The Crystal Structure of a Streptomyces thermoviolaceus Thermophilic Chitinase Known for Its Refolding Efficiency


Abstract
1. Introduction
2. Results
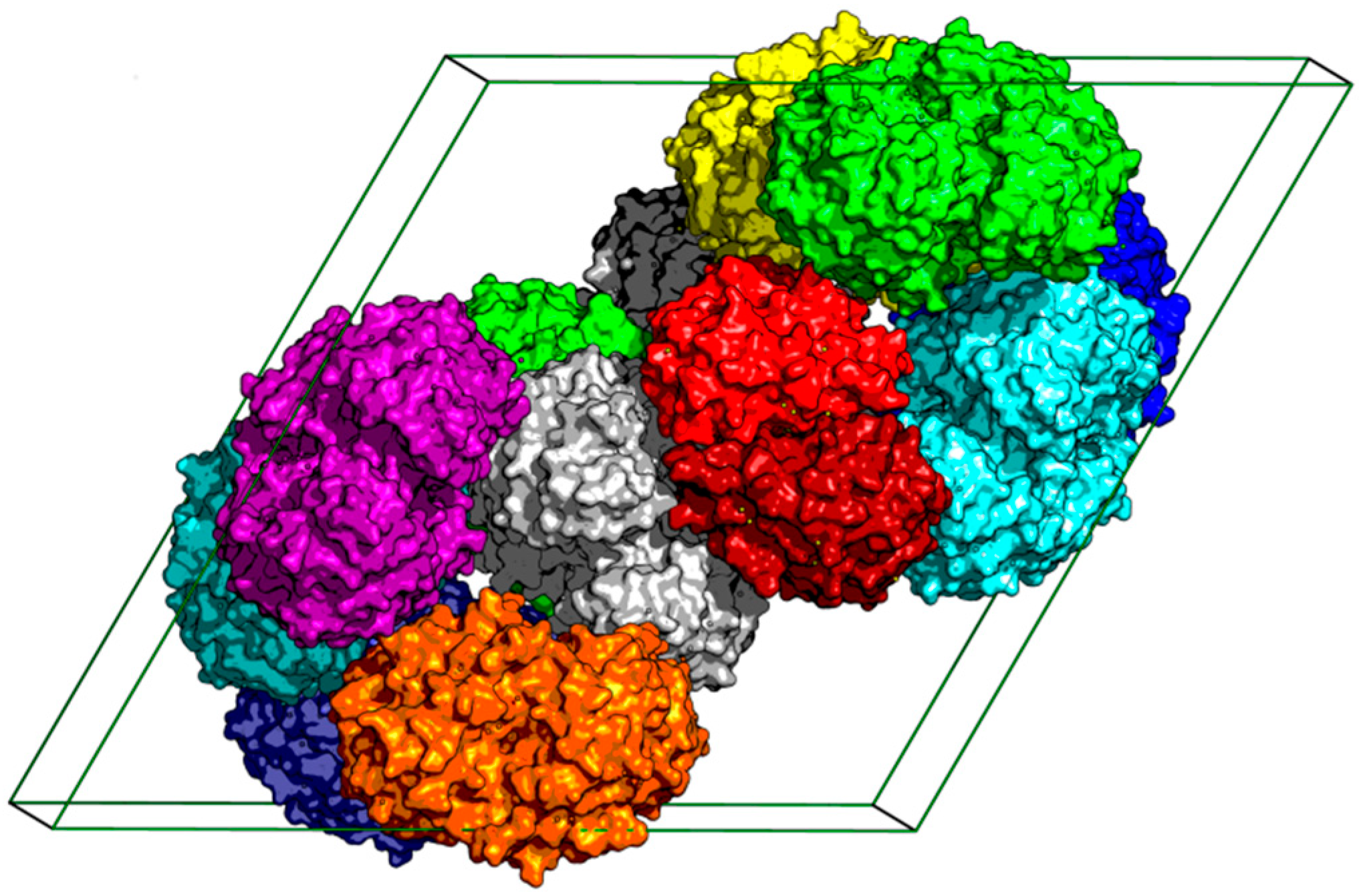
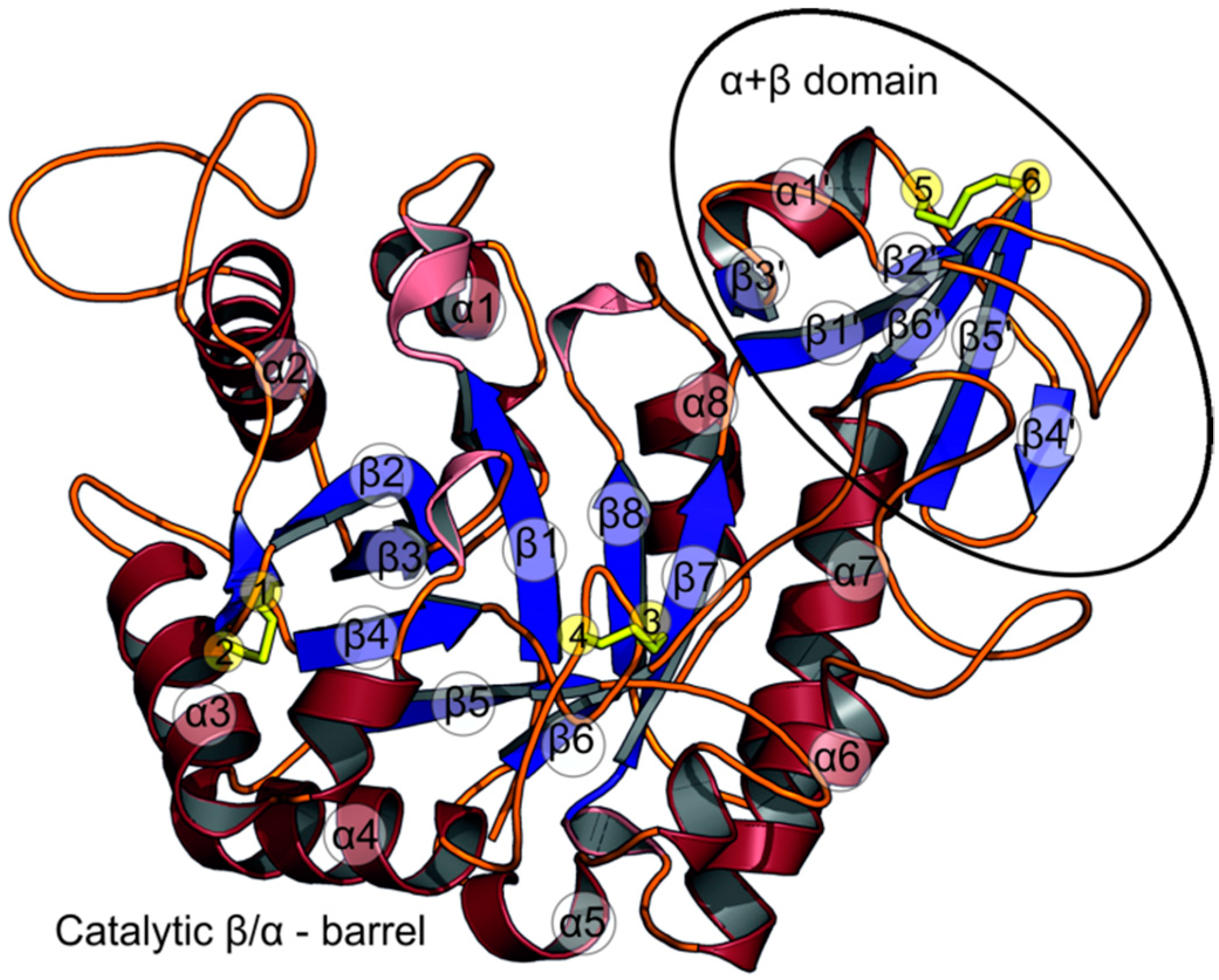
2.1. The Overall Structure of StChi40
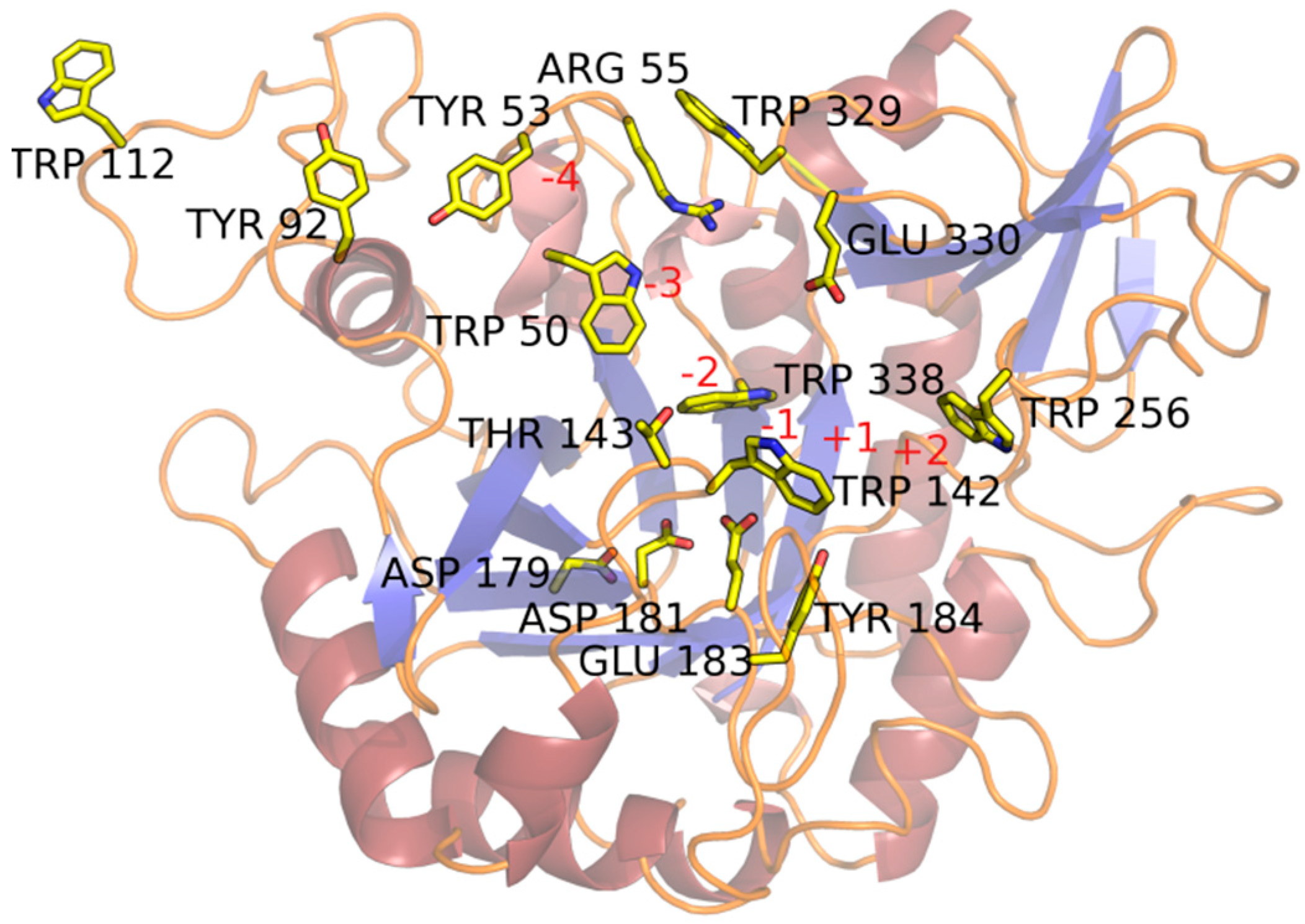
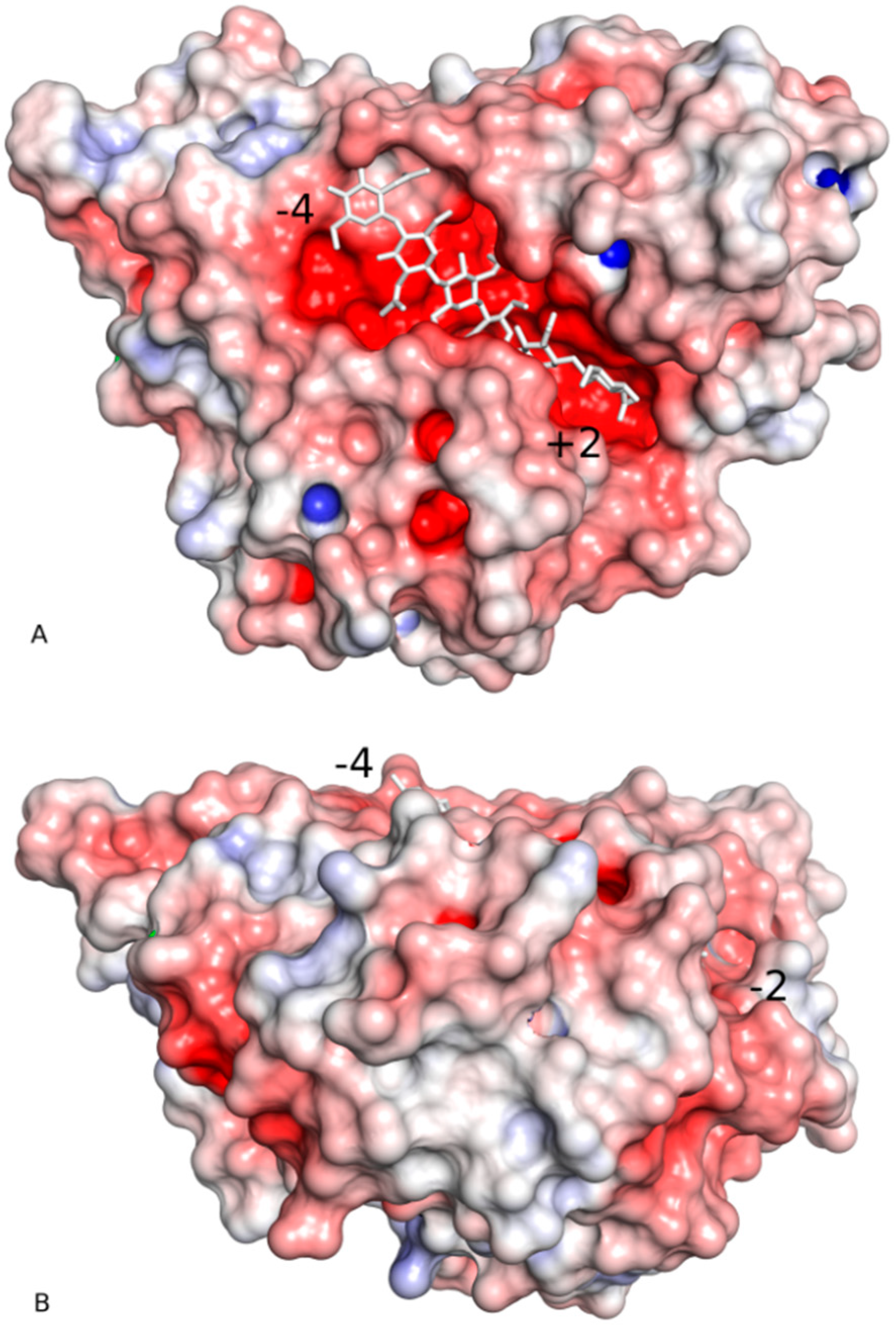
2.2. Substrate-Binding Groove
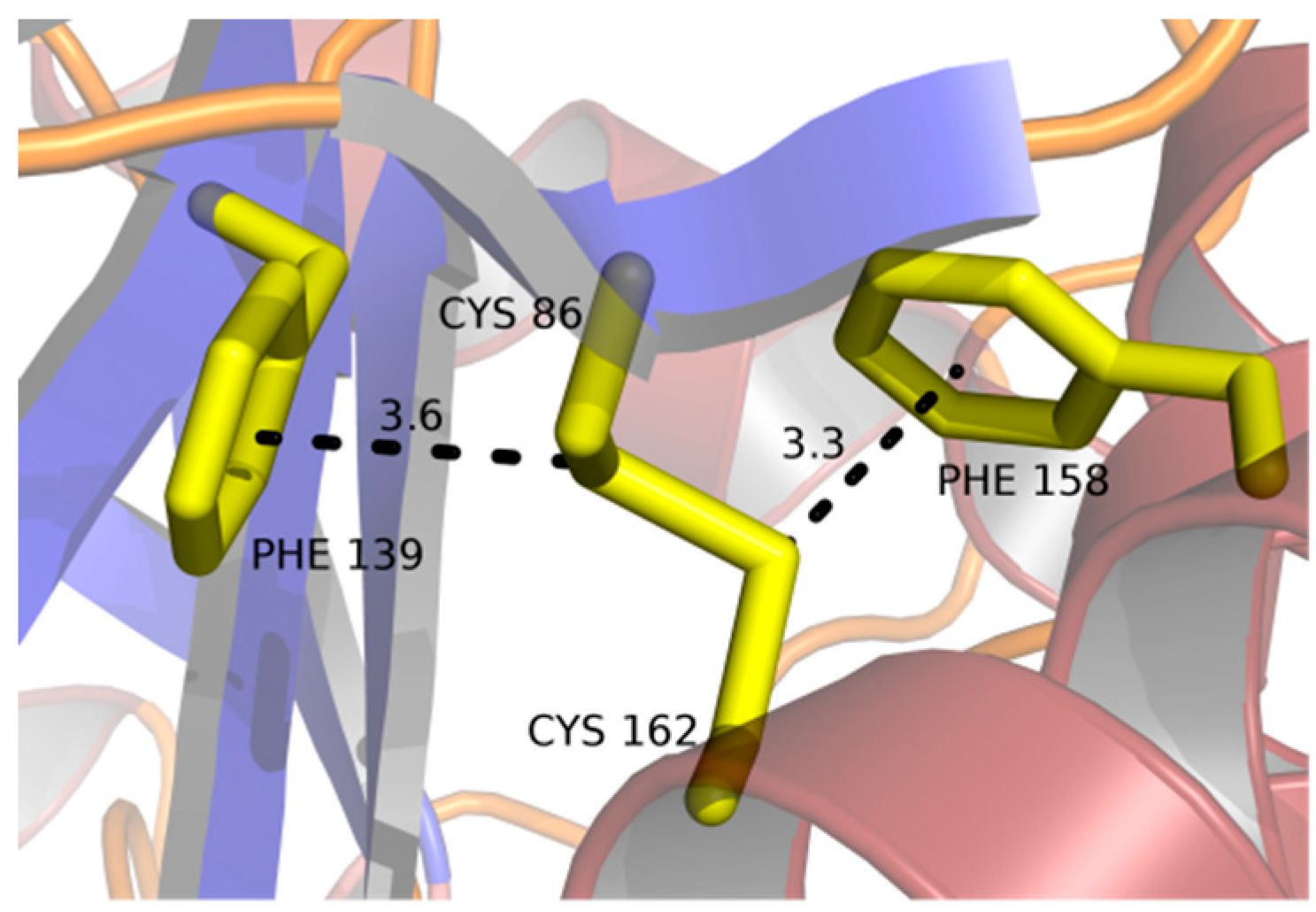
2.3. Disulfide Bridges
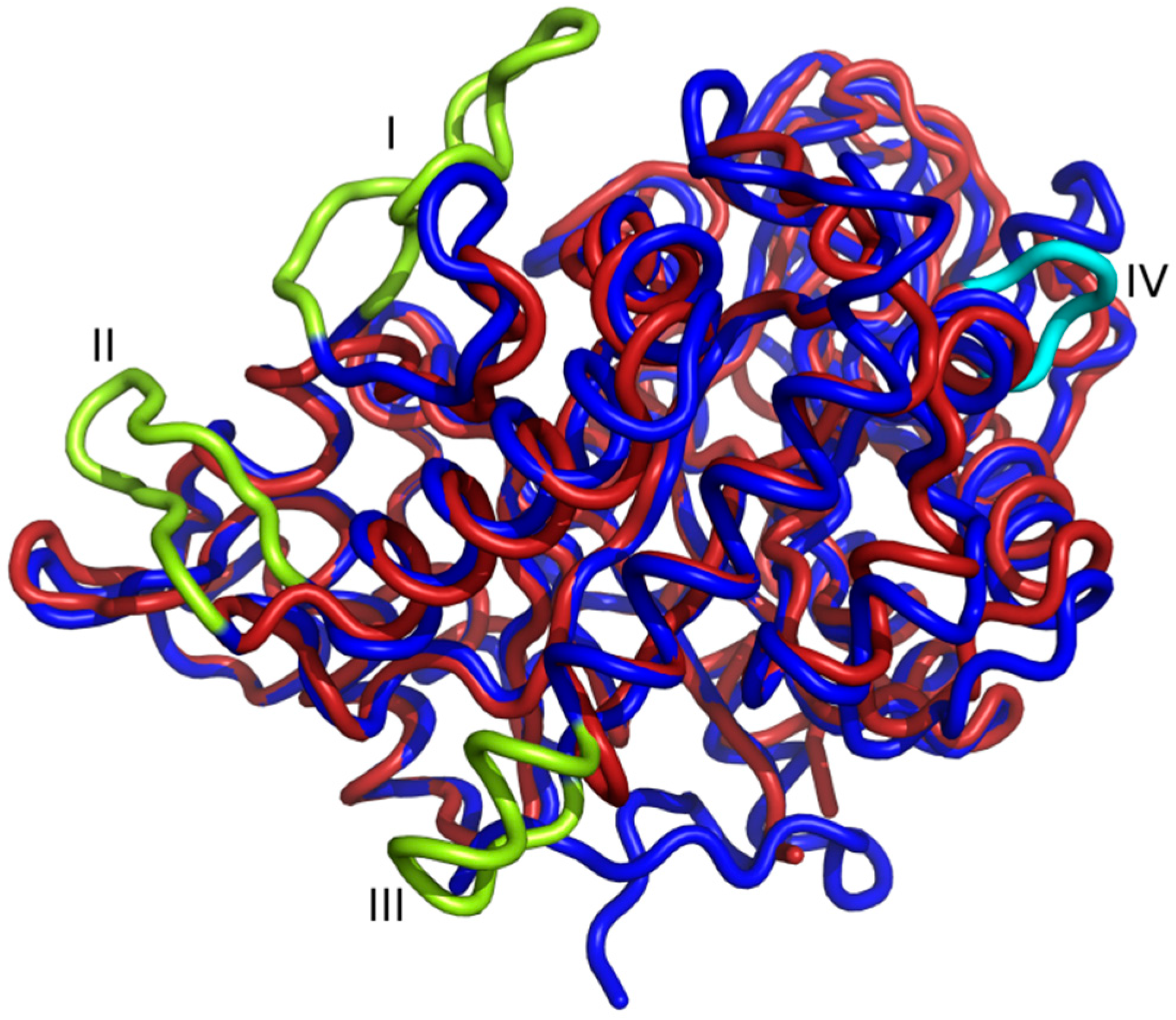
2.4. Amino Acid Substitutions and Their Structural Consequences
3. Discussion
4. Materials and Methods
4.1. Biological Source
4.2. Plasmid and DNA Manipulations
4.3. Crystallization and X-Ray Data Collection
4.4. Structure Determination and Refinement
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| StChi40 | Chitinase 40 from Streptomyces thermoviolaceus |
| GHJ | Glycoside hydrolase |
| ChBD | Chitin-binding domain |
| CBMs | Carbohydrate-binding modules |
| r.m.s. | Root-mean-square |
| TIM | Triose-phosphate isomerase |
| EC | Enzyme Commission (number) |
| ChiA | Chitinase A |
| ChiB | Chitinase B |
| NAG | N-Acetylglucosamine |
| PDB | Protein Data Bank |
| TLS | Translation/Libration/Screw |
| ATCC | American Type Culture Collection |
References
- Jolles, P.; Muzzarelli, R.A.A. Chitin and Chitinases; Springer: Basel, Switzerland, 1999. [Google Scholar]
- Boller, T. Plant–Microbe interactions: Molecular and Genetic Perspectives; Kosuge, T., Nester, E.W., Eds.; MacMillan: New York, NY, USA, 1987; Volume 2, pp. 384–414. [Google Scholar]
- Boot, R.G.; Renkema, G.H.; Strijland, A.; van Zonneveld, A.J.; Aerts, J.M. Cloning of a cDNA encoding chitotriosidase, a human chitinase produced by macrophages. J. Biol. Chem. 1995, 270, 26252–26256. [Google Scholar] [CrossRef]
- Gooday, G.W. Physiology of microbial degradation of chitin and chitosan. In Biochemistry of Microbial Degradation; Ratledge, C., Ed.; Kluwer: Dordrecht, The Netherlands, 1994; pp. 279–312. [Google Scholar]
- Henrissat, B.; Bairoch, A. Updating the sequence-based classification of glycosyl hydrolases. Biochem J. 1996, 316, 695–696. [Google Scholar] [CrossRef]
- Davies, G.; Henrissat, B. Structures and mechanisms of glycosyl hydrolases. Structure 1995, 3, 853–859. [Google Scholar] [CrossRef]
- Monreal, J.; Reese, E.T. The chitinase of Serratia marcescens. Can. J. Microbiol. 1969, 15, 689–696. [Google Scholar] [CrossRef]
- Vaaje-Kolstad, G.; Horn, S.J.; Sorlie, M.; Eijsink, V.G. The chitinolytic machinery of Serratia marcescens—A model system for enzymatic degradation of recalcitrant polysaccharides. FEBS J. 2013, 280, 3028–3049. [Google Scholar] [CrossRef] [PubMed]
- Tsujibo, H.; Minoura, K.; Miyamoto, K.; Endo, H.; Moriwaki, M.; Inamori, Y. Purification and properties of a thermostable chitinase from Streptomyces thermoviolaceus OPC-520. Appl. Environ. Microbiol. 1993, 59, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Tsujibo, H.; Hatano, N.; Endo, H.; Miyamoto, K.; Inamori, Y. Purification and characterization of a thermostable chitinase from Streptomyces thermoviolaceus OPC-520 and cloning of the encoding gene. Biosci. Biotechnol. Biochem. 2000, 64, 96–102. [Google Scholar] [CrossRef]
- Delic, I.; Robbins, P.; Westpheling, J. Direct repeat sequences are implicated in the regulation of two Streptomyces chitinase promoters that are subject to carbon catabolite control. Proc. Natl. Acad. Sci. USA 1992, 89, 1885–1889. [Google Scholar] [CrossRef]
- Tsujibo, H.; Hatano, N.; Okamoto, T.; Endo, H.; Miyamoto, K.; Inamori, Y. Synthesis of chitinase in Streptomyces thermoviolaceus is regulated by a two-component sensor-regulator system. FEMS. Microbiol. Lett. 1999, 181, 83–90. [Google Scholar] [CrossRef]
- Ni, X.; Westpheling, J. Direct repeat sequences in the Streptomyces chitinase-63 promoter direct both glucose repression and chitin induction. Proc. Natl. Acad. Sci. USA 1997, 94, 13116–13121. [Google Scholar] [CrossRef]
- Pyrpassopoulos, S.; Vlassi, M.; Tsortos, A.; Papanikolau, Y.; Petratos, K.; Vorgias, C.E.; Nounesis, G. Equilibrium heat-induced denaturation of chitinase 40 from Streptomyces thermoviolaceus. Proteins 2006, 64, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Zees, A.C.; Pyrpassopoulos, S.; Vorgias, C.E. Insights into the role of the (alpha+beta) insertion in the TIM-barrel catalytic domain, regarding the stability and the enzymatic activity of chitinase A from Serratia marcescens. Biochim. Biophys. Acta 2009, 1794, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Papanikolau, Y.; Prag, G.; Tavlas, G.; Vorgias, C.E.; Oppenheim, A.B.; Petratos, K. High resolution structural analyses of mutant chitinase A complexes with substrates provide new insight into the mechanism of catalysis. Biochemistry 2001, 40, 11338–11343. [Google Scholar] [CrossRef]
- Songsiriritthigul, C.; Pantoom, S.; Aguda, A.H.; Robinson, R.C.; Suginta, W. Crystal structures of Vibrio harveyi chitinase A complexed with chitooligosaccharides: Implications for the catalytic mechanism. J. Struct. Biol. 2008, 162, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Malecki, P.H.; Vorgias, C.E.; Petoukhov, M.V.; Svergun, D.I.; Rypniewski, W. Crystal structures of substrate-bound chitinase from the psychrophilic bacterium Moritella marina and its structure in solution. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gan, Z.; Lou, Z.; Tao, N.; Mi, Q.; Liang, L.; Sun, Y.; Guo, Y.; Huang, X.; Zou, C.; et al. Crystal structure and mutagenesis analysis of chitinase CrChi1 from the nematophagous fungus Clonostachys rosea in complex with the inhibitor caffeine. Microbiology 2010, 156, 3566–3574. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Perrakis, A.; Tews, I.; Dauter, Z.; Oppenheim, A.B.; Chet, I.; Wilson, K.S.; Vorgias, C.E. Crystal structure of a bacterial chitinase at 2.3 A resolution. Structure 1994, 2, 1169–1180. [Google Scholar] [CrossRef]
- Liu, T.; Chen, L.; Zhou, Y.; Jiang, X.; Duan, Y.W.; Yang, Q. Structure, Catalysis, and Inhibition of OfChi-h, the Lepidoptera-exclusive Insect Chitinase. J. Biol. Chem. 2017, 292, 2080–2088. [Google Scholar] [CrossRef]
- Juarez-Hernandez, E.O.; Casados-Vazquez, L.E.; Brieba, L.G.; Torres-Larios, A.; Jimenez-Sandoval, P.; Barboza-Corona, J.E. The crystal structure of the chitinase ChiA74 of Bacillus thuringiensis has a multidomain assembly. Sci Rep. 2019, 9. [Google Scholar] [CrossRef]
- Synstad, B.; Gaseidnes, S.; Van Aalten, D.M.; Vriend, G.; Nielsen, J.E.; Eijsink, V.G. Mutational and computational analysis of the role of conserved residues in the active site of a family 18 chitinase. Eur. J. Biochem. 2004, 271, 253–262. [Google Scholar] [CrossRef]
- Piovesan, D.; Minervini, G.; Tosatto, S.C.E. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016, 44, W367–W374. [Google Scholar] [CrossRef] [PubMed]
- van Aalten, D.M.; Komander, D.; Synstad, B.; Gaseidnes, S.; Peter, M.G.; Eijsink, V.G. Structural insights into the catalytic mechanism of a family 18 exo-chitinase. Proc. Natl. Acad. Sci. USA 2001, 98, 8979–8984. [Google Scholar] [CrossRef] [PubMed]
- Malecki, P.H.; Raczynska, J.E.; Vorgias, C.E.; Rypniewski, W. Structure of a complete four-domain chitinase from Moritella marina, a marine psychrophilic bacterium. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-X. Loops, linkages, rings, catenanes, cages, and crowders: Entropy-based strategies for stabilizing proteins. Acc. Chem. Res. 2004, 37, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.Y.; Song, H.K.; Chang, C.; Lee, J.; Lee, S.Y.; Kim, K.K.; Choe, S.; Sweet, R.M.; Suh, S.W. Crystal structure of thermostable alpha-amylase from Bacillus licheniformis refined at 1.7 A resolution. Mol. Cells 1997, 7, 251–258. [Google Scholar] [PubMed]
- Hardy, F.; Vriend, G.; van der Vinne, B.; Frigerio, F.; Grandi, G.; Venema, G.; Ejjsink, V.G.H. The effect of engineering surface loops on the thermal stability of Bacillus subtilis neutral protease. Protein Eng. 1994, 7, 425–430. [Google Scholar] [CrossRef]
- Toth, E.A.; Worby, C.; Dixon, J.E.; Goedken, E.R.; Marqusee, S.; Yeates, T.O. The crystal structure of adenylosuccinate lyase from Pyrobaculum aerophilum reveals an intracellular protein with three disulfide bonds. J. Mol. Biol. 2000, 301, 433–450. [Google Scholar] [CrossRef]
- Matsumura, M.; Signor, G.; Matthews, B.W. Substantial increase of protein stability by multiple disulphide bonds. Nature 1989, 342, 291–293. [Google Scholar] [CrossRef]
- Reid, K.S.C.; Lindley, P.F.; Thornton, J.M. Sulphur-aromatic interactions in proteins. FEBS J. 1985, 190, 209–213. [Google Scholar] [CrossRef]
- Ringer, A.L.; Senenko, A.; Sherrill, C.D. Models of S/pi interactions in protein structures: Comparison of the H2S benzene complex with PDB data. Protein Sci. 2007, 16, 2216–2223. [Google Scholar] [CrossRef]
- Matthews, B.W.; Nicholson, H.; Becktel, W.J. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc. Natl. Acad. Sci. USA 1987, 84, 6663–6667. [Google Scholar] [CrossRef]
- Davail, S.; Feller, G.; Narinx, E.; Gerday, C. Cold adaptation of proteins. Purification, characterization, and sequence of the heat-labile subtilisin from the antarctic psychrophile Bacillus TA41. J. Biol. Chem. 1994, 269, 17448–17453. [Google Scholar]
- Rutkiewicz, M.; Bujacz, A.; Wanarska, M.; Wierzbicka-Wos, A.; Cieslinski, H. Active Site Architecture and Reaction Mechanism Determination of Cold Adapted beta-d-galactosidase from Arthrobacter sp. 32cB. Int. J. Mol. Sci. 2019, 20, 4301. [Google Scholar] [CrossRef]
- Rutkiewicz, M.; Bujacz, A.; Bujacz, G. Structural features of cold-adapted dimeric GH2 beta-D-galactosidase from Arthrobacter sp. 32cB. Bba-Proteins Proteom. 2019, 1867, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Tsujibo, H.; Endo, H.; Minoura, K.; Miyamoto, K.; Inamori, Y. Cloning and sequence analysis of the gene encoding a thermostable chitinase from Streptomyces thermoviolaceus OPC-520. Gene 1993, 134, 113–117. [Google Scholar] [PubMed]
- Christodoulou, E.; Duffner, F.; Vorgias, C.E. Overexpression, purification, and characterization of a thermostable chitinase (Chi40) from Streptomyces thermoviolaceus OPC-520. Protein Expr. Purif. 2001, 23, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.P.; Lin, H.C.; Wang, S.S.; Callaway, J.; Wilcox, G. Characterization of the Erwinia carotovora pelB gene and its product pectate lyase. J. Bacteriol. 1987, 169, 4379–4383. [Google Scholar] [CrossRef]
- Poole, K.; Hancock, R.E. Phosphate transport in Pseudomonas aeruginosa. Involvement of a periplasmic phosphate-binding protein. Eur. J. Biochem. 1984, 144, 607–612. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Kurowski, M.A.; Bujnicki, J.M. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 2003, 31, 3305–3307. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Isupov, M.N.; Murshudov, G.N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 122–133. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name and Origin | Temperature Regimen | PDB Code | No. of Salt Bridges | StChi40 Residues Discussed in the Text and the Residues that Substitute Them in the Related Proteins | |||
|---|---|---|---|---|---|---|---|
| StChi40 from Streptomyces thermoviolaceus | thermophilic | 4w5u | 5 | His167 | Glu214 | Pro155 | Pro264 |
| Chitinase from fungus Rhizomucor miehei | thermophilic | 5xwq | 2 | Lys170 | Thr218 | Phe158 | Ala268 |
| Chitinase A1 from Bacillus circulans | mesophilic | 1itx | 12 | Arg192 | Tyr246 | Arg180 | His291 |
| Chitinase CrChi1 from Clonostachys rosea | mesophilic | 3g6l | 8 | Lys162 | Phe209 | Arg150 | His254 |
| ChiA from Serratia marcescens | mesophilic | 1ctn | 13 | Gln302 | Tyr357 | Arg290 | His403 |
| Chitinase B from fungus Aspergillus niger | mesophilic | 6igy | 10 | Ala137 | Phe184 | Arg125 | His229 |
| Chitinase-h from insect Ostrinia furnacalis | mesophilic | 5gpr | 13 | Gln295 | Tyr350 | Arg283 | His396 |
| ChiA74 from Bacillus thuringiensis | mesophilic | 6bt9 | 10 | Arg199 | Tyr253 | Arg187 | His298 |
| Chitinase from Vibrio harveyi | mesophilic | 3b8s | 9 | Lys302 | Tyr358 | Arg290 | His403 |
| Chitinase B from Arthrobacter TAD20 | psychrophilic | 1kfw | 13 | Lys165 | Tyr236 | Arg152 | His284 |
| Space group | P6122 |
| Unit cell parameters | |
| a = b (Å) | 183.2 |
| c (Å) | 130.8 |
| Beamline | MAX-lab I911-3 |
| Wavelength (Å) | 1.0000 |
| Resolution (Å) | 49–2.77 (2.94–2.77) * |
| Rmerge # | 0.15 (1.01) |
| Completeness (%) | 99.4 (98.1) |
| Observed reflections | 333,347 |
| Unique reflections | 33,240 |
| <I/σ(I)> | 13.5 (1.9) |
| Multiplicity | 10.0 |
| R §/Rfree $ | 0.15/0.20 |
| Protein atoms | 5572 |
| Ligand atoms | 14 |
| Water molecules | 74 |
| Average B factor (Å2) | 62 |
| Error in the Luzzati plot (Å) | 0.15 |
| R.m.s. deviation from ideal | |
| bond lengths (Å) | 0.017 |
| bond angles (°) | 1.63 |
| Ramachandran plot (%) | |
| favored | 96 |
| allowed | 4 |
| outliers | 0 |
| PDB code | 4w5u |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malecki, P.H.; Bejger, M.; Rypniewski, W.; Vorgias, C.E. The Crystal Structure of a Streptomyces thermoviolaceus Thermophilic Chitinase Known for Its Refolding Efficiency. Int. J. Mol. Sci. 2020, 21, 2892. https://doi.org/10.3390/ijms21082892
Malecki PH, Bejger M, Rypniewski W, Vorgias CE. The Crystal Structure of a Streptomyces thermoviolaceus Thermophilic Chitinase Known for Its Refolding Efficiency. International Journal of Molecular Sciences. 2020; 21(8):2892. https://doi.org/10.3390/ijms21082892
Chicago/Turabian StyleMalecki, Piotr H., Magdalena Bejger, Wojciech Rypniewski, and Constantinos E. Vorgias. 2020. "The Crystal Structure of a Streptomyces thermoviolaceus Thermophilic Chitinase Known for Its Refolding Efficiency" International Journal of Molecular Sciences 21, no. 8: 2892. https://doi.org/10.3390/ijms21082892
APA StyleMalecki, P. H., Bejger, M., Rypniewski, W., & Vorgias, C. E. (2020). The Crystal Structure of a Streptomyces thermoviolaceus Thermophilic Chitinase Known for Its Refolding Efficiency. International Journal of Molecular Sciences, 21(8), 2892. https://doi.org/10.3390/ijms21082892