Glycyrrhizin Inhibits PEDV Infection and Proinflammatory Cytokine Secretion via the HMGB1/TLR4-MAPK p38 Pathway

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
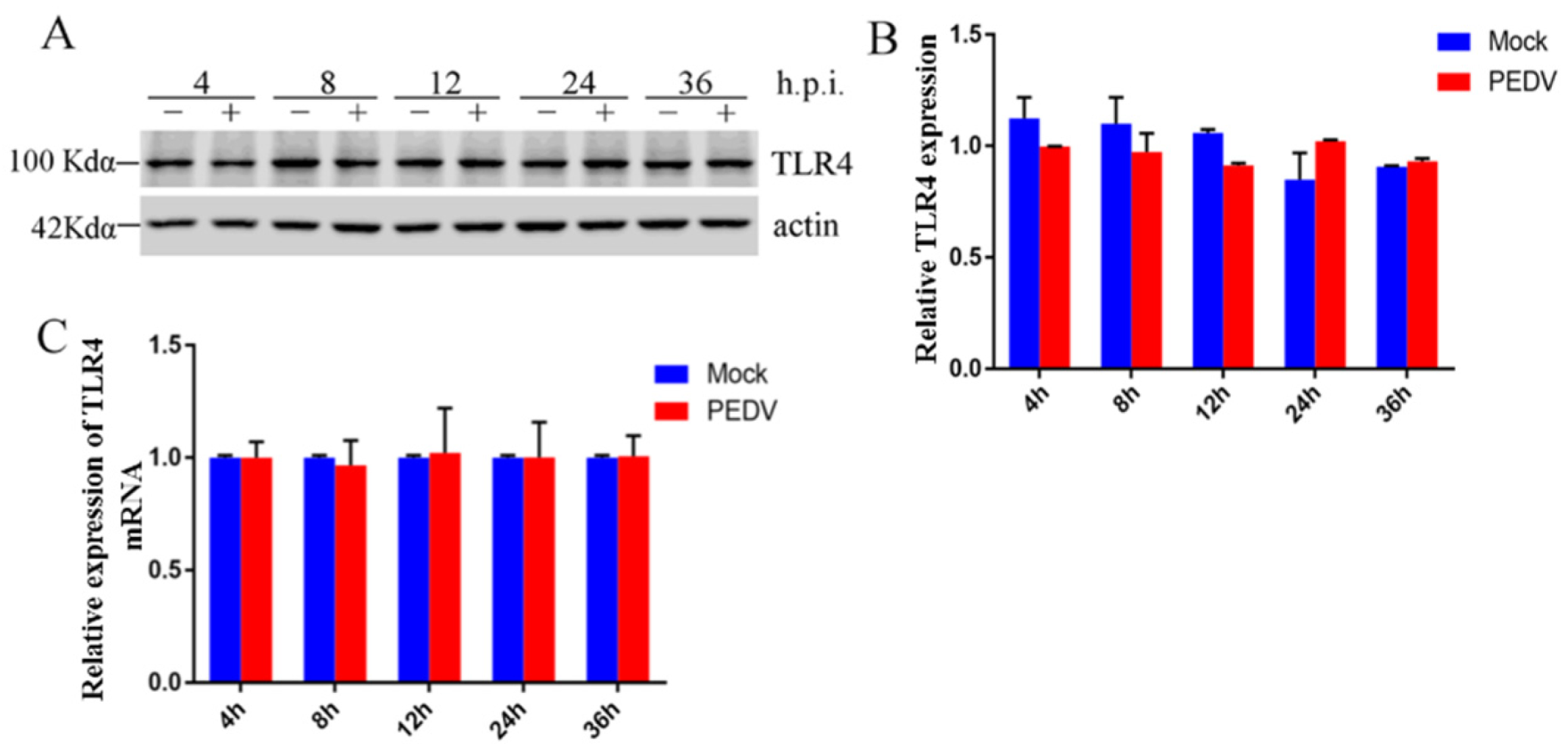
2.1. Effect of PEDV Infection on TLR4 in Vero Cells
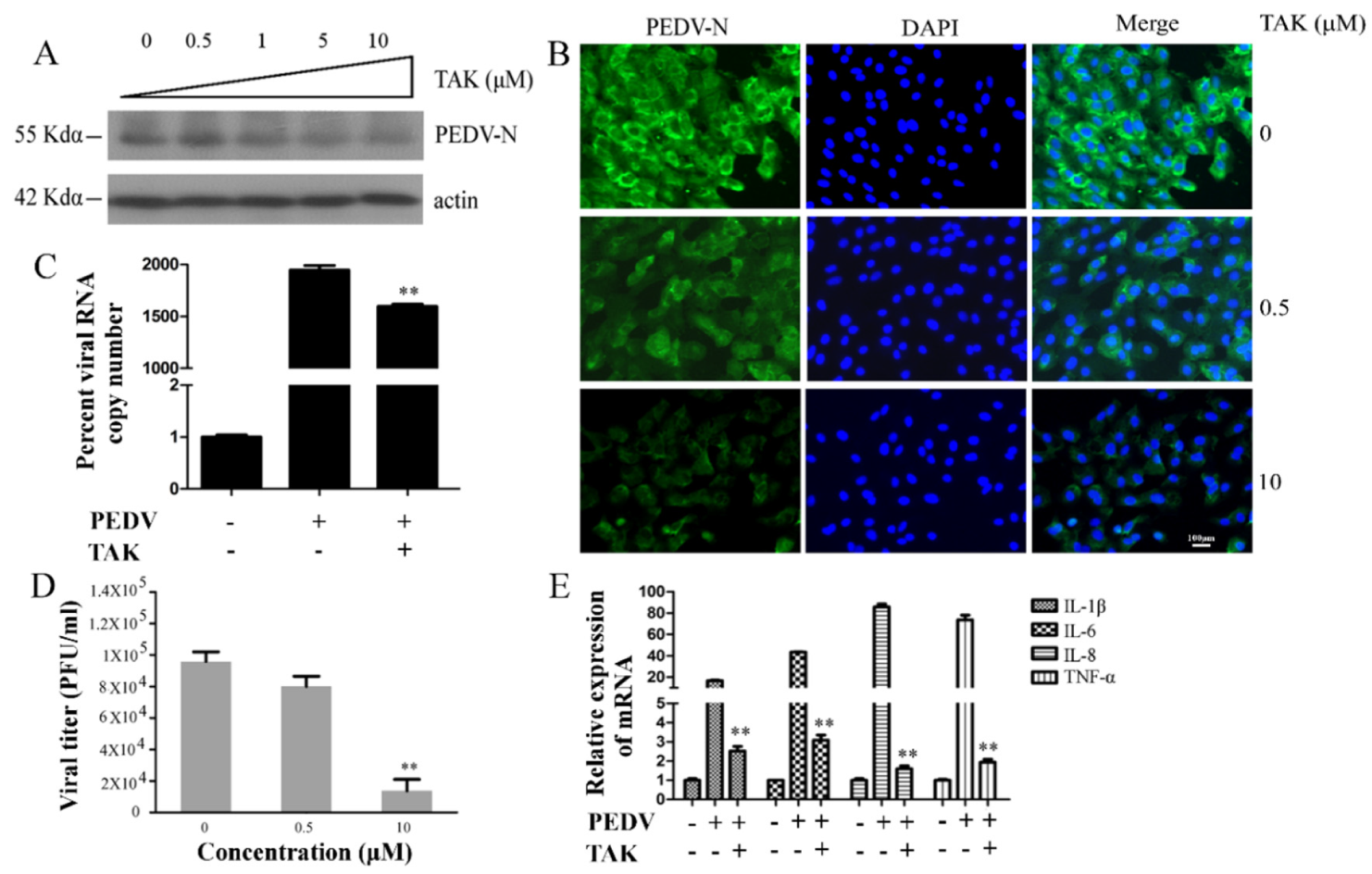
2.2. TLR4 Regulated PEDV Infection
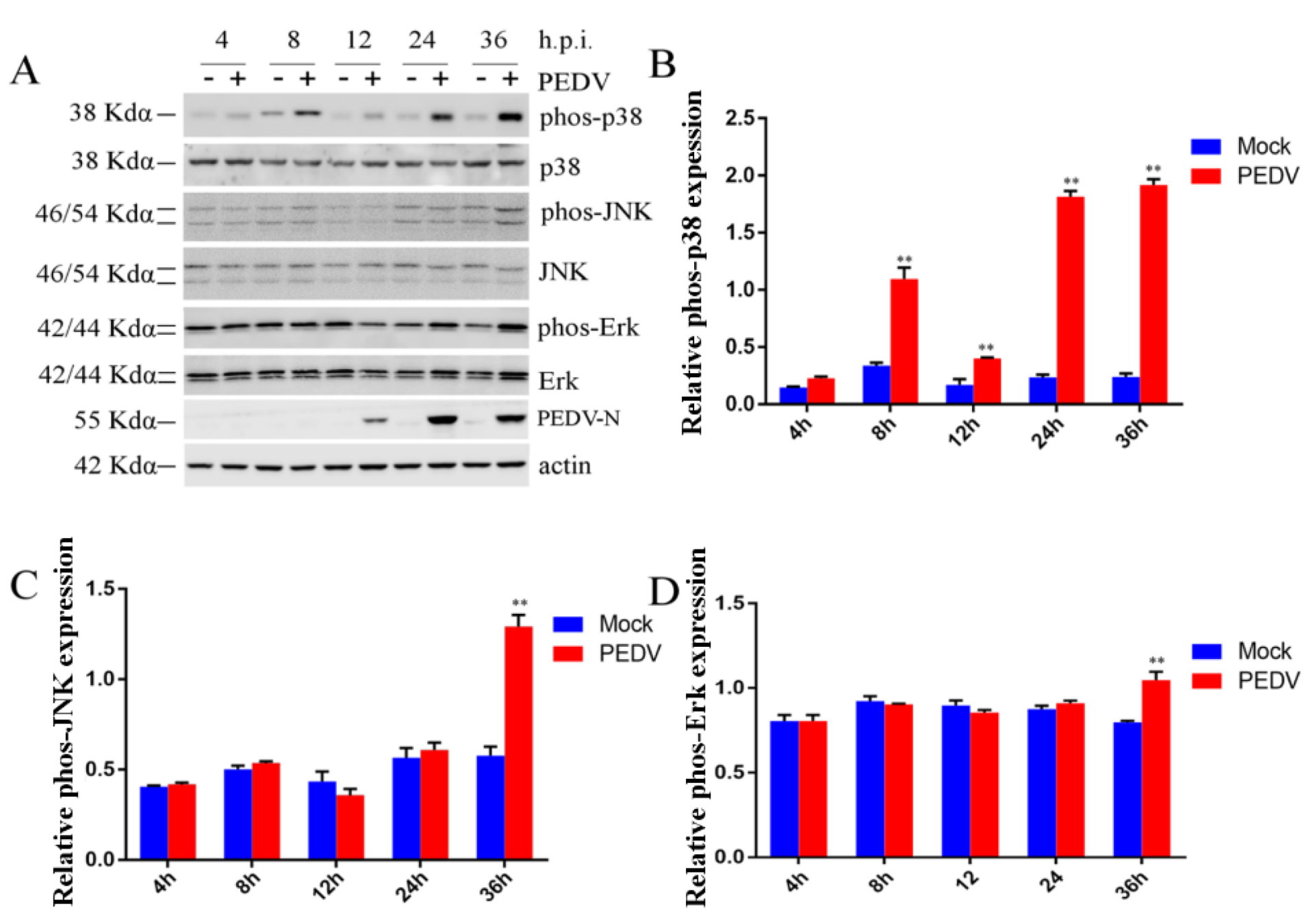
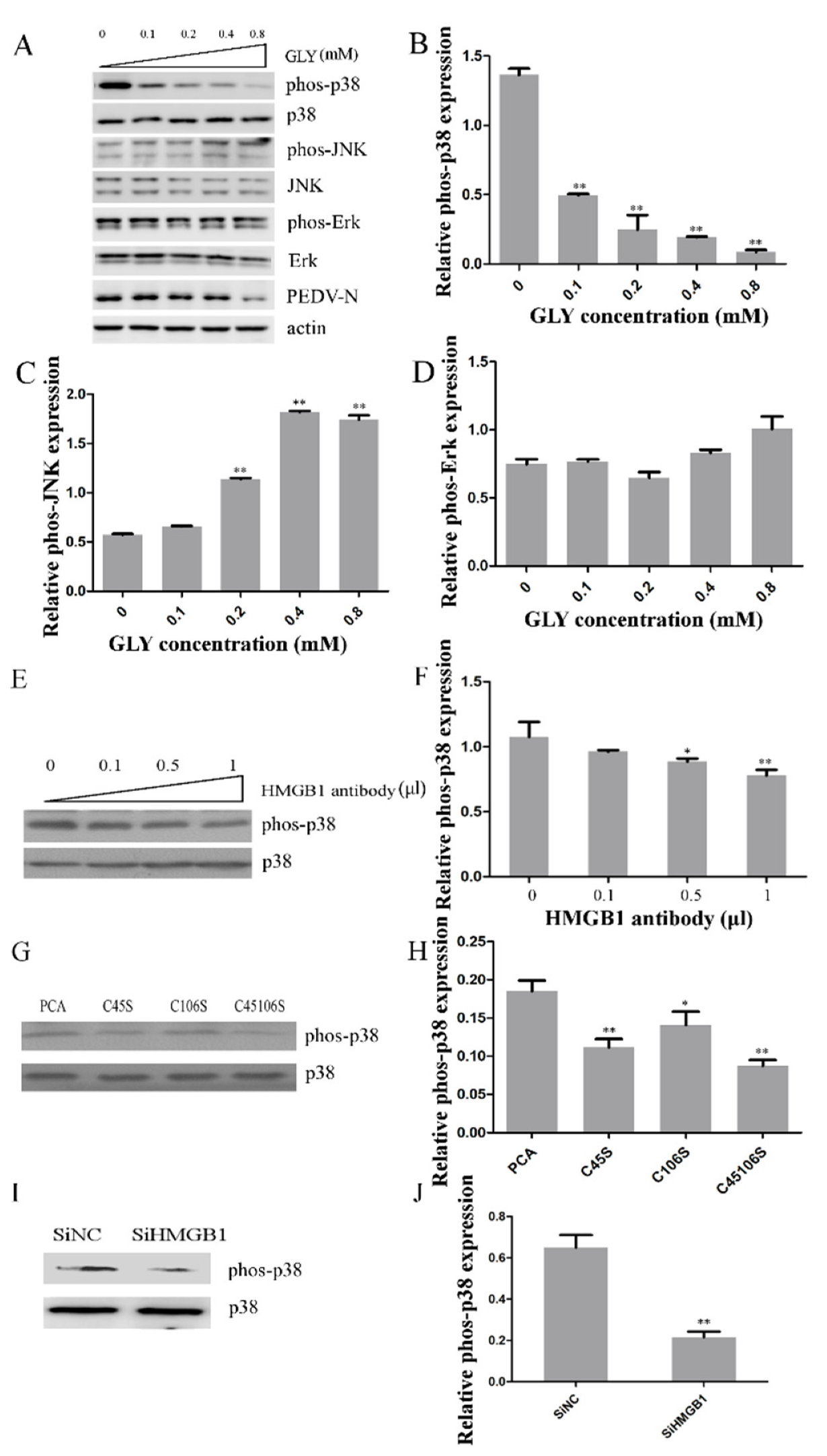
2.3. Effect of PEDV Infection on MAPK p38, Erk1/2, and JNK
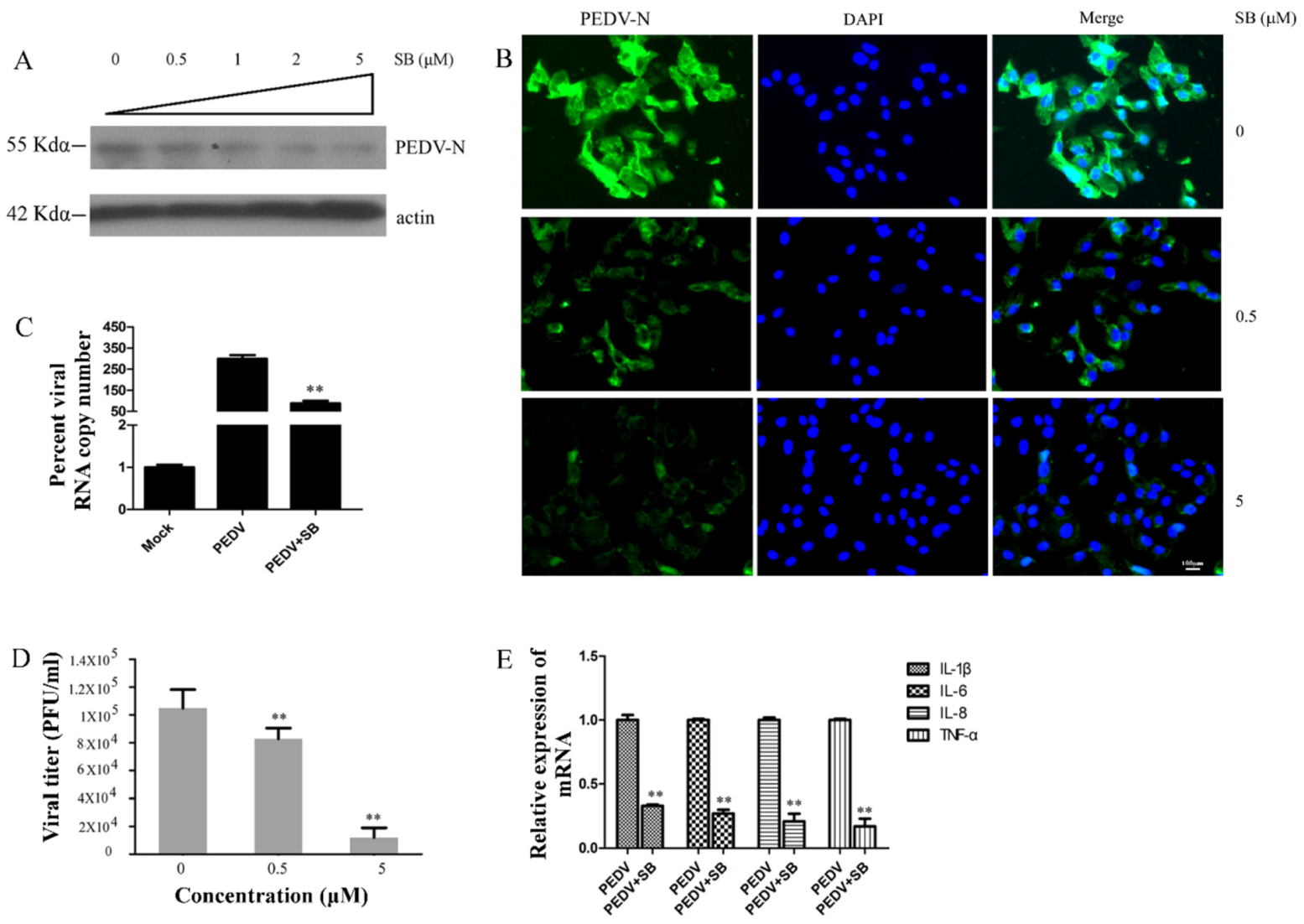
2.4. MAPK p38 Was Critical for PEDV Infection
2.5. Effects TAK on MAPK p38, JNK, and Erk during PEDV Infection
2.6. Effect of HMGB1 on MAPK p38 during PEDV Infection
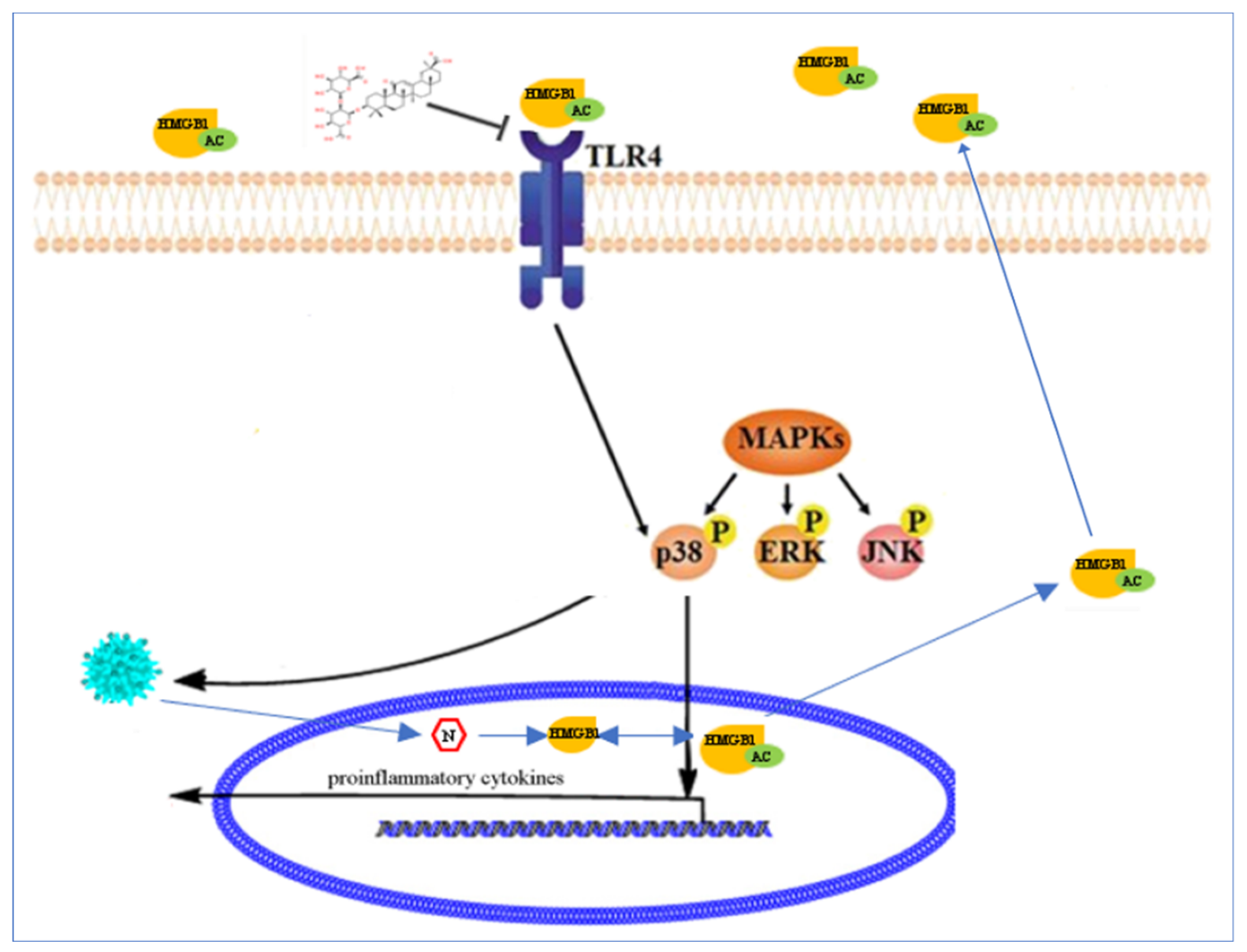
3. Discussion
4. Materials and Methods
4.1. Cells and Virus
4.2. Reagents and Antibodies
4.3. Cell Viability Assay
4.4. Western Blotting
4.5. Immuno Fluorescence Assay (IFA)
4.6. Quantitative Real-Time PCR (qRT-PCR)
4.7. Plaque Formation Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PEDV | porcine epidemic diarrhea virus |
| PED | porcine epidemic diarrhea |
| GLY | glycyrrhizin |
| HCV | hepatitis C virus |
| HMGB1 | high mobility group box-1 |
| TLR4 | toll-like receptor 4 |
| TAK | TAK-242 |
| SB | SB202190 |
| DMEM | Dulbecco’s modified Eagle’s medium |
| MAPK p38 | mitogen-activated protein kinase p38 |
| Erk1/2 | extracellular regulated protein kinases1/2 |
| JNK | c-Jun N-terminal kinase |
| CCK-8 | Cell Counting Kit-8 |
| IFA | immunofluorescence assay |
References
- Huang, Y.W.; Dickerman, A.W.; Pineyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. MBio 2013, 4, e00737. [Google Scholar] [CrossRef]
- Hanke, D.; Pohlmann, A.; Sauter-Louis, C.; Hoper, D.; Stadler, J.; Ritzmann, M.; Steinrigl, A.; Schwarz, B.A.; Akimkin, V.; Fux, R.; et al. Porcine Epidemic Diarrhea in Europe: In-Detail Analyses of Disease Dynamics and Molecular Epidemiology. Viruses 2017, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.Q.; Cai, R.J.; Chen, Y.Q.; Liang, P.S.; Chen, D.K.; Song, C.X. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg. Infect. Dis. 2012, 18, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, C.; Shi, H.; Qiu, H.; Liu, S.; Chen, X.; Zhang, Z.; Feng, L. Molecular epidemiology of porcine epidemic diarrhea virus in China. Arch. Virol. 2010, 155, 1471–1476. [Google Scholar] [CrossRef]
- Chen, Q.; Li, G.; Stasko, J.; Thomas, J.T.; Stensland, W.R.; Pillatzki, A.E.; Gauger, P.C.; Schwartz, K.J.; Madson, D.; Yoon, K.J.; et al. Isolation and characterization of porcine epidemic diarrhea viruses associated with the 2013 disease outbreak among swine in the United States. J. Clin. Microbiol. 2014, 52, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Ducatelle, R.; Coussement, W.; Pensaert, M.B.; Debouck, P.; Hoorens, J. In vivo morphogenesis of a new porcine enteric coronavirus, CV 777. Arch. Virol. 1981, 68, 35–44. [Google Scholar] [CrossRef]
- Pensaert, M.B.; de Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Tang, J.; Ma, Y.; Liang, X.; Yang, Y.; Peng, G.; Qi, Q.; Jiang, S.; Li, J.; Du, L.; et al. Receptor usage and cell entry of porcine epidemic diarrhea coronavirus. J. Virol. 2015, 89, 6121–6125. [Google Scholar] [CrossRef]
- Choudhury, B.; Dastjerdi, A.; Doyle, N.; Frossard, J.P.; Steinbach, F. From the field to the lab—An European view on the global spread of PEDV. Virus Res. 2016, 226, 40–49. [Google Scholar] [CrossRef]
- Lee, C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol. J. 2015, 12, 193. [Google Scholar] [CrossRef]
- Song, D.; Park, B. Porcine epidemic diarrhoea virus: A comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes 2012, 44, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, Y.; Sakamoto, M.; Matsushita, M.; Fujita, T.; Nishioka, K. Glycyrrhizin inhibits the lytic pathway of complement—Possible mechanism of its anti-inflammatory effect on liver cells in viral hepatitis. Microbiol. Immunol. 2000, 44, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Hibasami, H.; Iwase, H.; Yoshioka, K.; Takahashi, H. Glycyrrhetic acid (a metabolic substance and aglycon of glycyrrhizin) induces apoptosis in human hepatoma, promyelotic leukemia and stomach cancer cells. Int. J. Mol. Med. 2006, 17, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Mori, T.; Shibata, S.; Saito, H. Pharmacological activities of glycyrrhetinic acid derivatives: Analgesic and anti-type IV allergic effects. Chem. Pharm. Bull. 1987, 35, 3888–3893. [Google Scholar] [CrossRef]
- Pompei, R.; Flore, O.; Marccialis, M.A.; Pani, A.; Loddo, B. Glycyrrhizic acid inhibits virus growth and inactivates virus particles. Nature 1979, 281, 689–690. [Google Scholar] [CrossRef]
- Kimura, M.; Watanabe, H.; Abo, T. Selective activation of extrathymic T cells in the liver by glycyrrhizin. Biotherapy 1992, 5, 167–176. [Google Scholar] [CrossRef]
- Crance, J.M.; Leveque, F.; Biziagos, E.; van Cuyck-Gandre, H.; Jouan, A.; Deloince, R. Studies on mechanism of action of glycyrrhizin against hepatitis A virus replication in vitro. Antivir. Res. 1994, 23, 63–76. [Google Scholar] [CrossRef]
- Ashfaq, U.A.; Masoud, M.S.; Nawaz, Z.; Riazuddin, S. Glycyrrhizin as antiviral agent against Hepatitis C Virus. J. Transl. Med. 2011, 9, 112. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Matsuura, T.; Aoyagi, H.; Matsuda, M.; Hmwe, S.S.; Date, T.; Watanabe, N.; Watashi, K.; Suzuki, R.; Ichinose, S.; et al. Antiviral activity of glycyrrhizin against hepatitis C virus in vitro. PLoS ONE 2013, 8, e68992. [Google Scholar] [CrossRef]
- Sekizawa, T.; Yanagi, K.; Itoyama, Y. Glycyrrhizin increases survival of mice with herpes simplex encephalitis. Acta Virol. 2001, 45, 51–54. [Google Scholar]
- Utsunomiya, T.; Kobayashi, M.; Pollard, R.B.; Suzuki, F. Glycyrrhizin, an active component of licorice roots, reduces morbidity and mortality of mice infected with lethal doses of influenza virus. Antimicrob. Agents Chemother. 1997, 41, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lieberman, P.M. Mechanism of glycyrrhizic acid inhibition of Kaposi’s sarcoma-associated herpesvirus: Disruption of CTCF-cohesin-mediated RNA polymerase II pausing and sister chromatid cohesion. J. Virol. 2011, 85, 11159–11169. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Gamboni-Robertson, F.; He, Q.; Svetkauskaite, D.; Kim, J.Y.; Strassheim, D.; Sohn, J.W.; Yamada, S.; Maruyama, I.; Banerjee, A.; et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 2006, 290, C917–C924. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Huan, C.C.; Wang, H.X.; Sheng, X.X.; Wang, R.; Wang, X.; Mao, X. Glycyrrhizin inhibits porcine epidemic diarrhea virus infection and attenuates the proinflammatory responses by inhibition of high mobility group box-1 protein. Arch. Virol. 2017, 162, 1467–1476. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. MMBR 2004, 68, 320–344. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Sui, Y.; Zhao, Z.; Liu, R.; Cai, B.; Fan, D. Adenosine monophosphate-activated protein kinase activation enhances embryonic neural stem cell apoptosis in a mouse model of amyotrophic lateral sclerosis. Neural Regen. Res. 2014, 9, 1770–1778. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, W.; Zhang, X.; Wang, C.; Gao, L.; Yang, F.; Cao, W.; Li, K.; Tian, H.; Liu, X.; et al. Foot-and-Mouth Disease Virus Capsid Protein VP1 Interacts with Host Ribosomal Protein SA To Maintain Activation of the MAPK Signal Pathway and Promote Virus Replication. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, Y.; Wang, S.; Liu, W.; Hao, C.; Wang, W. Inhibition effects of patchouli alcohol against influenza a virus through targeting cellular PI3K/Akt and ERK/MAPK signaling pathways. Virol. J. 2019, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Varshney, B.; Lal, S.K. SARS-CoV accessory protein 3b induces AP-1 transcriptional activity through activation of JNK and ERK pathways. Biochemistry 2011, 50, 5419–5425. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Wang, Y. p38 MAP kinases in the heart. Gene 2016, 575, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Marber, M.S.; Rose, B.; Wang, Y. The p38 mitogen-activated protein kinase pathway—A potential target for intervention in infarction, hypertrophy, and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, M.; Zhang, J.; Xu, R. p38 MAPK: A potential target of chronic pain. Curr. Med. Chem. 2014, 21, 4405–4418. [Google Scholar] [CrossRef]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Kono, M.; Tatsumi, K.; Imai, A.M.; Saito, K.; Kuriyama, T.; Shirasawa, H. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: Involvement of p38 MAPK and ERK. Antivir. Res. 2008, 77, 150–152. [Google Scholar] [CrossRef]
- Dong, W.; Xie, W.; Liu, Y.; Sui, B.; Zhang, H.; Liu, L.; Tan, Y.; Tong, X.; Fu, Z.F.; Yin, P.; et al. Receptor tyrosine kinase inhibitors block proliferation of TGEV mainly through p38 mitogen-activated protein kinase pathways. Antivir. Res. 2020, 173, 104651. [Google Scholar] [CrossRef]
- Lai, J.L.; Liu, Y.H.; Liu, C.; Qi, M.P.; Liu, R.N.; Zhu, X.F.; Zhou, Q.G.; Chen, Y.Y.; Guo, A.Z.; Hu, C.M. Indirubin Inhibits LPS-Induced Inflammation via TLR4 Abrogation Mediated by the NF-kB and MAPK Signaling Pathways. Inflammation 2017, 40, 1–12. [Google Scholar] [CrossRef]
- Kim, D.O.; Byun, J.E.; Seong, H.A.; Yoon, S.R.; Choi, I.; Jung, H. Thioredoxin-interacting protein-derived peptide (TN13) inhibits LPS-induced inflammation by inhibiting p38 MAPK signaling. Biochem. Biophys. Res. Commun. 2018, 507, 489–495. [Google Scholar] [CrossRef]
- Dai, J.P.; Wang, Q.W.; Su, Y.; Gu, L.M.; Deng, H.X.; Chen, X.X.; Li, W.Z.; Li, K.S. Oxymatrine Inhibits Influenza A Virus Replication and Inflammation via TLR4, p38 MAPK and NF-kappaB Pathways. Int. J. Mol. Sci. 2018, 19, 965. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; You, H.; Li, X.M.; Liu, T.H.; Wang, P.; Wang, B.E. HMGB1 promotes the synthesis of pro-IL-1beta and pro-IL-18 by activation of p38 MAPK and NF-kappaB through receptors for advanced glycation end-products in macrophages. Asian Pac. J. Cancer Prev. 2012, 13, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- He, Z.W.; Qin, Y.H.; Wang, Z.W.; Chen, Y.; Shen, Q.; Dai, S.M. HMGB1 acts in synergy with lipopolysaccharide in activating rheumatoid synovial fibroblasts via p38 MAPK and NF-kappaB signaling pathways. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Huan, C.C.; Wang, H.X.; Sheng, X.X.; Wang, R.; Wang, X.; Liao, Y.; Liu, Q.F.; Tong, G.Z.; Ding, C.; Fan, H.J.; et al. Porcine epidemic diarrhea virus nucleoprotein contributes to HMGB1 transcription and release by interacting with C/EBP-beta. Oncotarget 2016, 7, 75064–75080. [Google Scholar] [CrossRef] [PubMed]
- Huan, C.C.; Guo, T.T.; Xu, W.Y.; Pan, H.C.; Gao, S. The role of MAPK pathways in porcine epidemic diarrhea virus infection. Arch. Virol. (under review).
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur. J. Biochem. 1973, 38, 14–19. [Google Scholar] [CrossRef]
- Joshi, S.R.; Sarpong, Y.C.; Peterson, R.C.; Scovell, W.M. Nucleosome dynamics: HMGB1 relaxes canonical nucleosome structure to facilitate estrogen receptor binding. Nucleic Acids Res. 2012, 40, 10161–10171. [Google Scholar] [CrossRef][Green Version]
- Khanolkar, A.; Hartwig, S.M.; Haag, B.A.; Meyerholz, D.K.; Harty, J.T.; Varga, S.M. Toll-like receptor 4 deficiency increases disease and mortality after mouse hepatitis virus type 1 infection of susceptible C3H mice. J. Virol. 2009, 83, 8946–8956. [Google Scholar] [CrossRef]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Banerjee, S.; Narayanan, K.; Mizutani, T.; Makino, S. Murine coronavirus replication-induced p38 mitogen-activated protein kinase activation promotes interleukin-6 production and virus replication in cultured cells. J. Virol. 2002, 76, 5937–5948. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Palese, P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J. Virol. 2006, 80, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Huan, C.C.; Wang, Y.; Ni, B.; Wang, R.; Huang, L.; Ren, X.F.; Tong, G.Z.; Ding, C.; Fan, H.J.; Mao, X. Porcine epidemic diarrhea virus uses cell-surface heparan sulfate as an attachment factor. Arch. Virol. 2015, 160, 1621–1628. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, C. Extracellular signal-regulated kinase (ERK) activation is required for porcine epidemic diarrhea virus replication. Virology 2015, 484, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.W.; Weng, S.S.; Cheng, Q.; Cui, P.; Li, Y.J.; Wu, H.L.; Zhu, Y.M.; Xu, B.; Zhang, W.H. Human Endophthalmitis Caused By Pseudorabies Virus Infection, China, 2017. Emerg. Infect. Dis. 2018, 24, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bao, L.; Zha, D.; Zhang, L.; Gao, P.; Zhang, J.; Wu, X. Oridonin protects against the inflammatory response in diabetic nephropathy by inhibiting the TLR4/p38-MAPK and TLR4/NF-kappaB signaling pathways. Int. Immunopharmacol. 2018, 55, 9–19. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, R.; Zhang, Y.; Kang, Y.; Xu, W.; Jiang, L.; Guo, T.; Huan, C. Glycyrrhizin Inhibits PEDV Infection and Proinflammatory Cytokine Secretion via the HMGB1/TLR4-MAPK p38 Pathway. Int. J. Mol. Sci. 2020, 21, 2961. https://doi.org/10.3390/ijms21082961
Gao R, Zhang Y, Kang Y, Xu W, Jiang L, Guo T, Huan C. Glycyrrhizin Inhibits PEDV Infection and Proinflammatory Cytokine Secretion via the HMGB1/TLR4-MAPK p38 Pathway. International Journal of Molecular Sciences. 2020; 21(8):2961. https://doi.org/10.3390/ijms21082961
Chicago/Turabian StyleGao, Ruyi, Yongshuai Zhang, Yuhui Kang, Weiyin Xu, Luyao Jiang, Tingting Guo, and Changchao Huan. 2020. "Glycyrrhizin Inhibits PEDV Infection and Proinflammatory Cytokine Secretion via the HMGB1/TLR4-MAPK p38 Pathway" International Journal of Molecular Sciences 21, no. 8: 2961. https://doi.org/10.3390/ijms21082961
APA StyleGao, R., Zhang, Y., Kang, Y., Xu, W., Jiang, L., Guo, T., & Huan, C. (2020). Glycyrrhizin Inhibits PEDV Infection and Proinflammatory Cytokine Secretion via the HMGB1/TLR4-MAPK p38 Pathway. International Journal of Molecular Sciences, 21(8), 2961. https://doi.org/10.3390/ijms21082961