Integrated Analysis of mRNA and microRNA Elucidates the Regulation of Glycyrrhizic Acid Biosynthesis in Glycyrrhiza uralensis Fisch

Abstract
1. Introduction
2. Results
2.1. Construction of RNA-Seq Libraries and Sequence Alignment
2.2. Gene Function Annotation
2.3. Structural and Transporter Genes Potentially Involved in GA Biosynthesis
2.4. Identification of Regulatory Proteins in G. uralensis
2.5. Identification of DEGs and qRT-PCR Validation
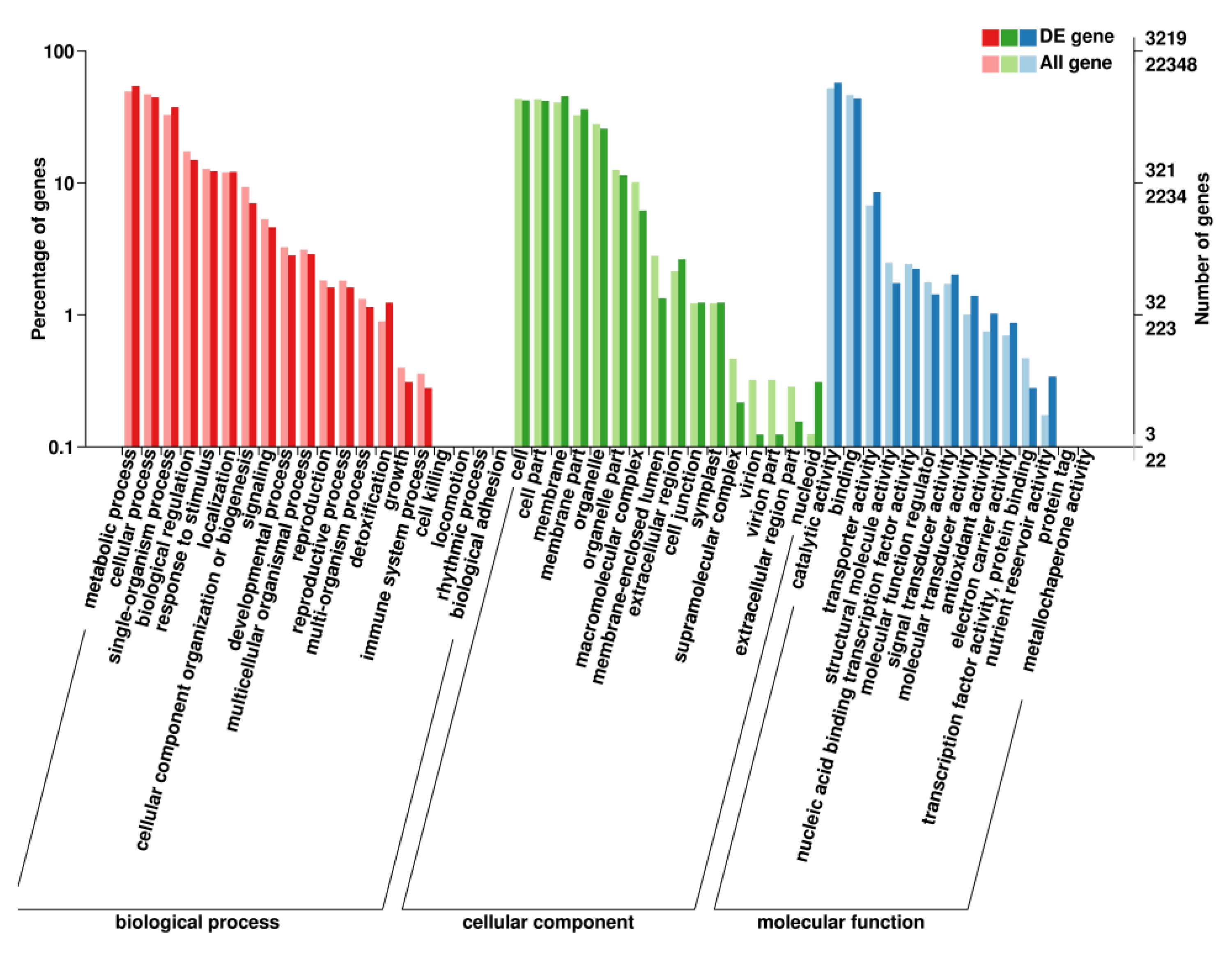
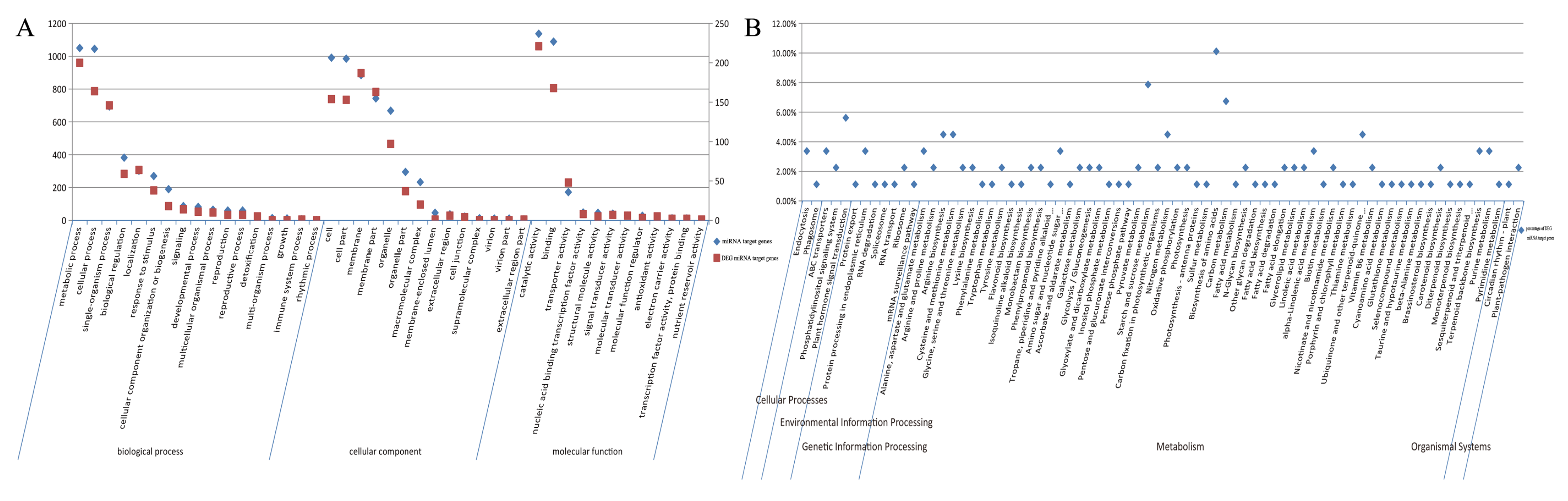
2.6. GO Enrichment Analysis of DEGs
2.7. KEGG Enrichment Analysis of DEGs
2.8. Correlation Analysis between Transporter and Structural Genes
2.9. Correlation Analysis between Regulatory Protein and the Structural Genes
2.10. Binding Site Analysis of R2R3-MYB in the Promoter Region of the Structural Gene
2.11. AS Analysis
2.12. Features of G. uralensis Small RNA Population
2.13. Identification and Validation of MiRNAs in G. uralensis
2.14. Target Prediction of MiRNAs in G. uralensis
2.15. Negative Correlation Expression between MiRNA and Target Transcripts
3. Discussion
4. Materials and Methods
4.1. Plant Materials and RNA Extraction
4.2. Illumina Transcriptome Library Construction and Sequencing
4.3. Illumina Data Mapping and Quantification of Gene Expression Levels
4.4. Gene Function Annotation
4.5. Identification of TF, TR, PK, and Transporter
4.6. Detection of AS Events
4.7. Validation of the Expression Profile of Transcripts by qRT-PCR
4.8. Small RNA Library Construction and High-Throughput Sequencing
4.9. MiRNA Identification and Target Prediction
4.10. Validation of the Authenticity of MiRNAs by Stem-Loop PCR
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- Chinese Pharmacopoeia Commission. Chinese Pharmacopoeia; The Chemical Industry Publishing House: Beijing, China, 2005; p. 203. [Google Scholar] [CrossRef]
- Montoro, P.; Maldini, M.; Russo, M.; Postorino, S.; Piacente, S.; Pizza, C. Metabolic profiling of roots of liquorice (Glycyrrhiza glabra) from different geographical areas by ESI/MS/MS and determination of major metabolites by LC-ESI/MS and LC-ESI/MS/MS. J. Pharm. Biomed. Anal. 2011, 54, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.H.; Li, T.; Tong, Y.G.; Chen, X.J.; Chen, X.P.; Wang, Y.T.; Lu, J.J. A Systematic Review of the Anticancer Properties of Compounds Isolated from Licorice (Gancao). Planta Med. 2015, 81, 1670–1687. [Google Scholar] [CrossRef] [PubMed]
- Ramilowski, J.A.; Sawai, S.; Seki, H.; Mochida, K.; Yoshida, T.; Sakurai, T.; Muranaka, T.; Saito, K.; Daub, C.O. Glycyrrhiza uralensis transcriptome landscape and study of phytochemicals. Plant Cell Physiol. 2013, 54, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Sakurai, T.; Seki, H.; Yoshida, T.; Takahagi, K.; Sawai, S.; Uchiyama, H.; Muranaka, T.; Saito, K. Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume. Plant J. 2017, 89, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yuan, B.C.; Ma, Y.S.; Wang, L.Q.; Liu, C.S.; Liu, Y. HMGR, SQS, β-AS, and Cytochrome P450 Monooxygenase Genes in Glycyrrhiza uralensis. Chin. Herb. Med. 2015, 7, 290–295. [Google Scholar] [CrossRef]
- Peacock, R.B.; Hicks, C.W.; Walker, A.M.; Dewing, S.M.; Lewis, K.M.; Abboud, J.C.; Stewart, S.W.A.; Kang, C.; Watson, J.M. Structural and Functional Characterization of Dynamic Oligomerization in Burkholderia cenocepacia HMG-CoA Reductase. Biochemistry 2019, 58, 3960–3970. [Google Scholar] [CrossRef]
- Qian, J.; Liu, Y.; Ma, C.; Chao, N.; Chen, Q.; Zhang, Y.; Luo, Y.; Cai, D.; Wu, Y. Positive Selection of Squalene Synthase in Cucurbitaceae Plants. Int. J. Genom. 2019, 2019, 5913491. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, H.H.; Wen, H.; Gao, Y.; Wang, L.Q.; Liu, C.S. Enhancing the accumulation of beta-amyrin in Saccharomyces cerevisiae by co-expression of Glycyrrhiza uralensis squalene synthase 1 and beta-amyrin synthase genes. Acta Pharm. Sin. 2014, 49, 734–741. [Google Scholar]
- Seki, H.; Sawai, S.; Ohyama, K.; Mizutani, M.; Ohnishi, T.; Sudo, H.; Fukushima, E.O.; Akashi, T.; Aoki, T.; Saito, K.; et al. Triterpene functional genomics in licorice for identification of CYP72A154 involved in the biosynthesis of glycyrrhizin. Plant Cell 2011, 23, 4112–4123. [Google Scholar] [CrossRef]
- Xu, G.; Cai, W.; Gao, W.; Liu, C. A novel glucuronosyltransferase has an unprecedented ability to catalyse continuous two-step glucuronosylation of glycyrrhetinic acid to yield glycyrrhizin. New Phytol. 2016, 212, 123–135. [Google Scholar] [CrossRef]
- Chu, S.; Wang, J.; Zhu, Y.; Liu, S.; Zhou, X.; Zhang, H.; Wang, C.E.; Yang, W.; Tian, Z.; Cheng, H.; et al. An R2R3-type MYB transcription factor, GmMYB29, regulates isoflavone biosynthesis in soybean. PLoS Genet. 2017, 13, e1006770. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef]
- Liu, X.; Yuan, L.; Xu, L.; Xu, Z. Over-expression ofGmMYB39leads to an inhibition of the isoflavonoid biosynthesis in soybean (Glycine max. L). Plant Biotechnol. Rep. 2013, 7, 445. [Google Scholar] [CrossRef]
- Wang, T.; Jia, Q.; Wang, W.; Hussain, S.; Ahmed, S.; Zhou, D.X.; Ni, Z.; Wang, S. GCN5 modulates trichome initiation in Arabidopsis by manipulating histone acetylation of core trichome initiation regulator genes. Plant Cell Rep. 2019, 38, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, Y.; Zhang, X.; Zha, P.; Lin, R. The SWI2/SNF2 Chromatin-Remodeling ATPase BRAHMA Regulates Chlorophyll Biosynthesis in Arabidopsis. Mol. Plant 2017, 10, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sung, S. Accelerated vernalization response by an altered PHD-finger protein in Arabidopsis. Plant Signal. Behav. 2017, 12, e1308619. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.W. Auxin response factors. Plant Cell Environ. 2016, 39, 1014–1028. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.J.; Choi, S.H.; Kim, J.H.; Kim, J.E.; Lee, M.H.; Chung, B.Y.; Woo, H.R. A Mutation in Plant-Specific SWI2/SNF2-Like Chromatin-Remodeling Proteins, DRD1 and DDM1, Delays Leaf Senescence in Arabidopsis thaliana. PLoS ONE 2016, 11, e0146826. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, Q.; Liu, Y.; Zhang, Q. Chromatin Remodeling and Plant Immunity. Adv. Protein Chem. Struct. Biol. 2017, 106, 243–260. [Google Scholar]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Garcia, A.V.; Al-Yousif, M.; Hirt, H. Role of AGC kinases in plant growth and stress responses. Cell Mol. Life Sci. 2012, 69, 3259–3267. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Zhang, D.; Zhou, H.; Zheng, T.; Yin, Y.; Lin, H. Transcription factor HAT1 is a substrate of SnRK2.3 kinase and negatively regulates ABA synthesis and signaling in Arabidopsis responding to drought. PLoS Genet. 2018, 14, e1007336. [Google Scholar] [CrossRef] [PubMed]
- Luang, S.; Sornaraj, P.; Bazanova, N.; Jia, W.; Eini, O.; Hussain, S.S.; Kovalchuk, N.; Agarwal, P.K.; Hrmova, M.; Lopato, S. The wheat TabZIP2 transcription factor is activated by the nutrient starvation-responsive SnRK3/CIPK protein kinase. Plant Mol. Biol. 2018, 96, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Jiao, C.; Gu, Z. Cyclic GMP mediates abscisic acid-stimulated isoflavone synthesis in soybean sprouts. Food Chem. 2019, 275, 439–445. [Google Scholar] [CrossRef]
- Veremeichik, G.N.; Grigorchuk, V.P.; Silanteva, S.A.; Shkryl, Y.N.; Bulgakov, D.V.; Brodovskaya, E.V.; Bulgakov, V.P. Increase in isoflavonoid content in Glycine max cells transformed by the constitutively active Ca(2+) independent form of the AtCPK1 gene. Phytochemistry 2019, 157, 111–120. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ben Chaabane, S.; Liu, R.; Chinnusamy, V.; Kwon, Y.; Park, J.H.; Kim, S.Y.; Zhu, J.K.; Yang, S.W.; Lee, B.H. STA1, an Arabidopsis pre-mRNA processing factor 6 homolog, is a new player involved in miRNA biogenesis. Nucleic Acids Res. 2013, 41, 1984–1997. [Google Scholar] [CrossRef]
- Ren, G.; Yu, B. Critical roles of RNA-binding proteins in miRNA biogenesis in Arabidopsis. RNA Biol. 2012, 9, 1424–1428. [Google Scholar] [CrossRef]
- Mallory, A.C.; Vaucheret, H. Functions of microRNAs and related small RNAs in plants. Nat. Genet. 2006, 38, S31–S36. [Google Scholar] [CrossRef]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread translational inhibition by plant miRNAs and siRNAs. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA methylation mediated by a microRNA pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.W.; Zhang, C.; Miller, M.; Palmer, G.; Whiteley, M.; Tholl, D.; Chen, Z.J. cis- and trans-Regulation of miR163 and target genes confers natural variation of secondary metabolites in two Arabidopsis species and their allopolyploids. Plant Cell 2011, 23, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Seo, H.D.; Hennighausen, L.; Lee, D.; Kang, K. Octopus-toolkit: A workflow to automate mining of public epigenomic and transcriptomic next-generation sequencing data. Nucleic Acids Res. 2018, 46, e53. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Reddy, V.A.; Wang, Q.; Dhar, N.; Kumar, N.; Venkatesh, P.N.; Rajan, C.; Panicker, D.; Sridhar, V.; Mao, H.Z.; Sarojam, R. Spearmint R2R3-MYB transcription factor MsMYB negatively regulates monoterpene production and suppresses the expression of geranyl diphosphate synthase large subunit (MsGPPS.LSU). Plant Biotechnol. J. 2017, 15, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, N.J.; Chen, X.; Wang, M.Y.; Matich, A.J.; Perez, R.L.; Allan, A.C.; Green, S.A.; Atkinson, R.G. Natural variation in monoterpene synthesis in kiwifruit: Transcriptional regulation of terpene synthases by NAC and ETHYLENE-INSENSITIVE3-like transcription factors. Plant Physiol. 2015, 167, 1243–1258. [Google Scholar] [CrossRef]
- Yan-Hua, X.; Jia-Wei, W.; Shui, W.; Jian-Ying, W.; Xiao-Ya, C. Characterization of GaWRKY1, a cotton transcription factor that regulates the sesquiterpene synthase gene (+)-delta-cadinene synthase-A. Plant Physiol. 2004, 135, 507–515. [Google Scholar]
- Miyamoto, K.; Nishizawa, Y.; Minami, E.; Nojiri, H.; Yamane, H.; Okada, K. Overexpression of the bZIP transcription factor OsbZIP79 suppresses the production of diterpenoid phytoalexin in rice cells. J. Plant Physiol. 2015, 173, 19–27. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Tej, S.S.; Luo, S.; Haudenschild, C.D.; Meyers, B.C.; Green, P.J. Elucidation of the small RNA component of the transcriptome. Science 2005, 309, 1567–1569. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Aksay, G.; Dolgosheina, E.; Ebhardt, H.A.; Magrini, V.; Mardis, E.R.; Sahinalp, S.C.; Unrau, P.J. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res. 2008, 18, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Identification and characterization of maize microRNAs involved in the very early stage of seed germination. BMC Genom. 2011, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.; Feyissa, B.A.; Amyot, L.; Aung, B.; Hannoufa, A. MicroRNA156 improves drought stress tolerance in alfalfa (Medicago sativa) by silencing SPL13. Plant Sci. 2017, 258, 122–136. [Google Scholar] [CrossRef]
- Aung, B.; Gruber, M.Y.; Amyot, L.; Omari, K.; Bertrand, A.; Hannoufa, A. MicroRNA156 as a promising tool for alfalfa improvement. Plant Biotechnol. J. 2015, 13, 779–790. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, B.; Zhao, D.; Li, C.; Shao, F.; Lu, S. Genome-wide analysis and molecular dissection of the SPL gene family in Salvia miltiorrhiza. J. Integr. Plant Biol. 2014, 56, 38–50. [Google Scholar] [CrossRef]
- Cui, G.; Duan, L.; Jin, B.; Qian, J.; Xue, Z.; Shen, G.; Snyder, J.H.; Song, J.; Chen, S.; Huang, L.; et al. Functional Divergence of Diterpene Syntheses in the Medicinal Plant Salvia miltiorrhiza. Plant Physiol. 2015, 169, 1607–1618. [Google Scholar] [CrossRef]
- Holland, N.; Holland, D.; Helentjaris, T.; Dhugga, K.S.; Xoconostle-Cazares, B.; Delmer, D.P. A comparative analysis of the plant cellulose synthase (CesA) gene family. Plant Physiol. 2000, 123, 1313–1324. [Google Scholar] [CrossRef]
- Taylor, N.G.; Howells, R.M.; Huttly, A.K.; Vickers, K.; Turner, S.R. Interactions among three distinct CesA proteins essential for cellulose synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Suo, F.; Lei, W.; Gu, L. Comprehensive analysis of alternative splicing in Digitalis purpurea by strand-specific RNA-Seq. PLoS ONE 2014, 9, e106001. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Zhang, K.; Qian, Q.; Huang, Z.; Wang, Y.; Li, M.; Hong, L.; Zeng, D.; Gu, M.; Chu, C.; Cheng, Z. GOLD HULL AND INTERNODE2 encodes a primarily multifunctional cinnamyl-alcohol dehydrogenase in rice. Plant Physiol. 2006, 140, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.K.; Chang, J.; Zhao, P.X. Prediction of membrane transport proteins and their substrate specificities using primary sequence information. PLoS ONE 2014, 9, e100278. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Yao, Y.; Li, Y.; Yang, M.; Wu, B.; Yu, D. Comprehensive transcriptome analysis reveals genes potentially involved in isoflavone biosynthesis in Pueraria thomsonii Benth. PLoS ONE 2019, 14, e0217593. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Deng, Y.; Jianqi, L.I.; Songfeng, W.U.; Zhu, Y.; Chen, Y.; Fuchu, H.E. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Rolf, A.; Amos, B.; Wu, C.H.; Barker, W.C.; Brigitte, B.; Serenella, F.; Elisabeth, G.; Hongzhan, H.; Rodrigo, L.; Michele, M. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Reddy, V.S.; Tsu, B.V.; Ahmed, M.S.; Li, C.; Moreno-Hagelsieb, G. The Transporter Classification Database (TCDB): Recent advances. Nucleic Acids Res. 2016, 44, D372–D379. [Google Scholar] [CrossRef]
- Hafner, M.; Landgraf, P.; Ludwig, J.; Rice, A.; Ojo, T.; Lin, C.; Holoch, D.; Lim, C.; Tuschl, T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 2008, 44, 3–12. [Google Scholar] [CrossRef]
- Wu, H.J.; Ma, Y.K.; Chen, T.; Wang, M.; Wang, X.J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Anderson, T.A. Identification of 188 conserved maize microRNAs and their targets. FEBS Lett. 2006, 580, 3753–3762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. CMLS 2006, 63, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotation | Transcript Number | Transcript ID | Predicted Subcellular Localization | Expression Profiles (FPKM) | Up-Regulated in Roots (Y/N) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| L1 | L2 | L3 | R1 | R2 | R3 | |||||
| AACT | 1 | Glyur000218s00011642.1 | Peroxisome | 38.32 | 42.15 | 47.59 | 105.87 | 152.71 | 143.58 | Y |
| HMGS | 1 | Glyur000195s00012841.1 | Cytoplasm | 2.35 | 2.81 | 7.56 | 173.37 | 280.56 | 276.05 | Y |
| HMGR | 4 | Glyur000037s00002618.1 | Endoplasmic Reticulum | 32.41 | 57.49 | 47.67 | 92.79 | 193.02 | 173.28 | Y |
| Glyur000135s00007303.1 | Endoplasmic Reticulum | 11.95 | 15.05 | 16.18 | 66.08 | 35.39 | 39.91 | Y | ||
| Glyur000203s00012900.1 | Endoplasmic Reticulum | 6.69 | 12.23 | 17.49 | 26.99 | 42.77 | 38.50 | Y | ||
| Glyur000682s00024324.1 | Endoplasmic Reticulum | 13.21 | 16.87 | 18.17 | 137.41 | 182.13 | 172.25 | Y | ||
| MK | 1 | Glyur000069s00004081.1 | Extracell | 5.15 | 6.75 | 6.73 | 9.61 | 6.62 | 6.40 | N |
| PMK | 1 | Glyur000343s00025703.1 | Cytoplasm | 16.23 | 9.45 | 10.92 | 5.40 | 13.19 | 13.26 | N |
| MPD | 1 | Glyur000002s00000233.1 | Cell Membrane and Extracell | 7.42 | 9.09 | 9.11 | 30.96 | 41.07 | 42.35 | Y |
| FPPS | 1 | Glyur000088s00007722.1 | Endoplasmic Reticulum and Nucleus | 5.14 | 6.08 | 11.84 | 35.58 | 40.90 | 40.94 | Y |
| SQS | 2 | Glyur000017s00002413.1 | Extracell | 21.40 | 19.05 | 24.26 | 18.33 | 48.71 | 46.67 | Y |
| Glyur000089s00008825.1 | Endoplasmic Reticulum. | 3.70 | 3.44 | 4.82 | 16.81 | 43.09 | 38.43 | Y | ||
| β-AS | 1 | Glyur001733s00027628.1 | Extracell and Nucleus | 0.97 | 2.28 | 10.35 | 45.99 | 189.49 | 162.89 | Y |
| CYP88D6 | 2 | Glyur000561s00023443.1 | Cytoplasm Endoplasmic Reticulum and Nucleus | 0.04 | 0.39 | 0.91 | 12.41 | 21.66 | 17.30 | Y |
| Glyur000561s00023451.1 | Endoplasmic Reticulum. | 0.25 | 0.64 | 2.67 | 183.15 | 327.26 | 297.26 | Y | ||
| CYP72A154 | 2 | Glyur000890s00019071.1 | Endoplasmic Reticulum. | 6.92 | 7.14 | 6.31 | 118.31 | 181.82 | 175.76 | Y |
| Glyur001936s00032203.1 | Extracell. | 10.00 | 1.51 | 7.57 | 103.78 | 167.71 | 142.77 | Y | ||
| GUCPR1 | 1 | Glyur000294s00011848.1 | Endoplasmic Reticulum. | 38.03 | 46.50 | 39.00 | 16.14 | 25.88 | 25.71 | Y |
| Gene ID | Structural Gene Name | Predicted Binding Site Upstream ATG (bp) |
|---|---|---|
| Glyur000218s00011642.1 | AACT | −1934 |
| Glyur000682s00024324.1 | HMGR | −1544 |
| Glyur000069s00004081.1 | MK | −1875 |
| Glyur000002s00000233.1 | MPD | −1748 |
| Glyur000089s00008825.1 | SQS1 | −696 |
| Glyur000561s00023451.1 | CYP88D6 | −340 and −347 |
| Gene ID | Gene Name | Event Type | Chrom | Event Pattern |
|---|---|---|---|---|
| Glyur000294s00011848 | CPR | TTS | Scaffold00294 | 18154 |
| TSS | Scaffold00294 | 10567 | ||
| Glyur000890s00019071 | CYP 72A154 | TTS | Scaffold00890 | 8048 |
| XIR_ON | Scaffold00890 | 8315–8798 | ||
| TSS | Scaffold00890 | 10471 | ||
| Glyur001936s00032203 | CYP 72A154 | TTS | Scaffold01936 | 49804 |
| TSS | Scaffold01936 | 52193 | ||
| Glyur000218s00011642 | AACT | TTS | Scaffold00218 | 225653 |
| TSS | Scaffold00218 | 229590 | ||
| Glyur000195s00012841 | HMGS | TTS | Scaffold00195 | 66634 |
| TSS | Scaffold00195 | 71499 | ||
| Glyur000682s00024324 | HMGR | TTS | Scaffold00682 | 96974 |
| TSS | Scaffold00682 | 95210 | ||
| Glyur000037s00002618 | TTS | Scaffold00037 | 147378 | |
| TSS | Scaffold00037 | 149004 | ||
| Glyur000135s00007303 | TTS | Scaffold00135 | 140846 | |
| TSS | Scaffold00135 | 142733 | ||
| Glyur000203s00012900 | TTS | Scaffold00203 | 184607 | |
| TSS | Scaffold00203 | 186414 | ||
| Glyur000069s00004081 | MK | TTS | Scaffold00069 | 351209 |
| TSS | Scaffold00069 | 345493 | ||
| IR_OFF | Scaffold00069 | 347728–347912,348276–348369 | ||
| IR_ON | Scaffold00069 | 347728–348369 | ||
| Glyur000343s00025703 | PMK | TTS | Scaffold00343 | 178004 |
| TSS | Scaffold00343 | 171667 | ||
| Glyur000002s00000233 | MPD | TTS | Scaffold00002 | 176640 |
| TSS | Scaffold00002 | 173112 | ||
| Glyur000088s00007722 | FPPS | TTS | Scaffold00088 | 174563 |
| TSS | Scaffold00088 | 178062 | ||
| Glyur000017s00002413 | SQS2 | TTS | Scaffold00017 | 277461 |
| TTS | Scaffold00017 | 277670 | ||
| TTS | Scaffold00017 | 277888 | ||
| TSS | Scaffold00017 | 271392 | ||
| Glyur000089s00008825 | SQS1 | SKIP_OFF | Scaffold00089 | 317631–318581 |
| SKIP_ON | Scaffold00089 | 317631,318018–318093,318581 | ||
| TTS | Scaffold00089 | 314669 | ||
| TSS | Scaffold00089 | 319057 | ||
| IR_OFF | Scaffold00089 | 315038–315129,315471–315559 | ||
| IR_ON | Scaffold00089 | 315038–315559 | ||
| Glyur001733s00027628 | β-AS | TTS | Scaffold01733 | 36913 |
| TSS | Scaffold01733 | 44011 | ||
| Glyur000561s00023451 | CYP88D6 | TTS | Scaffold00561 | 103653 |
| TTS | Scaffold00561 | 139473 | ||
| TSS | Scaffold00561 | 143458 |
| miRNA Sequence | miRNA Name | Target Gene ID | Target Annotation | Expectation |
|---|---|---|---|---|
| AGCAAAAAAGUACAUGUAUCUGGA | miRNA-n217c | Glyur000069s00004081.1 | MK | 3 |
| AGCAAAAAAGUACAUGUAUCUGGA | miRNA-n217b | Glyur000069s00004081.1 | MK | 3 |
| AGCAAAAAAGUACAUGUAUCUGGA | miRNA-n217a | Glyur000069s00004081.1 | MK | 3 |
| AGCAAAAAAGUACAUGUAUCUGGA | miRNA-n217e | Glyur000069s00004081.1 | MK | 3 |
| AGCAAAAAAGUACAUGUAUCUGGA | miRNA-n217d | Glyur000069s00004081.1 | MK | 3 |
| AAGAAAAAAAGUACAUGCAUCUGA | miRNA-n217f | Glyur000069s00004081.1 | MK | 2 |
| AGAAAAAAAAUACAUGUAUCUGGA | miRNA-n217i | Glyur000069s00004081.1 | MK | 1 |
| AGAAAAAAAAUACAUGUAUCUGGA | miRNA-n217h | Glyur000069s00004081.1 | MK | 1 |
| AGAAAAAAAAUACAUGUAUCUGGA | miRNA-n217g | Glyur000069s00004081.1 | MK | 1 |
| AUUCAGCUGGGGCUCACAUCCACC | miRNA-n244 | Glyur001733s00027628.1 | β-AS | 3 |
| CAAAUACACUGCAGAACUAAUGU | miRNA-n69 | Glyur000682s00024324.1 | HMGR | 3 |
| UCAAAGGGGGGUGUGGAGUAG | miR2119 | Glyur000294s00011848.1 | CPR1 | 2.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Chen, C.; Xie, Z.; Xu, J.; Wu, B.; Wang, W. Integrated Analysis of mRNA and microRNA Elucidates the Regulation of Glycyrrhizic Acid Biosynthesis in Glycyrrhiza uralensis Fisch. Int. J. Mol. Sci. 2020, 21, 3101. https://doi.org/10.3390/ijms21093101
Li Y, Chen C, Xie Z, Xu J, Wu B, Wang W. Integrated Analysis of mRNA and microRNA Elucidates the Regulation of Glycyrrhizic Acid Biosynthesis in Glycyrrhiza uralensis Fisch. International Journal of Molecular Sciences. 2020; 21(9):3101. https://doi.org/10.3390/ijms21093101
Chicago/Turabian StyleLi, Yanni, Caixia Chen, Zhenzhen Xie, Jing Xu, Bin Wu, and Wenquan Wang. 2020. "Integrated Analysis of mRNA and microRNA Elucidates the Regulation of Glycyrrhizic Acid Biosynthesis in Glycyrrhiza uralensis Fisch" International Journal of Molecular Sciences 21, no. 9: 3101. https://doi.org/10.3390/ijms21093101
APA StyleLi, Y., Chen, C., Xie, Z., Xu, J., Wu, B., & Wang, W. (2020). Integrated Analysis of mRNA and microRNA Elucidates the Regulation of Glycyrrhizic Acid Biosynthesis in Glycyrrhiza uralensis Fisch. International Journal of Molecular Sciences, 21(9), 3101. https://doi.org/10.3390/ijms21093101