Moss-Derived Human Recombinant GAA Provides an Optimized Enzyme Uptake in Differentiated Human Muscle Cells of Pompe Disease


Abstract
1. Introduction
2. Results
2.1. Production, Purification, and Characterization of moss-GAA Variants
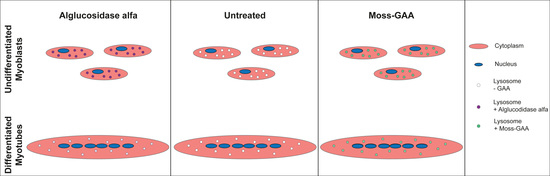
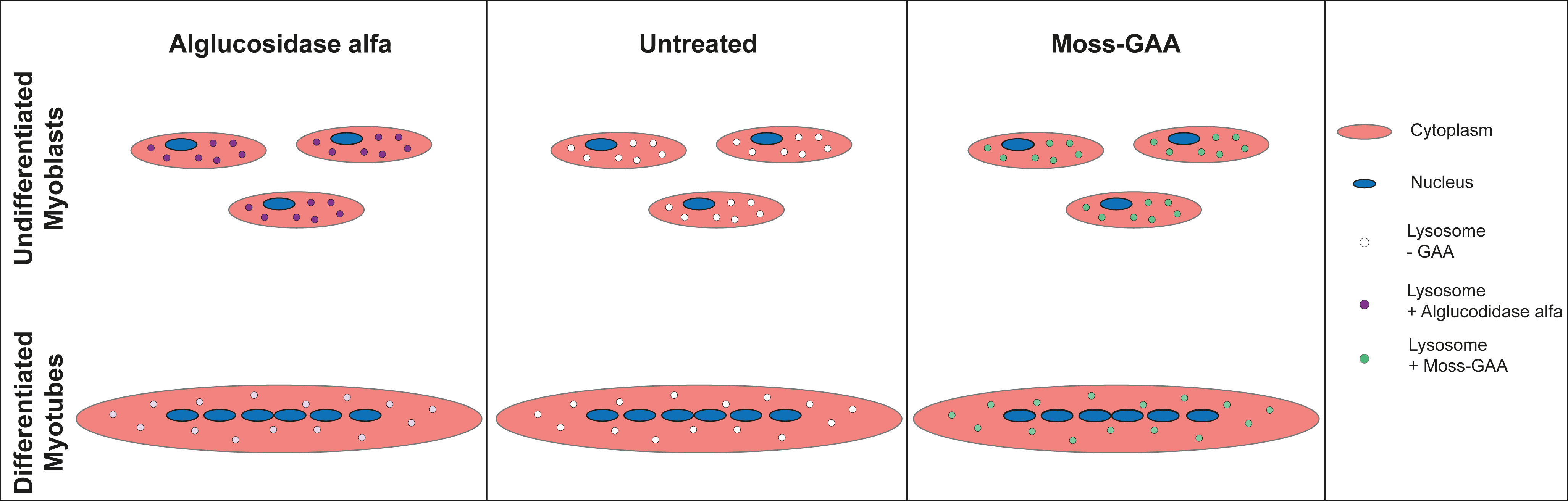
2.2. Uptake and Activity of rhGAA in Mouse Myoblasts
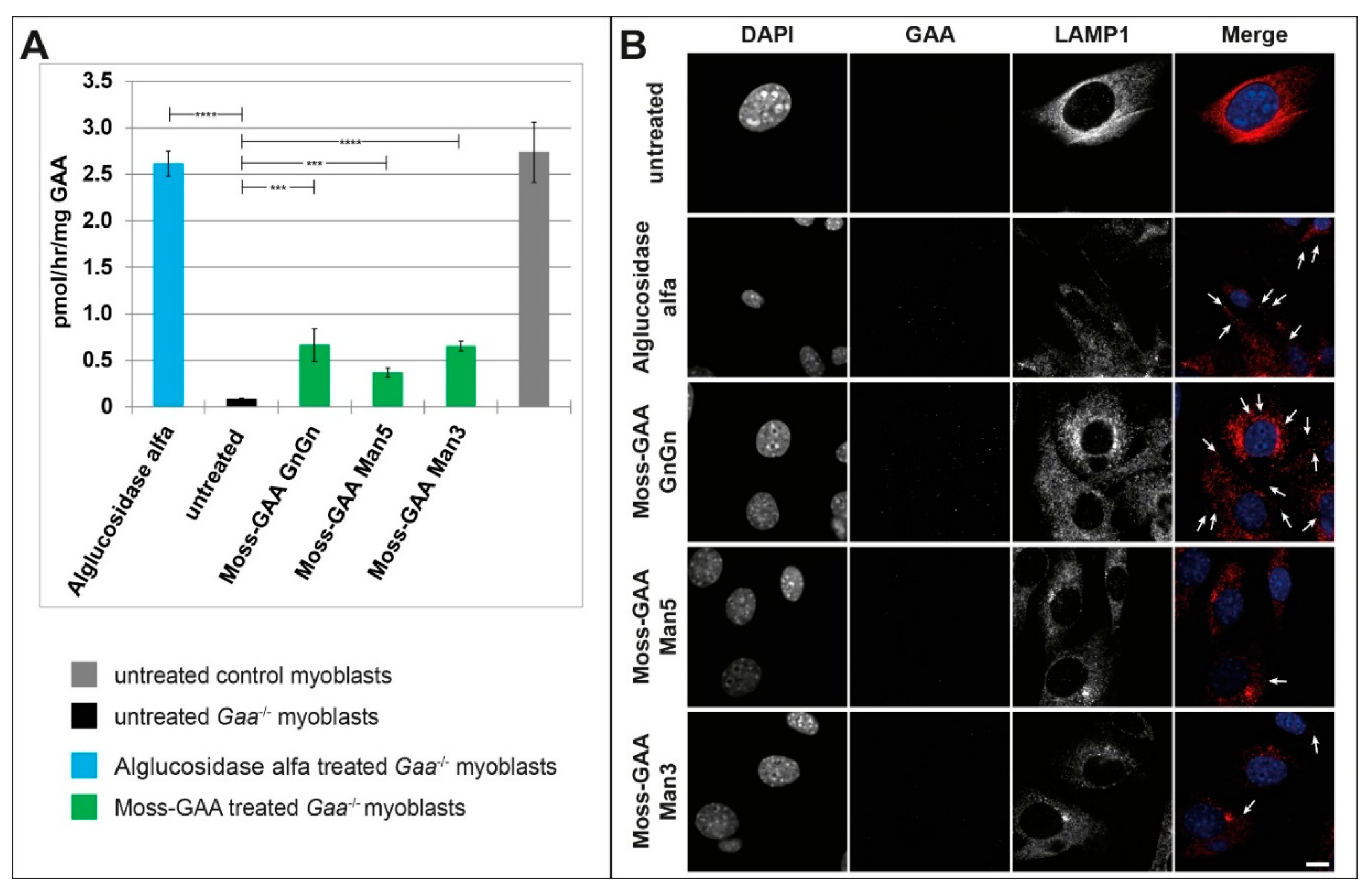
2.3. Uptake and Activity of rhGAA in Mouse Myotubes
2.4. Uptake and Activity of rhGAA in Human Myoblasts
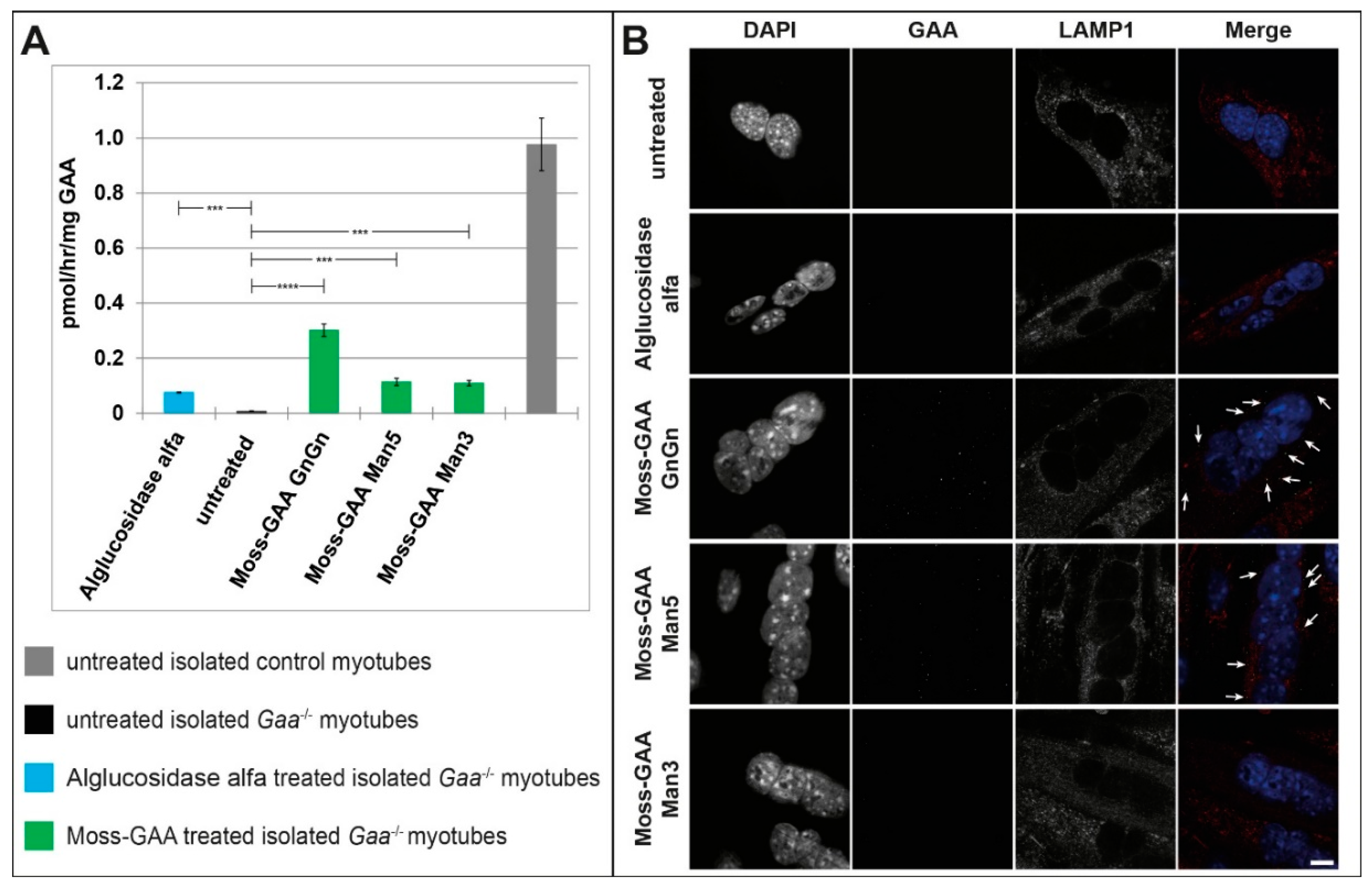
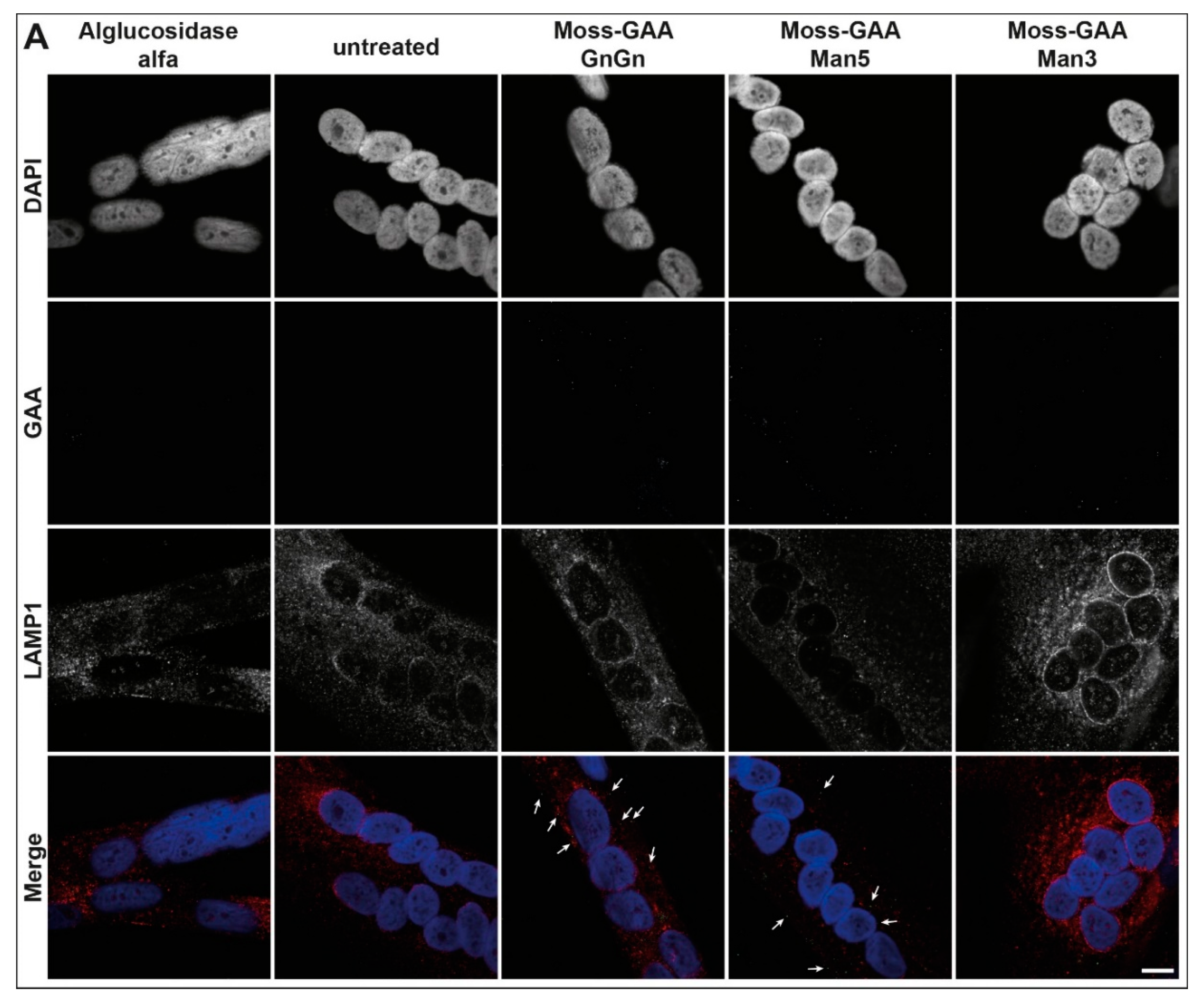
2.5. Uptake and Activity of rhGAA in Human Myotubes
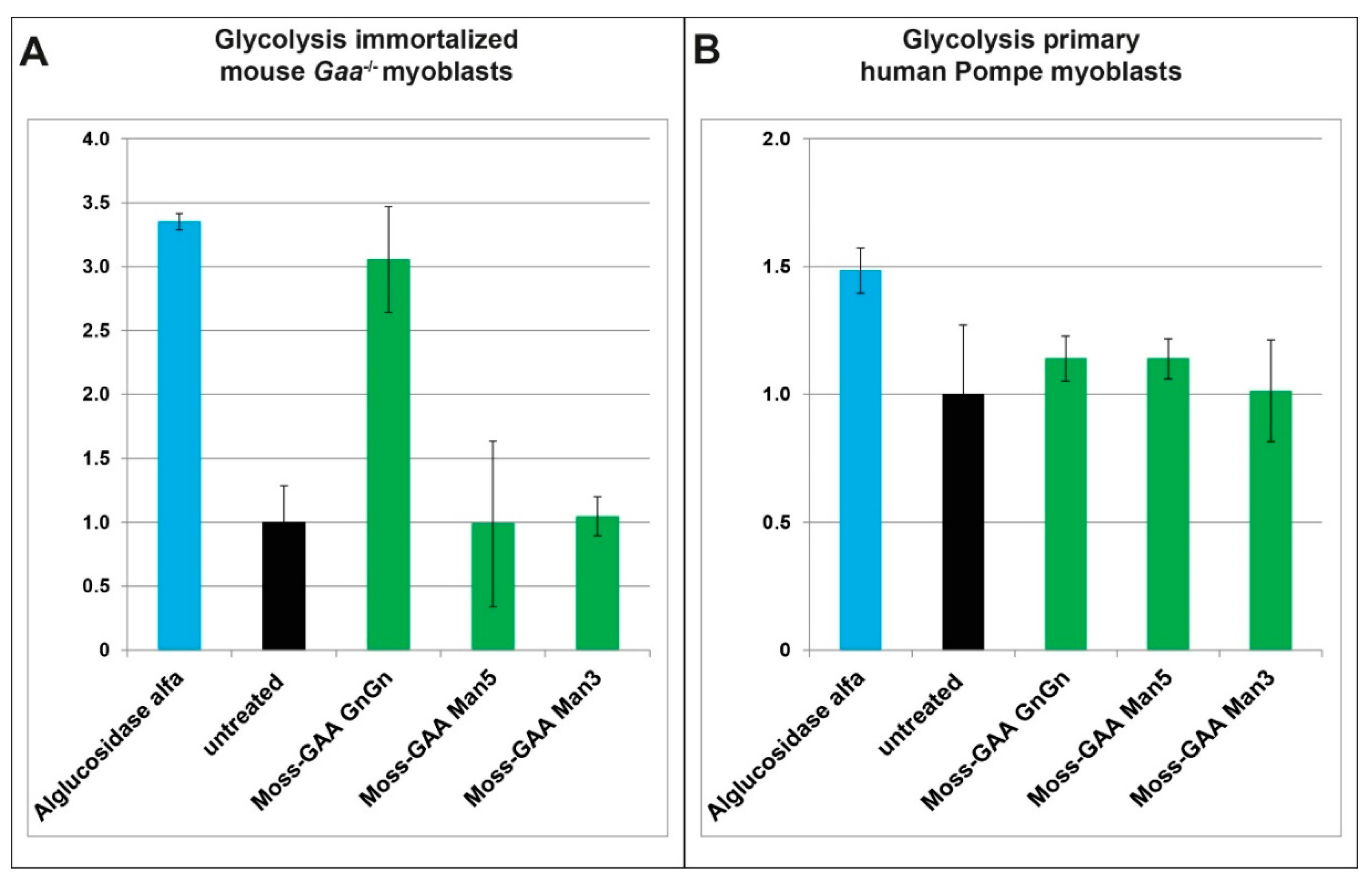
2.6. Metabolic Measurements
3. Discussion
4. Materials and Methods
4.1. Culture of Immortalized Mouse Cells
4.2. Patients and Controls
4.3. Culture of Primary Skeletal Muscle Cells
4.4. Enzymes
4.5. Moss-GAA Expression Strain Construction
4.6. Production and Purification of Moss-GAA
4.7. Preparation of Moss-GAA Man3 Variant
4.8. Analysis of Moss-GAA Variants
SDS-Page and Western Blot
4.9. GAA Uptake Assay
4.10. Preparation of Cell Lysates
4.11. GAA Activity Assay
4.12. Real Time Metabolic Measurements
4.13. PAS Staining
4.14. Immunohistochemistry
4.15. Microscopy and Image Analysis
4.16. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martiniuk, F.; Bodkin, M.; Tzall, S.; Hirschhorn, R. Identification of the base-pair substitution responsible for a human acid alpha glucosidase allele with lower “affinity” for glycogen (GAA 2) and transient gene expression in deficient cells. Am. J. Hum. Genet. 1990, 47, 440–445. [Google Scholar] [PubMed]
- Lam, C.W.; Yuen, Y.P.; Chan, K.Y.; Tong, S.F.; Lai, C.K.; Chow, T.C.; Lee, K.C.; Chan, Y.W.; Martiniuk, F. Juvenile-onset glycogen storage disease type II with novel mutations in acid alpha-glucosidase gene. Neurology 2003, 60, 715–717. [Google Scholar] [CrossRef]
- Matsuishi, T.; Yoshino, M.; Terasawa, K.; Nonaka, I. Childhood acid maltase deficiency. A clinical, biochemical, and morphologic study of three patients. Arch Neurol. 1984, 41, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, J.L.; Yang, H.W.; Wu, J.Y.; Brady, R.O.; Chen, Y.T. High-level production of recombinant human lysosomal acid alpha-glucosidase in Chinese hamster ovary cells which targets to heart muscle and corrects glycogen accumulation in fibroblasts from patients with Pompe disease. Proc. Natl. Acad. Sci. USA 1996, 93, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Kohler, L.; Puertollano, R.; Raben, N. Pompe Disease: From Basic Science to Therapy. Neurother. J. Am. Soc. Exp. Neurother. 2018, 15, 928–942. [Google Scholar] [CrossRef] [PubMed]
- El Cheikh, K.; Basile, I.; Da Silva, A.; Bernon, C.; Cerutti, P.; Salgues, F.; Perez, M.; Maynadier, M.; Gary-Bobo, M.; Caillaud, C.; et al. Design of Potent Mannose 6-Phosphate Analogues for the Functionalization of Lysosomal Enzymes To Improve the Treatment of Pompe Disease. Angew. Chem. 2016, 55, 14774–14777. [Google Scholar] [CrossRef]
- Basile, I.; Da Silva, A.; El Cheikh, K.; Godefroy, A.; Daurat, M.; Harmois, A.; Perez, M.; Caillaud, C.; Charbonne, H.V.; Pau, B.; et al. Efficient therapy for refractory Pompe disease by mannose 6-phosphate analogue grafting on acid alpha-glucosidase. J. Control. Rrelease Off. J. Control. Release Soc. 2018, 269, 15–23. [Google Scholar] [CrossRef]
- Zhu, Y.; Jiang, J.L.; Gumlaw, N.K.; Zhang, J.; Bercury, S.D.; Ziegler, R.J.; Lee, K.; Kudo, M.; Canfield, W.M.; Edmunds, T.; et al. Glycoengineered acid alpha-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 954–963. [Google Scholar] [CrossRef]
- LeBowitz, J.H.; Grubb, J.H.; Maga, J.A.; Schmiel, D.H.; Vogler, C.; Sly, W.S. Glycosylation-independent targeting enhances enzyme delivery to lysosomes and decreases storage in mucopolysaccharidosis type VII mice. Proc. Natl. Acad. Sci. USA 2004, 101, 3083–3088. [Google Scholar] [CrossRef]
- Maga, J.A.; Zhou, J.; Kambampati, R.; Peng, S.; Wang, X.; Bohnsack, R.N.; Thomm, A.; Golata, S.; Tom, P.; Dahms, N.M.; et al. Glycosylation-independent lysosomal targeting of acid alpha-glucosidase enhances muscle glycogen clearance in pompe mice. J. Biol. Chem. 2013, 288, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Downing, W.L.; Galpin, J.D.; Clemens, S.; Lauzon, S.M.; Samuels, A.L.; Pidkowich, M.S.; Clarke, L.A.; Kermode, A.R. Synthesis of enzymatically active human alpha-L-iduronidase in Arabidopsis cgl (complex glycan-deficient) seeds. Plant Biotechnol. J. 2006, 4, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Shaaltiel, Y.; Bartfeld, D.; Hashmueli, S.; Baum, G.; Brill-Almon, E.; Galili, G.; Dym, O.; Boldin-Adamsky, S.A.; Silman, I.; Sussman, J.L.; et al. Production of glucocerebrosidase with terminal mannose glycans for enzyme replacement therapy of Gaucher’s disease using a plant cell system. Plant Biotechnol. J. 2007, 5, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Kizhner, T.; Azulay, Y.; Hainrichson, M.; Tekoah, Y.; Arvatz, G.; Shulman, A.; Ruderfer, I.; Aviezer, D.; Shaaltiel, Y. Characterization of a chemically modified plant cell culture expressed human alpha-Galactosidase-A enzyme for treatment of Fabry disease. Mol. Genet. Metab. 2015, 114, 259–267. [Google Scholar] [CrossRef]
- Shen, J.S.; Busch, A.; Day, T.S.; Meng, X.L.; Yu, C.I.; Dabrowska-Schlepp, P.; Fode, B.; Niederkruger, H.; Forni, S.; Chen, S.; et al. Mannose receptor-mediated delivery of moss-made alpha-galactosidase A efficiently corrects enzyme deficiency in Fabry mice. J. Inherit. Metab. Dis. 2016, 39, 293–303. [Google Scholar] [CrossRef]
- Kermode, A.R.; McNair, G.; Pierce, O. Plant Recombinant Lysosomal Enzymes as Replacement Therapeutics for Lysosomal Storage Diseases. Mol. Pharming 2018, 2018, 181–215. [Google Scholar]
- Jung, J.W.; Kim, N.S.; Jang, S.H.; Shin, Y.J.; Yang, M.S. Production and characterization of recombinant human acid alpha-glucosidase in transgenic rice cell suspension culture. J. Biotechnol. 2016, 226, 44–53. [Google Scholar] [CrossRef]
- Martiniuk, F.; Reggi, S.; Tchou-Wong, K.M.; Rom, W.N.; Busconi, M.; Fogher, C. Production of a functional human acid maltase in tobacco seeds: Biochemical analysis, uptake by human GSDII cells, and in vivo studies in GAA knockout mice. Appl. Biochem. Biotechnol. 2013, 171, 916–926. [Google Scholar] [CrossRef]
- Hennermann, J.B.; Arash-Kaps, L.; Fekete, G.; Schaaf, A.; Busch, A.; Frischmuth, T. Pharmacokinetics, pharmacodynamics, and safety of moss-aGalactosidase A in patients with Fabry disease. J. Inherit. Metab. Dis. 2019, 42, 527–533. [Google Scholar] [CrossRef]
- Koprivova, A.; Stemmer, C.; Altmann, F.; Hoffmann, A.; Kopriva, S.; Gorr, G.; Reski, R.; Decker, E.L. Targeted knockouts of Physcomitrella lacking plant-specific immunogenic N-glycans. Plant Biotechnol. J. 2004, 2, 517–523. [Google Scholar] [CrossRef]
- Meinke, P.; Limmer, S.; Hintze, S.; Schoser, B. Assessing metabolic profiles in human myoblasts from patients with late-onset Pompe disease. Ann. Transl. Med. 2019, 7, 277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Brondyk, W.; Lydon, J.T.; Thurberg, B.L.; Piepenhagen, P.A. Biotherapeutic target or sink: Analysis of the macrophage mannose receptor tissue distribution in murine models of lysosomal storage diseases. J. Inherit. Metab. Dis. 2011, 34, 795–809. [Google Scholar] [CrossRef] [PubMed]
- van der Ploeg, A.T.; Kruijshaar, M.E.; Toscano, A.; Laforet, P.; Angelini, C.; Lachmann, R.H.; Pascual Pascual, S.I.; Roberts, M.; Rosler, K.; Stulnig, T.; et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: A 10-year experience. Eur. J. Neurol. 2017, 24, 768-e31. [Google Scholar] [CrossRef] [PubMed]
- Kuperus, E.; Kruijshaar, M.E.; Wens, S.C.A.; de Vries, J.M.; Favejee, M.M.; van der Meijden, J.C.; Rizopoulos, D.; Brusse, E.; van Doorn, P.A.; van der Ploeg, A.T.; et al. Long-term benefit of enzyme replacement therapy in Pompe disease: A 5-year prospective study. Neurology 2017, 89, 2365–2373. [Google Scholar] [CrossRef]
- van der Meijden, J.C.; Kruijshaar, M.E.; Harlaar, L.; Rizopoulos, D.; van der Beek, N.; van der Ploeg, A.T. Long-term follow-up of 17 patients with childhood Pompe disease treated with enzyme replacement therapy. J. Inherit. Metab. Dis. 2018, 41, 1205–1214. [Google Scholar] [CrossRef]
- Stahl, P.D.; Ezekowitz, R.A. The mannose receptor is a pattern recognition receptor involved in host defense. Curr. Opin. Immunol. 1998, 10, 50–55. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Prata, M.J.; Alves, S. A shortcut to the lysosome: The mannose-6-phosphate-independent pathway. Mol. Genet. Metab. 2012, 107, 257–266. [Google Scholar] [CrossRef]
- Staudt, C.; Puissant, E.; Boonen, M. Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. Int. J. Mol. Sci. 2016, 18, 47. [Google Scholar] [CrossRef]
- Raben, N.; Nagaraju, K.; Lee, E.; Kessler, P.; Byrne, B.; Lee, L.; LaMarca, M.; King, C.; Ward, J.; Sauer, B.; et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 1998, 273, 19086–19092. [Google Scholar] [CrossRef]
- Moreland, R.J.; Jin, X.; Zhang, X.K.; Decker, R.W.; Albee, K.L.; Lee, K.L.; Cauthron, R.D.; Brewer, K.; Edmunds, T.; Canfield, W.M. Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor. J. Biol. Chem. 2005, 280, 6780–6791. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GAA-Mutations | Sex | Age at Biopsy | Muscle Used | |
|---|---|---|---|---|
| Patient-1 | c.-32-13T>G/ c.1396delG | male | 43 | M. biceps brachii |
| Patient-2 | c.-32-13T>G/ c.1942G>A | female | 67 | M. quadriceps femoris |
| Patient-3 | c.-32-13T>G/ c.-32-13T>G | male | 55 | M. vastus lateralis |
| Patient-4 | c.-32-13T>G/ c.1446delC | male | 31 | unknown |
| Control-1 | --- | female | 49 | M. vastus lateralis |
| Control-2 | --- | male | 32 | M. gastrocnemius |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hintze, S.; Limmer, S.; Dabrowska-Schlepp, P.; Berg, B.; Krieghoff, N.; Busch, A.; Schaaf, A.; Meinke, P.; Schoser, B. Moss-Derived Human Recombinant GAA Provides an Optimized Enzyme Uptake in Differentiated Human Muscle Cells of Pompe Disease. Int. J. Mol. Sci. 2020, 21, 2642. https://doi.org/10.3390/ijms21072642
Hintze S, Limmer S, Dabrowska-Schlepp P, Berg B, Krieghoff N, Busch A, Schaaf A, Meinke P, Schoser B. Moss-Derived Human Recombinant GAA Provides an Optimized Enzyme Uptake in Differentiated Human Muscle Cells of Pompe Disease. International Journal of Molecular Sciences. 2020; 21(7):2642. https://doi.org/10.3390/ijms21072642
Chicago/Turabian StyleHintze, Stefan, Sarah Limmer, Paulina Dabrowska-Schlepp, Birgit Berg, Nicola Krieghoff, Andreas Busch, Andreas Schaaf, Peter Meinke, and Benedikt Schoser. 2020. "Moss-Derived Human Recombinant GAA Provides an Optimized Enzyme Uptake in Differentiated Human Muscle Cells of Pompe Disease" International Journal of Molecular Sciences 21, no. 7: 2642. https://doi.org/10.3390/ijms21072642
APA StyleHintze, S., Limmer, S., Dabrowska-Schlepp, P., Berg, B., Krieghoff, N., Busch, A., Schaaf, A., Meinke, P., & Schoser, B. (2020). Moss-Derived Human Recombinant GAA Provides an Optimized Enzyme Uptake in Differentiated Human Muscle Cells of Pompe Disease. International Journal of Molecular Sciences, 21(7), 2642. https://doi.org/10.3390/ijms21072642