Abstract
Due to the increasing resistance of currently used antimicrobial drugs, there is an urgent problem for the treatment of cholera disease, selective inhibition of the α-class carbonic anhydrases (CA, EC 4.2.1.1) from the pathogenic bacterium Vibrio cholerae (VcCA) presents an alternative therapeutic target. In this study, a series of hydrazone derivatives, carrying the 2-(hydrazinocarbonyl)-3-phenyl-1H-indole-5-sulfonamide scaffold, have been evaluated as inhibitors of the VcCA with molecular modeling studies. The results suggest that these compounds may bind to the active site of VcCA. To verify this, VcCA enzyme inhibition studies were performed and as predicted most of the tested compounds displayed potent inhibitory activities against VcCA with three compounds showing KI values lower than 30 nM. In addition, all these compounds showed selectivity for VcCA and the off-targets hCA I and II.
1. Introduction
Cholera is an acute diarrheal infection caused by pathogenic Gram-negative comma-shaped bacterium Vibrio cholerae and spreads by the ingestion of contaminated water or food. Vibrio cholerae is divided into 200 serogroups based on O antigen, but only the O1 or O139 serogroups are responsible for epidemic or pandemic cholera [1,2,3]. This highly virulent bacteria causes characteristic rice-water stool, typical clinical symptoms like dehydration, hypovolemic shock, acidosis, and if not well-treated leads to death [4,5]. Every year, there are roughly 1.3–4.0 million cases, and 21,000–143,000 deaths worldwide due to cholera and the last estimated global burden of cholera was approximately 1.2 million cases and 5654 deaths in 34 endemic countries, reported in 2017 by the World Health Organization (WHO) [6,7]. Asia has been cited as the starting point for seven cholera epidemics that have spread to most of Africa, Europe, and America since 1817 [8]. Between April 2017 and 2018, Yemen faced the world’s largest cholera outbreak, with 1,090,280 cases and 2275 deaths [9]. Cholera continues to be a global public health problem that often affects the nations with lower economic status and indicates the inequality and lack of social development between the countries [10,11].
Today, cholera is a preventable disease that can be eliminated by whole-cell oral cholera vaccines (OCV). There are three WHO-prequalified OCVs to date: Dukoral (Valneva, Sweden), Shanchol (Sanofi/Shanta Biotechnics, India), and Euvichol (Eubiologics, South Korea), which can be used in endemic cholera, situations with a high risk of cholera and cholera outbreaks [12,13]. For cholera treatment, the first step is stopping dehydration and the correction of ongoing electrolyte loss through fluid replacement with intravenous fluids like Ringer’s lactate and then oral rehydration solutions (ORS), which is readily prepared sugar and salt solution [14]. Antimicrobial agents can also be used as adjunct therapy to hydration and help to reduce the bacterial shedding and volume of diarrhea [15]. Antibiotics such as tetracycline, doxycycline, ciprofloxacin are the most common agents in therapy. In addition, macrolides like erythromycin or azithromycin present an option for children and pregnant women [16]. Considering the fact that the resistance of the current antimicrobial agents for cholera treatment is a growing problem hard to overcome, the development of new strategies and drugs with different mechanisms of action become an important approach to fight this mortal disease [17,18,19].
As an alternative drug target, three main classes of carbonic anhydrase enzymes (CA) from Vibrio cholerae, α, β, and γ were investigated for their potential for new antimicrobial drugs [20,21,22,23,24,25,26,27,28,29]. α-CA from Vibrio cholerae (VcCA), like all other carbonic anhydrase families (α-, β-, γ-, δ-, ζ-, η-, θ-, and ι-CAs), catalyzes the reversible conversion of carbon dioxide to a bicarbonate ion and a proton, and play an important role for the homeostasis and the virulence of Vibrio cholerae [30,31,32]. This Gram-negative bacterium colonizes the upper small intestine, where the amount of the sodium bicarbonate is at a high concentration. Bicarbonate is a potential inducer of virulence gene expression. If there are not enough transporter proteins, V. cholerae can increase cytosolic bicarbonate levels through the action of VcCA. Therefore, survival and virulence of this pathogen seems to be related to the utilization of the CA system [22,33]. Thus, inhibition of VcCA with selective carbonic anhydrase inhibitors offers a new aspect for cholera treatment. In our previous work, a new series of 2-(hydrazinocarbonyl)-3-phenyl-1H-indole-5-sulfonamide based hydrazones were synthesized and tested for their inhibitory profiles against tumor-related human carbonic anhydrase isozymes hCA IX/XII and against widely distributed off-targeted isoforms hCA I/II. In enzyme inhibition assays, these new molecules showed good inhibitory activity in the low nanomolar range and expressed selectivity over hCA I/II [34]. In this study, we first suggest that these compounds are potential inhibitors of VcCA with docking and molecular dynamics simulations. Subsequently, VcCA enzyme inhibition assays were performed to verify these compounds as novel inhibitors of VcCA, with selectivity over the widely distributed hCA I and II enzymes. These compounds may be used as leads in the search of new potent antimicrobial compounds for cholera treatment.
2. Results
2.1. VcCA Homology Model Construction and Molecular Dynamics Simulation
No crystal structure exists of the Vibrio cholerae α-CA enzyme (VcCA; Uniprot: A0A0X1L2C8). Therefore, a homology model was constructed. To this end, a BLAST search for protein crystal structures with similar sequences as the VcCA sequence was performed. This identified the α-CA from Photobacterium profundum (PpCA; pdb: 5hpj; 1.5 Å) as a template for the construction of VcCA homology models (query cover: 90%; identity: 59.45%).
Prior to the homology modeling, the PpCA structure was superposed upon the hCA IX structure in complex with acetazolamide (azm) (3iai) to obtain insight into the differences between the two αCAs. These two structures show 53.6% sequence identity. Both structures share a reasonable structural similarity overall (RMSD: 2.221 Å; Cα-atoms; 211 residues) and between the active sites overall (RMSD: 0.402 Å; Cα-atoms; 13 residues). The loop between Ser124–Gly140 (hCA IX numbering), which is lining the active site and is located within 4.0 Å from azm, is much shorter and differently folded in the PpCA structure (Figure 1).
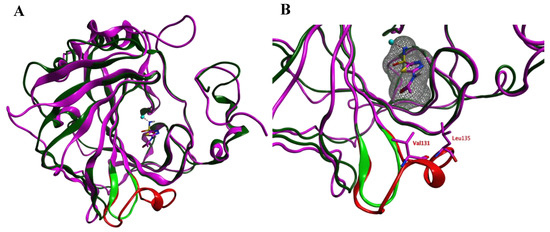
Figure 1.
(A) The overlay of the hCA IX (purple and red) and PpCA (dark and light green) structures. The carbonic anhydrase enzymes (CA) inhibitor acetazolamide (purple) that is cocrystallized in the hCA IX structure is shown. (B) A zoom-in of the active sites of both enzymes reveals that the hCA IX loop (Ser124–Gly140; red) is larger compared to the PcCA loop (green) and approaches the cocrystallized ligand. The ligand surface is indicated in a grey mesh.
The RMSD values of the pocket residues of both enzymes (defined as all amino acids within 4.5 Å distance to azm, except for Val131 and Leu135) is rather low (0.404 Å over 14 residues; Figure 2). Visual inspection furthermore revealed that the active site zinc ion, the three zinc-binding His residues (His94, 96, and 119; hCA IX numbering), and the residues that are involved in the binding of azm (Leu198, Thr199, Thr200; hCA IX numbering) superpose well. Interestingly, these pocket amino acids are also conserved within the VcCA sequence (Figure 2). Therefore, the azm coordinates were copied into the PpCA structure.

Figure 2.
Sequence alignment of VcCA (UniProt: A0A0X1L2C8), PpCA (pdb: 5hpj) and hCA IX (pdb: 3iai). The residues indicated in blue belong to the binding pocket (within 4.5 Å of azm). The residues indicated in grey are part of the different folded loop near the pocket (Ser124–Gly140; hCA IX numbering). The RMSD per residue between PpCA and hCA IX is shown.
A total of 100 homology models were constructed of VcCA using the PpCA crystal structure as a template that also contained the azm coordinates from the hCA IX structure. The VcCA homology model with the lowest contact energy was selected for further optimization after the binding pose of azm was confirmed to be identical to the PpCa-azm template. Subsequently, all backbone atoms, all atoms of His104, 106, and 123 (counterparts of hCA IX His94, 96, and 119), and azm atoms were restrained and the VcCA-azm model was minimized (AMBER14:EHT). This minimization was repeated with a controlled release of restrains that allowed the restrained atoms to move further away from their original coordinates (standard deviation tether of 0.5, 1.0, and 1.5 Å, respectively). Finally, an energy minimization without restraints was performed to yield the final VcCA-azm model that was used for docking and molecular dynamics simulations.
A 10 ns MD simulation was performed on the newly constructed VcCA homology model in complex with azm (Figure 3). The intermolecular interactions between the enzyme active site and azm were analyzed using the protein–ligand interaction fingerprint (PLIF) tool of MOE (v2019.01.02, Chemical Computing Group Inc., Montreal, Quebec, Canada). The main observed interactions during the simulation (represented as 100 snapshots; one snapshot per 100 ps) were with Gln102 (“A”: side chain acceptor interaction), Leu188 (“R”: H-Arene interaction), Thr189 (“D”: side chain donor interaction, “a”: backbone acceptor interaction), and Thr190 (“A”, “a”). The interactions of azm with the binding pocket of the VcCA model remained stable during the simulation, especially between the ligand’s sulfonamide group and the sidechain of Thr189 and Zn2+ (all snapshots). The hydrogen bond between azm and the sidechain of Thr190 was observed in 48% of the snapshots. The H–arene interaction between the ligand and the sidechain of Leu188 was observed in 37% of the snapshots. The hydrogen bond with the sidechain of Gln102 was only observed in 6% of the frames, including the first (0 ns) and the last (10 ns). The calculated average binding energy between azm and VcCA was 57.3 kJ/mol (Figure 3D).
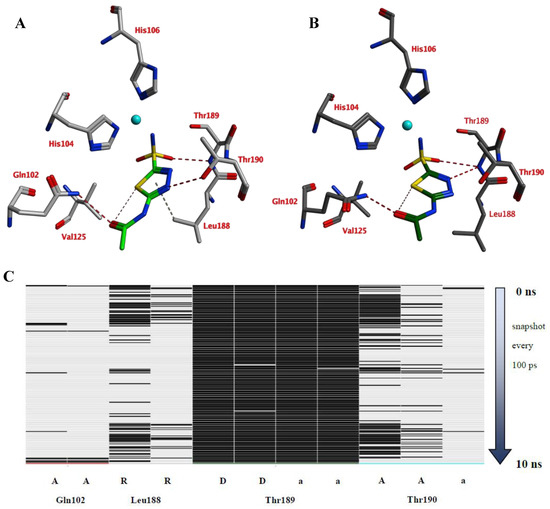
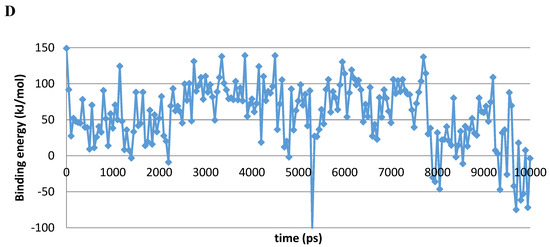
Figure 3.
(A) The VcCA-azm model (t = 0 ns). (B) The VcCA-azm model after a 10 ns MD simulation. (C) The protein–ligand interaction fingerprint (PLIF) showing the interaction of azm with the VcCA binding pocket residues during the simulation as barcodes (a black line indicating the presence of the interaction at snapshot). (D) The binding energy (kJ/mol) between azm and VcCA. Hydrogen bonds are indicated in red dashed lines. H–arene interactions are indicated in yellow dashed lines. “A” indicates sidechain acceptor interactions, “a” indicates backbone acceptor interactions, “D” indicates side chain donor interactions, and “R” indicates H–arene interactions. Two interactions of the same type per residue (for example “A” for Gln102) indicates that multiple interactions of the specified type between ligand and residue are formed. The arrow indicates the sequence of snapshots of every 100 ps from 0 to 10 ns.
2.2. Investigation of Compounds 4–24 as Possible Inhibitors of VcCA
Compounds of series 4–24 have been docked into the active site of the VcCA homology model. All ligand sulfonamides interacted with the active site Zn2+ ion as required by our docking protocol. As such, the common 3-phenyl-1H-indole-5-sulfonamide moiety of all compounds interacted in a similar way with the VcCA active site. The phenyl group may form hydrogen–arene interactions with the side chains of Asn77 and Thr190 (Figure 4A). Compound 23, which has the top-ranked docking score, shows a hydrogen bond with the sidechain of Gln82. The aliphatic ring forms hydrophobic interactions with the sidechains of Trp23 and His79. The linker between the aliphatic ring and the indole ring has an extended flat conformation. The indole NH group and the cationic N atom of the aliphatic ring are solvent exposed and most likely could form hydrogen bonds with water.
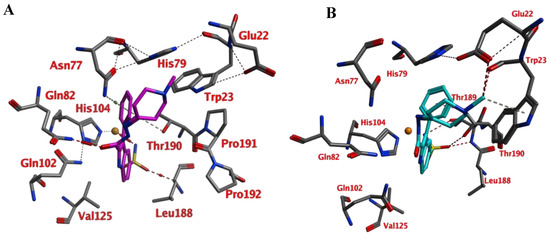
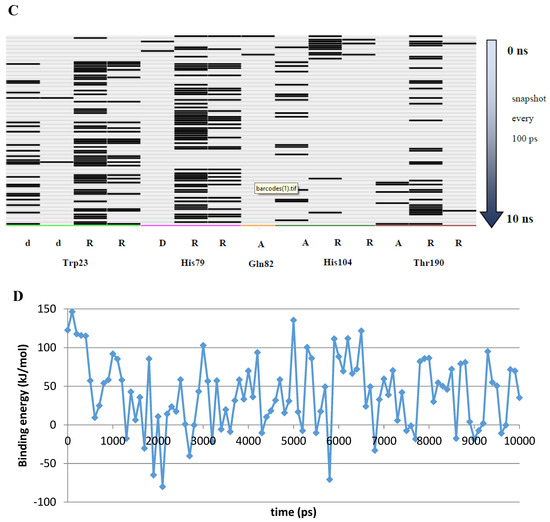
Figure 4.
(A) The VcCA-23 structure obtained from docking (t = 0 ns). (B) The VcCA-23 model after a 10 ns MD simulation. (C) The protein–ligand interaction fingerprint (PLIF) showing the interaction of 23 with the VcCA binding pocket residues during the simulation as barcodes (a black line indicating the presence of the interaction at snapshot). (D) The binding energy (kJ/mol) between 23 and VcCA. Hydrogen bonds are indicated in red dashed lines. H–arene interactions are indicated in yellow dashed lines. “A” indicates sidechain acceptor interactions, “a” indicates backbone acceptor interactions, “D” indicates side chain donor interactions, and “R” indicates H–arene interactions. Two interactions of the same type per residue (for example “A” for Gln102) indicates that multiple interactions of the specified type between ligand and residue are formed. The arrow indicates the sequence of snapshots of every 100 ps.
During a 10 ns MD simulation the interaction between the negatively charged nitrogen atom and the active site zinc ion remain stable even though no restraints between these two atoms were used (Figure 4). However, the hydrogen bond between the ligand carbonyl group and the Gln82 sidechain was not stable and lost early in the simulation. Instead, the sidechains of Trp23 and His79 adopt a different conformation that allows for a better accommodation of the ligands cationic aliphatic ring. This ring of the ligand forms H–arene interactions with mainly with the sidechains of Trp23 (50% of snapshots) and His79 sidechain (53% of snapshots). The distance between the cationic nitrogen atom to Trp23 and His79 is smaller than 5 Å for almost 90% of the time, thus, cation–π interactions are also possible. In addition, Glu22 moves closer to the cationic nitrogen atom during the simulation and 90% of the time the negatively charged oxygen atom of Glu22 is closer than 5.5Å to the cationic nitrogen atom. As such, a strong and long-ranged electrostatic binding interaction is present between 23 and the active site of VcCA.
The calculated average binding energy between 23 and VcCA was 40.2 kJ/mol (Figure 4D), this is lower compared to the binding energy observed for the VcCA-azm simulation. In contrast to the VcCA-azm MD simulation (Figure 3), a reorganization of the binding pocket was observed in the VcCA-23 simulation to allow for interactions with the ligand. As such, compound 23 was expected to bind to VcCA possibly with a higher Ki value.
As noted above, the docking studies suggest that the common 3-phenyl-1H-indole-5-sulfonamide moiety of all compounds interact in a similar way with the VcCA active site. In light of the MD results, it may be expected that the phenyl ring of this scaffold may form hydrogen–arene interactions with the side chain of Thr190. Smaller and neutral R groups may also allow for the formation of hydrogen bonds with Gln82. As such, compound 23 and possibly other compounds from this series are expected to show affinity for the VcCA active site.
2.3. Enzyme Inhibition Assays
The compounds 4–24 and azm were tested in enzyme inhibition assays against VcCA and the data was compared to the previously obtained data of these compounds for the off-targets hCA I and II [34] (Figure 5, Table 1). A total of 19 of 21 compounds have KI values lower than 100 nM against VcCA, including three compounds with a KI values of less than 30 nM (i.e., compounds 11, 12, and 23). It is remarkable that for all compounds, the KI values against hCA I and II are much higher, with the lowest KI values being 3134.9 and 309 nM, respectively. Compared with VcCA, all compounds showed at least ≈10-fold and ≈4-fold selectivity over hCA I and hCA II.
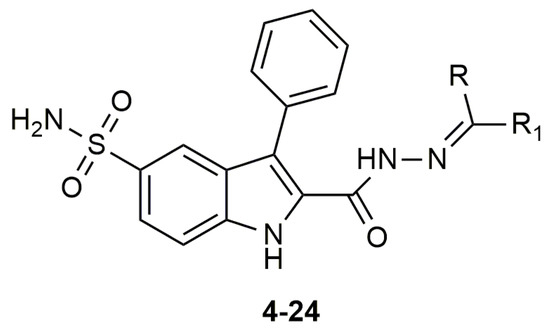
Figure 5.
The molecular structure of compounds 4–24.

Table 1.
Inhibition data against human (h) isoforms hCA I, II (cytosolic), and bacterial enzyme VcCA of derivatives 4–24 and azm by a stopped-flow CO2 hydrase assay.
Compound 23, carrying a piperidine ring with a methyl moiety formed strong interactions with the active site of the enzyme and was predicted as a potent inhibitor of VcCA in docking and molecular dynamic studies (Figure 4). In enzyme inhibition tests, compound 23 inhibited the VcCA enzyme with high selectivity and shows one of lowest inhibition values KI = 25.2 nM.
Compound 12 with two methyl moieties at the tail part, has the best inhibitory activity (KI = 22.8 nM) for VcCA and also has the best selectivity ratios for both off-targeted enzymes hCA I/II. Similarly compounds 10 and 11 that possessed small structures like methyl or hydrogen showed also good inhibition for VcCA (KIs = 46.6 and 27.1 nM, respectively). On the other hand, it is not beneficial to extend the aliphatic chain with ethyl or isobutyl for the VcCA inhibition, like compounds 13–15.
In comparison, alicyclic or heteroaromatic substituted derivatives showed a moderate inhibition for VcCA, but cyclohexane with a tert-butyl (i.e., compound 22) or piperidine ring with a methyl group (i.e., compound 23 in Figure 4) was also well tolerated and led to increasing of the enzyme inhibitory activity.
Generally, all tested compounds showed good inhibitory activity towards VcCA in low nanomolar range and significant selectivity over hCA I and II. Compared to azm, new hydrazones had lower selectivity ratios for inhibiting VcCA against hCA I and II (36.7 and 1.76, respectively) (Table 1).
3. Materials and Methods
3.1. Construction of VcCA Homology Model
Homology models (100 models) of Vibrio cholerae α-CA enzyme (VcCA; Uniprot: A0A0X1L2C8) were constructed using the crystal structure of the α-CA from Photobacterium profundum (PpCA; pdb: 5hpj; 1.5 Å) including the azm coordinates from the hCA IX structure using the MOE software package (v2019.01.02, Chemical Computing Group Inc., Montreal, QC, Canada). The homology model with the highest contact score was selected and further optimized by steepest-descent energy minimization protocols (AMBER14:EHT force field). All heavy atoms of azm (acquired from hCA IX structure), the active site residues (all residues within 4.5 Å of azm), the zinc ion, the zinc-binding His residues, and the protein backbone were restrained, while the other protein atoms were unrestrained. Consequent minimizations with a stepwise release of the restraints was then performed. In the final step, the system was minimized without any restraints.
3.2. Preparation of Ligand Structures
Three-dimensional structures of the investigated ligands were prepared with MOE in low-energy conformations. The most prevalent protonation state of the ligands at pH 7 was calculated. The sulfonamide nitrogen atom was assigned a negative charge as this is the form in which this groups binds to the active site zinc ion. Subsequently, the ligands were energy minimized using the MMFF94x force field.
3.3. Docking Studies into VcCA Homology Model
Docking studies were performed using the FlexX docking tool (v2.3.2; BioSolveIT GmbH, St. Augustin, Germany) within MOE. The binding pocket was described as all residues within 10 Å of azm (VcCA homology model). All ligands were docked 50 times and the highest scoring three poses were subjected to refinement calculations. To this end, the docked ligand and the binding pocket (defined as all residues within 6.5 Å of the docked ligand) were energy minimized and rescored using the GBVI/WSA force field [35].
3.4. Molecular Dynamics Simulations of VcCA-Ligand Complexes
The Yasara Structure software package (v18.8.9, YASARA Biosciences GmbH) was used for the molecular dynamics simulations with the PME method [36,37,38]. The selected docked poses (ligand-VcCA complexes) were first placed into the center of a cuboid box with periodic boundary conditions (minimal distance of 10 Å between protein and boundary). Afterwards, both water molecules (0.997 gr/mL; TIP3P) and counter ions (NaCl) were added to generate a solvated and neutral system. The system was energy minimized using a steepest-descent protocol (AMBER14) [39,40]. The system was energy minimization using steepest descent (100 cycles). Subsequently, the system was simulated for 10 ns at constant temperature (300 K, Berendsen, default values) and pressure (1 bar, Berendsen, default values), without any position restrains (production run). The only restraints applied were distance restraints to keep the zinc ion in the correct orientation towards nitrogen atoms of the zinc binding His residues (force: 100 N/m). The timestep was set to 2 × 1.25 fs and all bonds were constrained using the LINCS and SETTLE algorithms [41,42]. Snapshots were taken every 100 ps of the 10 ns production run. The binding energy (MM/PBSA) was calculated with Yasara Structure, while the RMSD values as well as the binding interactions (protein–ligand interaction fingerprint) were calculated with MOE.
3.5. Enzyme Inhibition Studies
As previously reported, a stopped-flow instrument (SX.18 MV-R Applied Photophysics model) was used for assaying the CO2 hydration activity of various CA isozymes [43]. The 0.2 mm phenol red was used as indicator, working at the absorbance maximum of 557 nm with 10 mM Hepes (pH 7.4) as a buffer and 0.1 M NaClO4 (for constantly maintaining the ionic strength; this anion was not inhibitory in the used concentration) following the CA-catalyzed CO2 hydration reaction for a period of 5–10 s. Saturated CO2 solutions in water at 25 °C were used as substrates. Stock solutions of inhibitors were prepared at a concentration of 10 mM (in DMSO/water 1:1, v/v) and dilutions up to 0.01 nM were done with the assay buffer mentioned above. For allowing the complete formation of the enzyme-inhibitor adduct, the inhibitor and the enzyme were pre-incubated for 15 min. IC50 values were obtained from dose response curves working at seven different concentrations of the test compound (from 0.1 nM to 50 mM) by fitting the curves using PRISM (www.graphpad.com) and non-linear least squares methods, the obtained values representing the mean of at least three different determinations. The inhibition constants (KI) were derived from the IC50 values by using the Cheng–Prusoff equation as follows: KI = IC50/(1+[S]/KM) where [S] represents the CO2 concentration at which the measurement was carried out, and KM represents the concentration of the substrate at which the enzyme activity was at half maximal. All CA isoforms were recombinant ones obtained in-house as reported earlier [44,45,46,47,48].
4. Conclusions
In the present work, a novel series of 2-(hydrazinocarbonyl)-3-phenyl-1H-indole-5-sulfonamide incorporating hydrazone derivatives were investigated with docking and molecular dynamics studies for the purpose of discovering new potent inhibitors of α-class carbonic anhydrase of the pathogenic bacterium Vibrio cholerae (VcCA). Molecular modeling studies suggested that these compounds (especially compound 23) may bind to the active site of VcCA. Subsequently, all compounds were tested in VcCA enzyme inhibition assays. These molecules show strong inhibitory activity towards VcCA in low nanomolar range and display significant selectivity over hCA I/II. It is also remarkable that compound 23 which was suggested as a strong binder, inhibited the VcCA enzyme with one of the lowest KI value, 25.2 nM and with high selectivity.
Author Contributions
Conceptualization, Ö.G.-A. and C.T.S.; Synthesis, K.D.-Y. and Ö.G.-A.; Molecular modeling, A.A. (Atilla Akdemir) and K.D.-Y.; Enzyme inhibition assays, A.A. (Andrea Angeli) and C.T.S.; Writing—Original Draft Preparation, K.D.-Y., A.A. (Atilla Akdemir) and Ö.G.-A.; Writing—Review and Editing, A.A. (Atilla Akdemir) and Ö.G.-A.; Supervision, Ö.G.-A.; Project Administration, Ö.G.-A.; Funding Acquisition, Ö.G.-A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funding by the Istanbul University Scientific Research Projects Department under project numbers TSA-2019-30535.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| CA | Carbonic anhydrases |
| VcCA | Vibrio cholerae carbonic anhydrase |
| hCA I/II/IX | Human carbonic anhydrases I/II/IX |
| PpCA | Photobacterium profundum carbonic anhydrase |
| azm | Acetazolamide |
| MD | Molecular dynamics |
| RMSD | Root-mean-square deviation |
| MOE | Molecular operating environment |
| KI | Inhibition constant |
References
- Harris, J.B.; LaRocque, R.C.; Qadri, F.; Ryan, E.T.; Calderwood, S.B. Cholera. In Proceedings of the The Lancet; Lancet Publishing Group: London, UK, 2012; Volume 379, pp. 2466–2476. [Google Scholar]
- Dorman, M.J.; Domman, D.; Uddin, M.I.; Sharmin, S.; Afrad, M.H.; Begum, Y.A.; Qadri, F.; Thomson, N.R. High quality reference genomes for toxigenic and non-toxigenic Vibrio cholerae serogroup O139. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Morris, J.G.; Acheson, D. Cholera and other types of vibriosis: A story of human pandemics and oysters on the half shell. Clin. Infect. Dis. 2003, 37, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B.; Morris, J.G.; Levine, M.M. Cholera. Clin. Microbiol. Rev. 1995, 8, 48–86. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Mood, B.; Metanat, M. Diagnosis, clinical management, prevention, and control of cholera. A review study. Int. J. Infect. 2014, 1, e18303. [Google Scholar] [CrossRef]
- Ali, M.; Nelson, A.R.; Lopez, A.L.; Sack, D.A. Updated global burden of cholera in endemic countries. PLoS Negl. Trop. Dis. 2015, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Weekly Epidemiological Record; World Health Organization: Geneva, Switzerland, 2018; Volume 38, pp. 489–500. [Google Scholar]
- Deen, J.; Mengel, M.A.; Clemens, J.D. Epidemiology of cholera. Vaccine 2020, 38, A31–A40. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The Global Task Force on Cholera Control. Available online: https://www.who.int/news-room/fact-sheets/detail/cholera (accessed on 31 March 2020).
- Kelvin, A.A. Cholera outbreak in the republic of congo, the Democratic Republic of Congo, and cholera worldwide. J. Infect. Dev. Ctries. 2011, 5, 688–691. [Google Scholar] [CrossRef]
- World Health Organization. Cholera Homepage. Available online: https://www.who.int/news-room/fact-sheets/detail/cholera (accessed on 2 April 2020).
- Qadri, F.; Clemens, J.D.; Holmgren, J. Cholera immunity and development and use of oral cholera vaccines for disease control. In Mucosal Vaccines: Innovation for Preventing Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2019; pp. 537–561. ISBN 9780128119242. [Google Scholar]
- World Health Organization. Cholera vaccine: WHO position paper, August 2017–Recommendations. Vaccine 2018, 36, 3418–3420. [Google Scholar] [CrossRef]
- Saha, D.; LaRocque, R.C. Cholera and other vibrios. Hunter’s Tropical Medicine and Emerging Infectious Disease, 10th ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; pp. 448–453. ISBN 9781416043904. [Google Scholar]
- Leibovici-Weissman, Y.; Neuberger, A.; Bitterman, R.; Sinclair, D.; Salam, M.A.; Paul, M. Antimicrobial drugs for treating cholera. Cochrane Database Syst. Rev. 2014, 6, 1–184. [Google Scholar] [CrossRef]
- Hiremath, N.S.; Cattamanchi, S.; Vidyalakshmi, P.R.; Trounce, M. Vibrio cholerae (Cholera) Attack. In Ciottone’s Disaster Medicine; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 724–725. ISBN 9780323286657. [Google Scholar]
- Kitaoka, M.; Miyata, S.T.; Unterweger, D.; Pukatzki, S. Antibiotic resistance mechanisms of Vibrio cholerae. J. Med. Microbiol. 2011, 60, 397–407. [Google Scholar] [CrossRef]
- López-Igual, R.; Bernal-Bayard, J.; Rodríguez-Patón, A.; Ghigo, J.M.; Mazel, D. Engineered toxin–intein antimicrobials can selectively target and kill antibiotic-resistant bacteria in mixed populations. Nat. Biotechnol. 2019, 37, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lou, J.; Liu, J.; Zhang, L.; Li, J.; Kan, B. Antibiotic resistance of Vibrio cholerae O1 El Tor strains from the seventh pandemic in China, 1961–2010. Int. J. Antimicrob. Agents 2012, 40, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Capasso, C. Carbonic anhydrases from pathogens: Bacterial carbonic anhydrases and their inhibitors as potential antiinfectives. In Carbonic Anhydrases: Biochemistry and Pharmacology of an Evergreen Pharmaceutical Target; Elsevier: Amsterdam, The Netherlands, 2019; pp. 387–417. ISBN 9780128164761. [Google Scholar]
- Smith, K.S.; Ferry, J.G. Prokaryotic carbonic anhydrases. FEMS Microbiol. Rev. 2000, 24, 335–366. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, S.; De Luca, V.; Scozzafava, A.; Carginale, V.; Supuran, C.T.; Capasso, C. Biochemical properties of a new α-carbonic anhydrase from the human pathogenic bacterium, Vibrio cholerae. J. Enzyme Inhib. Med. Chem. 2014, 29, 23–27. [Google Scholar] [CrossRef]
- Alafeefy, A.M.; Ceruso, M.; Al-Tamimi, A.M.S.; Del Prete, S.; Capasso, C.; Supuran, C.T. Quinazoline-sulfonamides with potent inhibitory activity against the α-carbonic anhydrase from Vibrio cholerae. Bioorganic Med. Chem. 2014, 22, 5133–5140. [Google Scholar] [CrossRef]
- Carta, F.; Osman, S.M.; Vullo, D.; AlOthman, Z.; Del Prete, S.; Capasso, C.; Supuran, C.T. Poly(amidoamine) dendrimers show carbonic anhydrase inhibitory activity against α-, β-, γ- and η-class enzymes. Bioorganic Med. Chem. 2015, 23, 6794–6798. [Google Scholar] [CrossRef]
- De Vita, D.; Angeli, A.; Pandolfi, F.; Bortolami, M.; Costi, R.; Di Santo, R.; Suffredini, E.; Ceruso, M.; Del Prete, S.; Capasso, C.; et al. Inhibition of the α-carbonic anhydrase from Vibrio cholerae with amides and sulfonamides incorporating imidazole moieties. J. Enzyme Inhib. Med. Chem. 2017, 32, 798–804. [Google Scholar] [CrossRef]
- Angeli, A.; Abbas, G.; del Prete, S.; Capasso, C.; Supuran, C.T. Selenides bearing benzenesulfonamide show potent inhibition activity against carbonic anhydrases from pathogenic bacteria Vibrio cholerae and Burkholderia pseudomallei. Bioorg. Chem. 2018, 79, 319–322. [Google Scholar] [CrossRef]
- Bua, S.; Osman, S.M.; Del Prete, S.; Capasso, C.; AlOthman, Z.; Nocentini, A.; Supuran, C.T. Click-tailed benzenesulfonamides as potent bacterial carbonic anhydrase inhibitors for targeting Mycobacterium tuberculosis and Vibrio cholerae. Bioorg. Chem. 2019, 86, 183–186. [Google Scholar] [CrossRef]
- Nocentini, A.; Osman, S.M.; Del Prete, S.; Capasso, C.; ALOthman, Z.A.; Supuran, C.T. Extending the γ-class carbonic anhydrases inhibition profiles with phenolic compounds. Bioorg. Chem. 2019, 93, 103336. [Google Scholar] [CrossRef]
- Del Prete, S.; Vullo, D.; De Luca, V.; Carginale, V.; Osman, S.M.; Alothman, Z.; Supuran, C.T.; Capasso, C. Comparison of the sulfonamide inhibition profiles of the α-, β- And γ-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorganic Med. Chem. Lett. 2016, 26, 1941–1946. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases and metabolism. Metabolites 2018, 8, 25. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Capasso, C.; Supuran, C.T. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin. Ther. Targets. 2015, 19, 1689–1704. [Google Scholar] [CrossRef] [PubMed]
- Abuaita, B.H.; Withey, J.H. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect. Immun. 2009, 77, 4111–4120. [Google Scholar] [CrossRef]
- Demir-Yazıcı, K.; Bua, S.; Akgüneş, N.M.; Akdemir, A.; Supuran, C.T.; Güzel-Akdemir, Ö. Indole-based hydrazones containing a sulfonamide moiety as selective inhibitors of tumor-associated human carbonic anhydrase isoforms IX and XII. Int. J. Mol. Sci. 2019, 20, 2354. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. The generalized born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. Yasara view—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar]
- Angeli, A.; Abbas, G.; Del Prete, S.; Carta, F.; Capasso, C.; Supuran, C.T. Acyl selenoureido benzensulfonamides show potent inhibitory activity against carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg. Chem. 2017, 75, 170–172. [Google Scholar] [CrossRef]
- D’ascenzio, M.; Guglielmi, P.; Carradori, S.; Secci, D.; Florio, R.; Mollica, A.; Ceruso, M.; Akdemir, A.; Sobolev, A.P.; Supuran, C.T. Open saccharin-based secondary sulfonamides as potent and selective inhibitors of cancer-related carbonic anhydrase IX and XII isoforms. J. Enzyme Inhib. Med. Chem. 2017, 32, 51–59. [Google Scholar] [CrossRef]
- Mishra, C.B.; Kumari, S.; Angeli, A.; Monti, S.M.; Buonanno, M.; Tiwari, M.; Supuran, C.T. Discovery of benzenesulfonamides with potent human carbonic anhydrase inhibitory and effective anticonvulsant action: Design, synthesis, and pharmacological assessment. J. Med. Chem. 2017, 60, 2456–2469. [Google Scholar] [CrossRef]
- Abdoli, M.; Angeli, A.; Bozdag, M.; Carta, F.; Kakanejadifard, A.; Saeidian, H.; Supuran, C.T. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo[d]thiazole-5- and 6-sulfonamides. J. Enzyme Inhib. Med. Chem. 2017, 32, 1071–1078. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J. Enzyme Inhib. Med. Chem. 2018, 33, 485–495. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).