Biomarkers for Lysosomal Storage Disorders with an Emphasis on Mass Spectrometry

Abstract
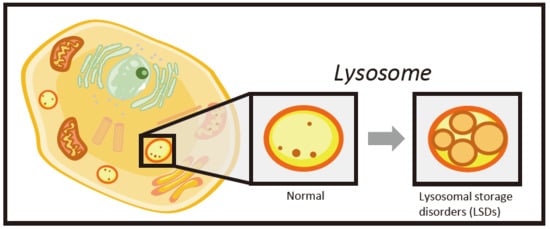
1. Introduction
2. LysoGb3
3. N-palmitoyl-O-phosphocholineserine (PPCS)/LysoSM-509
4. Heparan Sulfate
5. Oligosaccharides
6. Enzyme Activity
7. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50, v4–v12. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-Sulfatase with Anti-human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019, 27, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Thurberg, B.L.; Chiang, S.C.; Zhang, X.K.; Keutzer, J.; Huang, A.-C.; Wu, M.-H.; Huang, P.-H.; Chen, Y.T.; et al. Pompe Disease in Infants: Improving the Prognosis by Newborn Screening and Early Treatment. Pediatrics 2009, 124, e1116–e1125. [Google Scholar] [CrossRef]
- Liao, H.C.; Chiang, C.C.; Niu, D.M.; Wang, C.H.; Kao, S.M.; Tsai, F.J.; Huang, Y.H.; Liu, H.C.; Huang, C.K.; Yang, C.F.; et al. Detecting multiple lysosomal storage diseases by tandem mass spectrometry--a national newborn screening program in Taiwan. Clin. Chim. Acta 2014, 431, 80–86. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working Group; Atkinson, A.J., Jr.; Colburn, W.A.; DeGruttola, V.G.; DeMets, D.L.; Downing, G.J.; Hoth, D.F.; Oates, J.A.; Peck, C.C.; Spilker, B.A.; et al. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Desnick, R.J.; Brady, R.; Barranger, J.; Collins, A.J.; Germain, D.P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W.R. Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann. Intern. Med. 2003, 138, 338–346. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Vedder, A.C.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Boutin, M.; Gagnon, R.; Lavoie, P.; Auray-Blais, C. LC-MS/MS analysis of plasma lyso-Gb3 in Fabry disease. Clin. Chim. Acta 2012, 414, 273–280. [Google Scholar] [CrossRef]
- Lukas, J.; Scalia, S.; Eichler, S.; Pockrandt, A.M.; Dehn, N.; Cozma, C.; Giese, A.K.; Rolfs, A. Functional and Clinical Consequences of Novel α-Galactosidase A Mutations in Fabry Disease. Hum. Mutat. 2016, 37, 43–51. [Google Scholar] [CrossRef]
- Nowak, A.; Mechtler, T.P.; Desnick, R.J.; Kasper, D.C. Plasma LysoGb3: A useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol. Genet. Metab. 2017, 120, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Smid, B.E.; van der Tol, L.; Biegstraaten, M.; Linthorst, G.E.; Hollak, C.E.; Poorthuis, B.J. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J. Med. Genet. 2015, 52, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Auray-Blais, C.; Boutin, M. Novel gb(3) isoforms detected in urine of fabry disease patients: A metabolomic study. Curr. Med. Chem. 2012, 19, 3241–3252. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R.; Okuyama, T.; Ohira, M. Biosynthesis of long chain base in sphingolipids in animals, plants and fungi. Future Sci. OA 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Manwaring, V.; Boutin, M.; Auray-Blais, C. A metabolomic study to identify new globotriaosylceramide-related biomarkers in the plasma of Fabry disease patients. Anal. Chem. 2013, 85, 9039–9048. [Google Scholar] [CrossRef]
- Sueoka, H.; Aoki, M.; Tsukimura, T.; Togawa, T.; Sakuraba, H. Distributions of Globotriaosylceramide Isoforms, and Globotriaosylsphingosine and Its Analogues in an α-Galactosidase A Knockout Mouse, a Model of Fabry Disease. PLoS ONE 2015, 10, e0144958. [Google Scholar] [CrossRef]
- Hong, X.; Gelb, M.H. One-step synthesis of carbon-13-labeled globotriaosylsphingosine (lyso-Gb3), an internal standard for biomarker analysis of Fabry disease. Mol. Genet. Metab. 2018, 125, 292–294. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef]
- Alvelius, G.; Hjalmarson, O.; Griffiths, W.J.; Björkhem, I.; Sjövall, J. Identification of unusual 7-oxygenated bile acid sulfates in a patient with Niemann-Pick disease, type C. J. Lipid Res. 2001, 42, 1571–1577. [Google Scholar]
- Maekawa, M.; Jinnoh, I.; Narita, A.; Iida, T.; Saigusa, D.; Iwahori, A.; Nittono, H.; Okuyama, T.; Eto, Y.; Clayton, P.T.; et al. Investigation of diagnostic performance of five urinary cholesterol metabolites for Niemann-Pick disease type C. J. Lipid Res. 2019, 60, 2074–2081. [Google Scholar] [CrossRef]
- Mashima, R.; Maekawa, M. Lipid biomarkers for the peroxisomal and lysosomal disorders: Their formation, metabolism and measurement. Biomark. Med. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Mazzacuva, F.; Mills, P.; Mills, K.; Camuzeaux, S.; Gissen, P.; Nicoli, E.R.; Wassif, C.; Porter, F.D.; Maekawa, M.; Mano, N.; et al. Identification of novel bile acids as biomarkers for the early diagnosis of Niemann-Pick C disease. FEBS Lett. 2016, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sidhu, R.; Mydock-McGrane, L.; Hsu, F.F.; Covey, D.F.; Scherrer, D.E.; Earley, B.; Gale, S.E.; Farhat, N.Y.; Dietzen, D.J.; et al. Development of a bile acid-based newborn screen for Niemann-Pick disease type C. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sidhu, R.; Orsini, J.J.; Farhat, N.Y.; Porter, F.D.; Berry-Kravis, E.; Schaffer, J.E.; Ory, D.S. Diagnosis of niemann-pick C1 by measurement of bile acid biomarkers in archived newborn dried blood spots. Mol. Genet. Metab. 2019, 126, 183–187. [Google Scholar] [CrossRef]
- Giese, A.K.; Mascher, H.; Grittner, U.; Eichler, S.; Kramp, G.; Lukas, J.; Eisa, N.A.; Cortina-Borja, M.; Porter, F.D.; Platt, F.M.; et al. A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease. Orphanet J. Rare Dis. 2015, 10, 78. [Google Scholar] [CrossRef]
- Kuchar, L.; Sikora, J.; Gulinello, M.E.; Poupetova, H.; Lugowska, A.; Malinova, V.; Jahnova, H.; Asfaw, B.; Ledvinova, J. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal. Biochem. 2017, 525, 73–77. [Google Scholar] [CrossRef]
- Polo, G.; Burlina, A.P.; Kolamunnage, T.B.; Zampieri, M.; Dionisi-Vici, C.; Strisciuglio, P.; Zaninotto, M.; Plebani, M.; Burlina, A.B. Diagnosis of sphingolipidoses: A new simultaneous measurement of lysosphingolipids by LC-MS/MS. Clin. Chem. Lab. Med. 2017, 55, 403–414. [Google Scholar] [CrossRef]
- Sidhu, R.; Mondjinou, Y.; Qian, M.; Song, H.; Kumar, A.B.; Hong, X.; Hsu, F.F.; Dietzen, D.J.; Yanjanin, N.M.; Berry-Kravis, E.; et al. N-acyl-O-phosphocholineserines: Structures of a novel class of lipids that are biomarkers for Niemann-Pick C1 disease. J. Lipid Res. 2019, 60, 1410–1424. [Google Scholar] [CrossRef]
- Maekawa, M.; Jinnoh, I.; Matsumoto, Y.; Narita, A.; Mashima, R.; Takahashi, H.; Iwahori, A.; Saigusa, D.; Fujii, K.; Higaki, K.; et al. Structural Determination of Lysosphingomyelin-509 and Discovery of Novel Class Lipids from Patients with Niemann-Pick Disease Type C. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Stone, J.E. Urine analysis in the diagnosis of mucopolysaccharide disorders. Ann. Clin. Biochem. 1998, 35, 207–225. [Google Scholar] [CrossRef]
- Saville, J.T.; McDermott, B.K.; Fletcher, J.M.; Fuller, M. Disease and subtype specific signatures enable precise diagnosis of the mucopolysaccharidoses. Genet. Med. 2019, 21, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Young, S.P.; Auray-Blais, C.; Orchard, P.J.; Tolar, J.; Millington, D.S. Analysis of Glycosaminoglycans in Cerebrospinal Fluid from Patients with Mucopolysaccharidoses by Isotope-Dilution Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry. Clin. Chem. 2011, 57, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Kida, S.; Kinoshita, M.; Morimoto, H.; Shibasaki, T.; Tachibana, K.; Yamamoto, R. Evaluation of cerebrospinal fluid heparan sulfate as a biomarker of neuropathology in a murine model of mucopolysaccharidosis type II using high-sensitivity LC/MS/MS. Mol. Genet. Metab. 2018, 125, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Auray-Blais, C.; Lavoie, P.; Tomatsu, S.; Valayannopoulos, V.; Mitchell, J.J.; Raiman, J.; Beaudoin, M.; Maranda, B.; Clarke, J.T.R. UPLC-MS/MS detection of disaccharides derived from glycosaminoglycans as biomarkers of mucopolysaccharidoses. Anal. Chim. Acta 2016, 936, 139–148. [Google Scholar] [CrossRef]
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Tsai, C.C.; Liu, H.L.; Lin, S.P. A modified liquid chromatography/tandem mass spectrometry method for predominant disaccharide units of urinary glycosaminoglycans in patients with mucopolysaccharidoses. Orphanet J. Rare Dis. 2014, 9. [Google Scholar] [CrossRef]
- He, Q.Q.; Trim, P.J.; Snel, M.F.; Hopwood, J.J.; Ferro, V. Synthesis and mass spectrometric analysis of disaccharides from methanolysis of heparan sulfate. Org. Biomol. Chem. 2018, 16, 8791–8803. [Google Scholar] [CrossRef]
- Trim, P.J.; Lau, A.A.; Hopwood, J.J.; Snel, M.F. A simple method for early age phenotype confirmation using toe tissue from a mouse model of MPS IIIA. Rapid Commun. Mass Spectrom. 2014, 28, 933–938. [Google Scholar] [CrossRef]
- Lawrence, R.; Van Vleet, J.L.; Mangini, L.; Harris, A.; Martin, N.; Clark, W.; Chandriani, S.; LeBowitz, J.H.; Giugliani, R.; Yogalingam, G.; et al. Characterization of glycan substrates accumulating in GM1 Gangliosidosis. Mol. Genet. Metab. Reports 2019, 21. [Google Scholar] [CrossRef]
- Huang, R.; Cathey, S.; Pollard, L.; Wood, T. UPLC-MS/MS Analysis of Urinary Free Oligosaccharides for Lysosomal Storage Diseases: Diagnosis and Potential Treatment Monitoring. Clin. Chem. 2018, 64, 1772–1779. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shimojima, K.; Matsufuji, M.; Mashima, R.; Sakai, E.; Okuyama, T. Aspartylglucosaminuria caused by a novel homozygous mutation in the AGA gene was identified by an exome-first approach in a patient from Japan. Brain Dev. 2017, 39. [Google Scholar] [CrossRef]
- Xia, B.; Asif, G.; Arthur, L.; Pervaiz, M.A.; Li, X.; Liu, R.; Cummings, R.D.; He, M. Oligosaccharide analysis in urine by maldi-tof mass spectrometry for the diagnosis of lysosomal storage diseases. Clin. Chem. 2013, 59, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Bonesso, L.; Piraud, M.; Caruba, C.; Van Obberghen, E.; Mengual, R.; Hinault, C. Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry. Orphanet J. Rare Dis. 2014, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Lowden, J.A.; O’brien, J.S. Sialidosis: A Review of Human Neuraminidase Deficiency. Am. J. Hum. Genet. 1979, 31, 1. [Google Scholar] [PubMed]
- Piraud, M.; Pettazzoni, M.; Menegaut, L.; Caillaud, C.; Nadjar, Y.; Vianey-Saban, C.; Froissart, R. Development of a new tandem mass spectrometry method for urine and amniotic fluid screening of oligosaccharidoses. Rapid Commun. Mass Spectrom. 2017, 31, 951–963. [Google Scholar] [CrossRef]
- Wang, Q.; Yin, B.; Chung, C.Y.; Betenbaugh, M.J. Glycoengineering of CHO cells to improve product quality. In Heterologous Protein Production in CHO Cells; Humana Press Inc.: New York, NY, USA, 2017; pp. 25–44. [Google Scholar] [CrossRef]
- de Geest, N.; Bonten, E.; Mann, L.; de Sousa-Hitzler, J.; Hahn, C.; d’Azzo, A. Systemic and neurologic abnormalities distinguish the lysosomal disorders sialidosis and galactosialidosis in mice. Hum. Mol. Genet. 2002, 11, 1455–1464. [Google Scholar] [CrossRef]
- Young, S.P.; Stevens, R.D.; An, Y.; Chen, Y.-T.; Millington, D.S. Analysis of a glucose tetrasaccharide elevated in Pompe disease by stable isotope dilution-electrospray ionization tandem mass spectrometry. Anal. Biochem. 2003, 316, 175–180. [Google Scholar] [CrossRef]
- Chien, Y.H.; Goldstein, J.L.; Hwu, W.L.; Smith, P.B.; Lee, N.C.; Chiang, S.C.; Tolun, A.A.; Zhang, H.; Vaisnins, A.E.; Kishnani, P.S.; et al. Baseline urinary glucose tetrasaccharide concentrations in patients with infantile- and late-onset pompe disease identified by newborn screening. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2015; pp. 67–73. [Google Scholar] [CrossRef]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Diagnosis of α-L-iduronidase deficiency in dried blood spots on filter paper: The possibility of newborn diagnosis. Clin. Chem. 2001, 47, 780–781. [Google Scholar] [CrossRef]
- Gelb, M.H.; Lukacs, Z.; Ranieri, E.; Schielen, P.C.J.I. Newborn screening for lysosomal storage disorders: Methodologies for measurement of enzymatic activities in dried blood spots. Int. J. Neonatal Screen. 2019, 5. [Google Scholar] [CrossRef]
- Scott, C.R.; Elliott, S.; Hong, X.; Huang, J.Y.; Kumar, A.B.; Yi, F.; Pendem, N.; Chennamaneni, N.K.; Gelb, M.H. Newborn Screening for Mucopolysaccharidoses: Results of a Pilot Study with 100 000 Dried Blood Spots. J. Pediatr. 2019. [Google Scholar] [CrossRef]
- Gelb, M.H.; Scott, C.R.; Turecek, F. Newborn screening for lysosomal storage diseases. Clin. Chem. 2015, 61, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Masi, S.; Chennamaneni, N.; Turecek, F.; Scott, C.R.; Gelb, M.H. Specific Substrate for the Assay of Lysosomal Acid Lipase. Clin. Chem. 2018, 64, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Hong, X.; Kumar, A.B.; Zong, C.; Boons, G.J.; Scott, C.R.; Turecek, F.; Robinson, B.H.; Gelb, M.H. Detection of mucopolysaccharidosis III-A (Sanfilippo Syndrome-A) in dried blood spots (DBS) by tandem mass spectrometry. Mol. Genet. Metab. 2018, 125, 59–63. [Google Scholar] [CrossRef]
- Kumar, A.B.; Hong, X.; Yi, F.; Wood, T.; Gelb, M.H. Tandem mass spectrometry-based multiplex assays for α-mannosidosis and fucosidosis. Mol. Genet. Metab. 2019, 127, 207–211. [Google Scholar] [CrossRef]
- Chan, M.J.; Liao, H.C.; Gelb, M.H.; Chuang, C.K.; Liu, M.Y.; Chen, H.J.; Kao, S.M.; Lin, H.Y.; Huang, Y.H.; Chennamaneni, N.K.; et al. Taiwan National Newborn Screening Program by Tandem Mass Spectrometry for mucopolysaccharidoses Types I, II, and VI. J. Pediatr. 2019, 205, 176–182. [Google Scholar] [CrossRef]
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Huang, Y.H.; Chan, M.J.; Liao, H.C.; Lo, Y.T.; Wang, L.Y.; Tu, R.Y.; Chen, T.L.; et al. Status of newborn screening and follow up investigations for Mucopolysaccharidoses i and II in Taiwan. Orphanet J. Rare Dis. 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Raskovalova, T.; Deegan, P.B.; Mistry, P.K.; Pavlova, E.; Yang, R.; Zimran, A.; Berger, J.; Bourgne, C.; Pereira, B.; Berger, M.G.; et al. Accuracy of chitotriosidase activity and CCL18 concentration in assessing type I Gaucher disease severity. A systematic review with meta-analysis of individual participant data. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPS III Disease-Subtype | OMIM | Enzyme | Abbreviation | Biomarker | Number of Sulfate | Other MPS |
|---|---|---|---|---|---|---|
| MPS IIIA | 252900 | N-sulfoglucosamine sulfohydrolase | SGSH | HN-UA | 1 | Not reported |
| MPS IIIB | 252920 | N-acetyl-α-glucosaminidase | NAGLU | (HNAc-UA)2 | 1 | Not reported |
| MPS IIIC | 252930 | Heparan-α-glucosaminide N-acetyltransferase | HGSNAT | (HN-UA)2-NAc | 2 | Not reported |
| MPS IIID | 252940 | Glucosamine (N-acetyl)-6-sulfatase | GNS | (HNAc-UA)2 | 2 | IVA, VI |
| HNAc | 1 | IVA, VI |
| Scott CR et al. (2019) (n = 105,214) [51] | Chuang C-K et al. (2018) (n = 153,032) [57] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| cDNA | Protein | Interpretation | n | Frequency | cDNA | Protein | GAG | n | Frequency |
| (1/million) | (1/million) | ||||||||
| c.851C>T | p.P284L | Benign | 7 | 66.5 | c.103+34_56dup | Intronic mutation | Negative | 49 | 320.2 |
| c.1090C>T | p.P364S | Benign | 2 | 19.0 | c.1499C>T | p.T500I | Negative | 10 | 65.3 |
| c.1499C>T | p.T500I | Benign | 1 | 9.5 | c.301C>T | p.R101C | Negative | 7 | 45.7 |
| c.684A>G/ c.851C>T | p.P228P+ p.P284L | Benign | 1 | 9.5 | c.1478G>A | p.R493H | Negative | 4 | 26.1 |
| c.1409C>T | p.S470L | At risk | 1 | 9.5 | c.103+34_56dup/ c.851C>T | p.P284L | Negative | 1 | 6.5 |
| c.159T>C | p.C53W | At risk | 1 | 9.5 | c.103+34_56dup/ c.851C>T/ c.1180+184T>C | p.P284L | Negative | 1 | 6.5 |
| c.817C>T | p.R273W | Affected | 1 | 9.5 | c.589C>T | p.P197S | Negative | 1 | 6.5 |
| c.851C>T | p.P284L | Negative | 1 | 6.5 | |||||
| c.890G>A | p.R297H | Negative | 1 | 6.5 | |||||
| c.1025A>G | p.H342R | Positive | 1 | 6.5 | |||||
| c.311A>T | p.D104V | Positive | 1 | 6.5 | |||||
| c.817C>T | p.R273W | Positive | 1 | 6.5 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mashima, R.; Okuyama, T.; Ohira, M. Biomarkers for Lysosomal Storage Disorders with an Emphasis on Mass Spectrometry. Int. J. Mol. Sci. 2020, 21, 2704. https://doi.org/10.3390/ijms21082704
Mashima R, Okuyama T, Ohira M. Biomarkers for Lysosomal Storage Disorders with an Emphasis on Mass Spectrometry. International Journal of Molecular Sciences. 2020; 21(8):2704. https://doi.org/10.3390/ijms21082704
Chicago/Turabian StyleMashima, Ryuichi, Torayuki Okuyama, and Mari Ohira. 2020. "Biomarkers for Lysosomal Storage Disorders with an Emphasis on Mass Spectrometry" International Journal of Molecular Sciences 21, no. 8: 2704. https://doi.org/10.3390/ijms21082704
APA StyleMashima, R., Okuyama, T., & Ohira, M. (2020). Biomarkers for Lysosomal Storage Disorders with an Emphasis on Mass Spectrometry. International Journal of Molecular Sciences, 21(8), 2704. https://doi.org/10.3390/ijms21082704