- Omary, M.B.; Ku, N.O.; Liao, J.; Price, D. Keratin modifications and solubility properties in epithelial cells and in vitro. Sub-Cell. Biochem. 1998, 31, 105–140. [Google Scholar]
- Leube, R.E.; Moch, M.; Windoffer, R. Intracellular motility of intermediate filaments. Cold Spring Harb. Perspect. Biol. 2017, 9, a021980. [Google Scholar] [CrossRef]
- Snider, N.T.; Omary, M.B. Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 163–177. [Google Scholar] [CrossRef]
- Wong, P.; Coulombe, P. Loss of keratin 6 (K6) proteins reveals a function for intermediate filaments during wound repair. J. Cell Biol. 2003, 163, 327–337. [Google Scholar] [CrossRef]
- Wang, F.R.; Chen, S.; Liu, H.B.; Parent, C.; Coulomb, P. Keratin 6 regulates collective keratinocyte migration by altering cell–cell and cell–matrix adhesion. J. Cell Biol. 2018, 217, 4314–4330. [Google Scholar] [CrossRef]
- Boguslawski, G.; Lyons, D.; Harvey, K.A.; Kovala, A.T.; English, D. Sphingosylphosphorylcholine induces endothelial cell migration and morphogenesis. Biochem. Biophys. Res. Commun. 2000, 272, 603–609. [Google Scholar] [CrossRef]
- Pan, X.; Hobbs, R.P.; Coulombe, P.A. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr. Opin. Cell Biol. 2013, 25, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Coulombe, P.; Fuchs, E. The roles of k5 and k14 head, tail, and R/K L L E G E domains in keratin filament assembly in vitro. J. Cell Biol. 1992, 119, 401–414. [Google Scholar] [CrossRef]
- Hatzfeld, M.; Burba, M. Function of type I and type II keratin head domains: Their role in dimer, tetramer and filament formation. J. Cell Sci. 1994, 107, 1959–1972. [Google Scholar]
- Omary, M.B.; Ku, N.O.; Strnad, P.; Hanada, S. Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Investig. 2009, 119, 1794–1805. [Google Scholar] [CrossRef]
- Klymkowsky, M.; Maynell, L.; Nislow, C. Cytokeratin phosphorylation, cytokeratin filament severing and the solubilization of the maternal mRNA Vg1. J. Cell Biol. 1991, 114, 787–797. [Google Scholar] [CrossRef]
- He, T.; Stepulak, A.; Holmstro, T.H.; Omary, M.B.; Eriksson, J.E. The intermediate filament protein keratin 8 is a novel cytoplasmic substrate for c-Jun N-terminal kinase. J. Biol. Chem. 2002, 277, 10767–10774. [Google Scholar] [CrossRef]
- Tiwari, R.; Sahu, I.; Soni, B.L.; Sathe, G.J.; Datta, K.K.; Thapa, P.; Sinha, S.; Vadivel, C.K.; Dhaka, B.; Gowda, H.; et al. Quantitative phosphoproteomic analysis reveals system-wide signaling pathways regulated by site-specific phosphorylation of Keratin-8 in skin squamous cell carcinoma derived cell line. Proteomics 2017, 17, 182–190. [Google Scholar] [CrossRef]
- Huang, C.; Jacobson, K.; Schaller, M.D. MAP kinases and cell migration. J. Cell Sci. 2004, 117, 4619–4628. [Google Scholar] [CrossRef] [PubMed]
- Guldiken, N.; Zhou, Q.; Kucukoglu, O.; Rehm, M.; Levada, K.; Gross, A.; Kwan, R.; James, L.P.; Trautwein, C.; Omary, M.B.; et al. Human keratin 8 variants promote mouse acetaminophen hepatotoxicity coupled with c-jun amino-terminal kinase activation and protein adduct formation. Hepatology 2015, 62, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Baskic, D.; Popovic, S.; Ristic, P.; Arsenijevic, N.N. Analysis of cycloheximide-induced apoptosis in human leukocytes: Fluorescence microscopy using annexin V/propidium iodide versus acridin orange/ethidium bromide. Cell Biol. Int. 2006, 30, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Li, L.F.; Zhang, Q.; Yang, X.; Zhang, C.; Zhang, X.Y.; Zhang, D.X.; Lv, Y.L.; Song, H.P.; Chen, B.; et al. Phosphorylation of microtubule-associated protein 4 promotes hypoxic endothelial cell migration and proliferation. Front. Pharmacol. 2019, 10, 368. [Google Scholar] [CrossRef]
- Ding, L.; Liu, G.A.; Wang, L.; Han, W. Cytotoxic ent-kaurane diterpenoids from Isodon racemosa (Hemsl) Hara. Indial Chem. 2006, 45, 548–551. [Google Scholar] [CrossRef]
- Magina, T.M.; Vijayaraja, P.; Leube, R.E. Structural and regulatory functions of keratins. Exp. Cell Res. 2007, 313, 2021–2032. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, B.; Zhang, H.; Yang, M.; Du, S.; Wei, J.; Ding, L. Leukamenin E Induces K8/18 Phosphorylation and Blocks the Assembly of Keratin Filament Networks Through ERK Activation. Int. J. Mol. Sci. 2020, 21, 3164. https://doi.org/10.3390/ijms21093164
Xia B, Zhang H, Yang M, Du S, Wei J, Ding L. Leukamenin E Induces K8/18 Phosphorylation and Blocks the Assembly of Keratin Filament Networks Through ERK Activation. International Journal of Molecular Sciences. 2020; 21(9):3164. https://doi.org/10.3390/ijms21093164
Chicago/Turabian StyleXia, Bo, Hui Zhang, Minghui Yang, Shilong Du, Jingxin Wei, and Lan Ding. 2020. "Leukamenin E Induces K8/18 Phosphorylation and Blocks the Assembly of Keratin Filament Networks Through ERK Activation" International Journal of Molecular Sciences 21, no. 9: 3164. https://doi.org/10.3390/ijms21093164
APA StyleXia, B., Zhang, H., Yang, M., Du, S., Wei, J., & Ding, L. (2020). Leukamenin E Induces K8/18 Phosphorylation and Blocks the Assembly of Keratin Filament Networks Through ERK Activation. International Journal of Molecular Sciences, 21(9), 3164. https://doi.org/10.3390/ijms21093164