Conditional Up-Regulation of SERCA2a Exacerbates RyR2-Dependent Ventricular and Atrial Arrhythmias

 and
and
Abstract
1. Introduction
2. Results
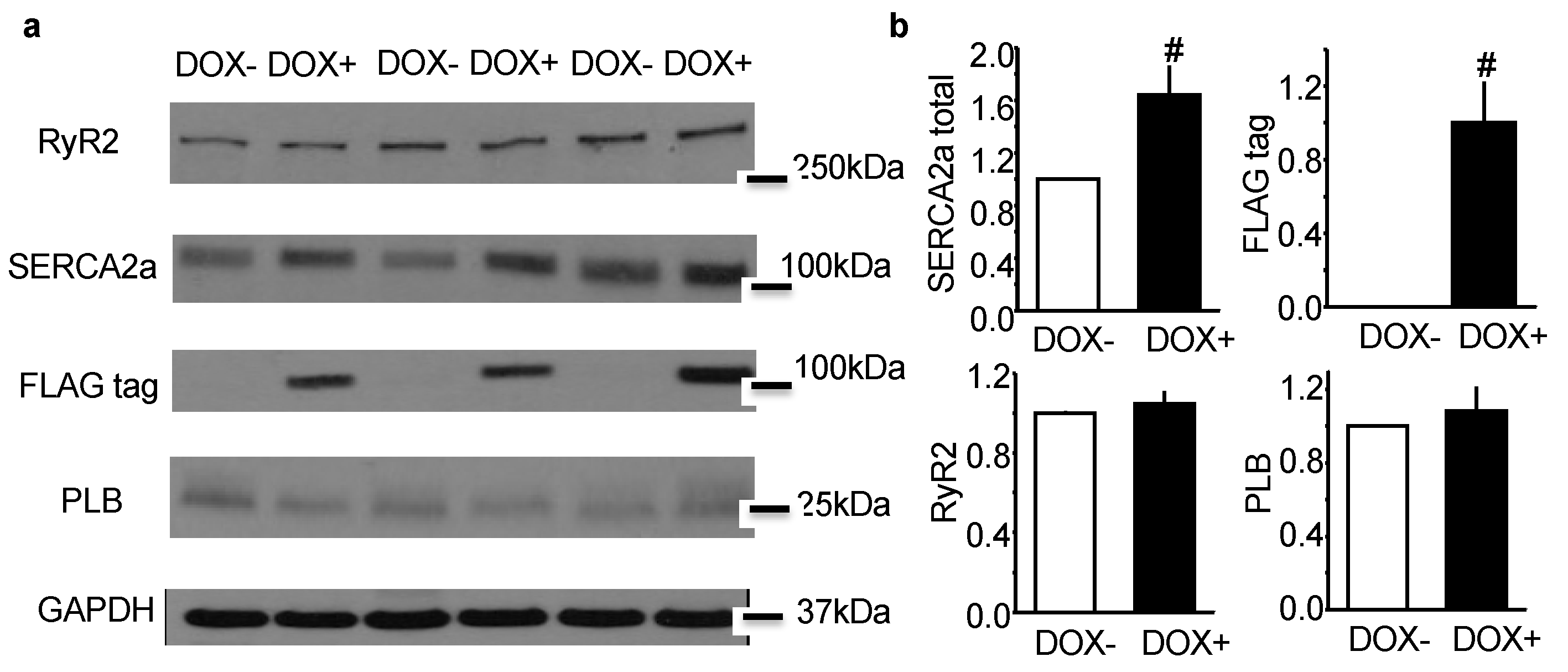
2.1. Acute Upregulation of SERCA2a in the CASQ2 KO Mice Did Not Alter Expression of RyR2 and Phospholamban (PLB)
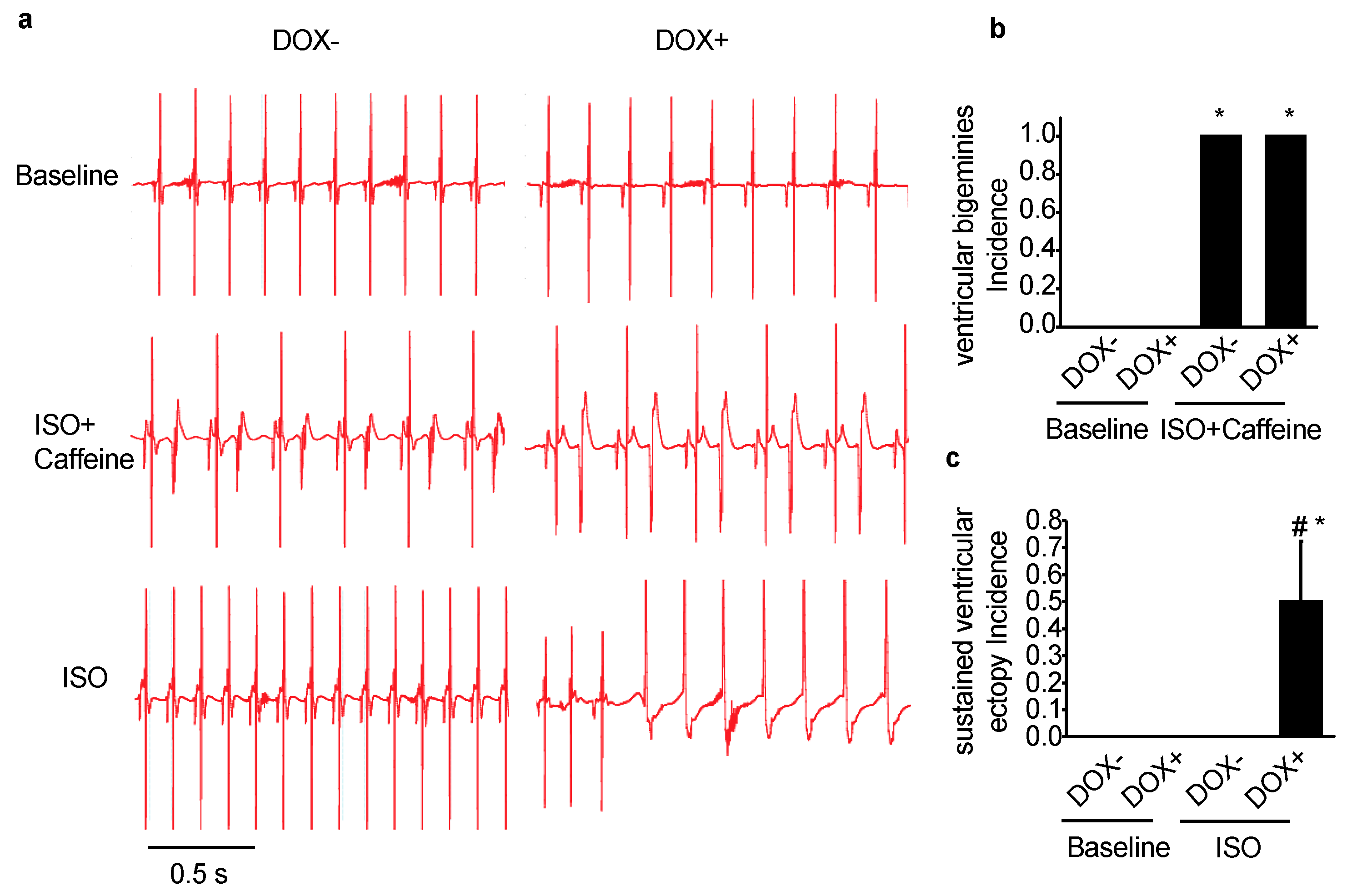
2.2. Acute Upregulation of SERCA2a in the CASQ2 KO Mice Exacerbated Ventricular Arrhythmias
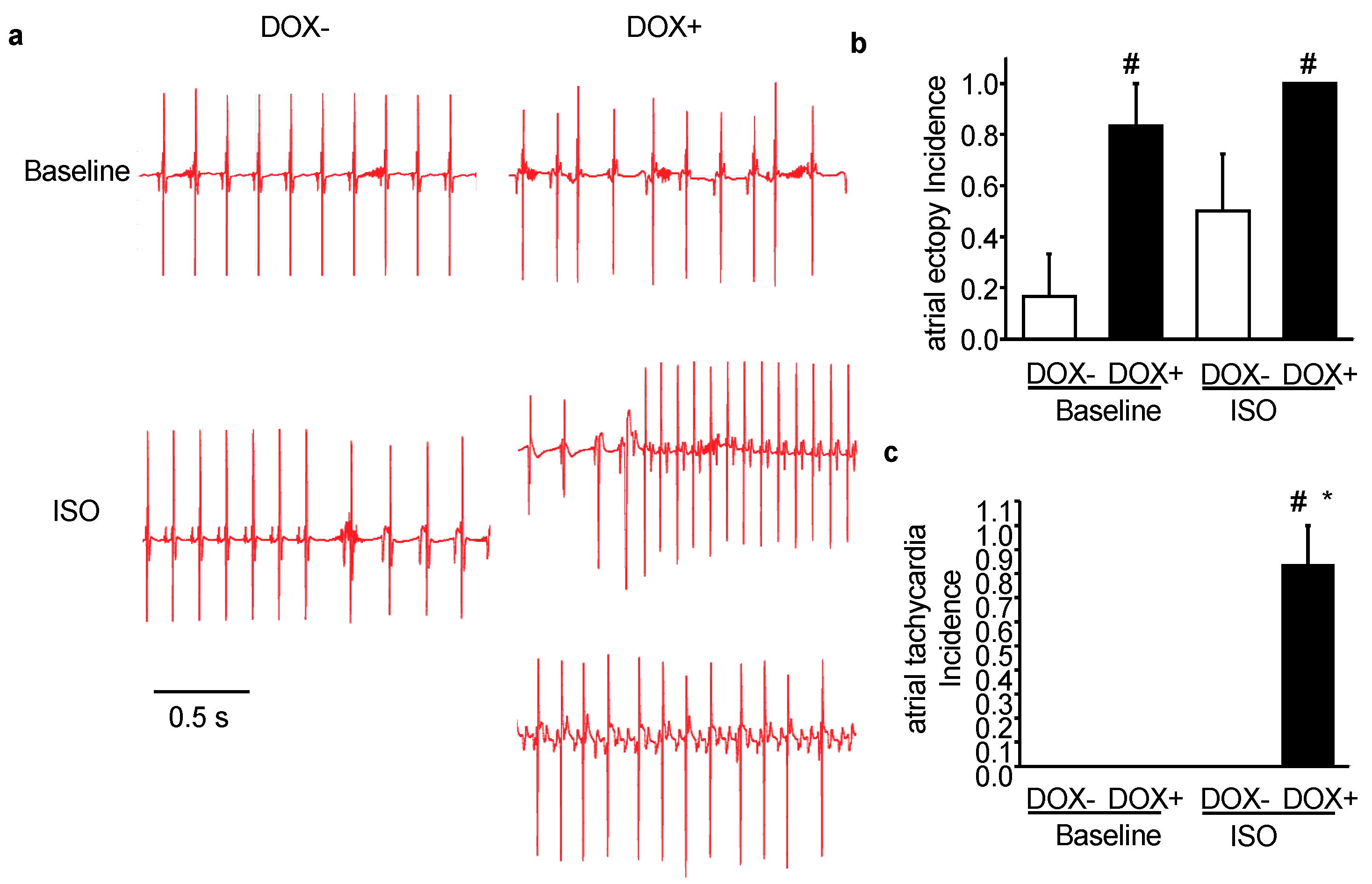
2.3. Acute Upregulation of SERCA2a in the CASQ2 KO Mice Exacerbated Atrial Arrhythmias
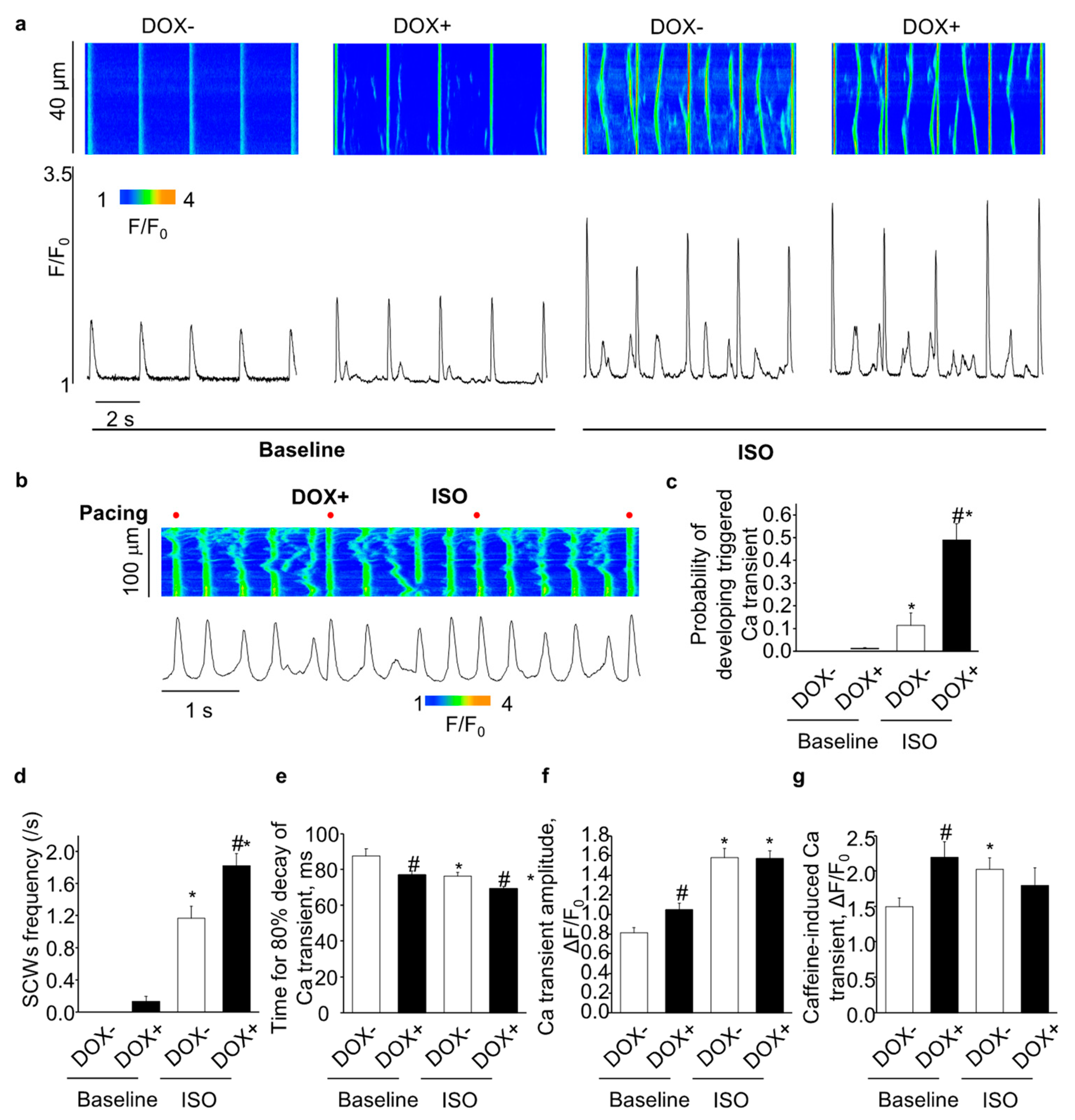
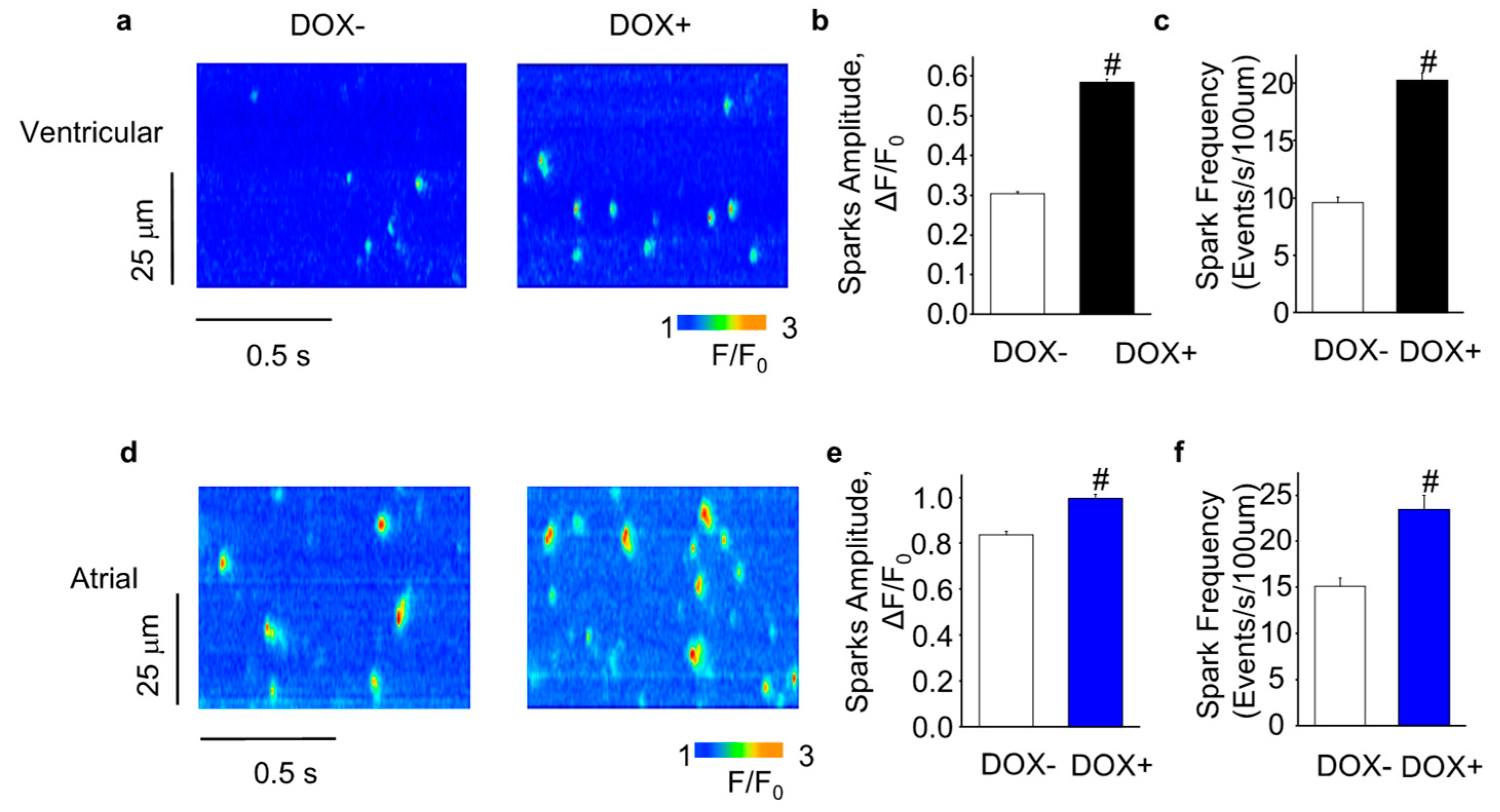
2.4. Acute Upregulation of SERCA2a Increased Diastolic Ca Release in Intact CASQ2 KO Ventricular Myocytes
2.5. Acute Upregulation of SERCA2a Increased Diastolic Ca Release in Intact CASQ2 KO Atrial Myocytes
2.6. Acute SERCA2a Overexpression Increased Diastolic SR Ca Release in Permeabilized CASQ2 KO Myocytes
2.7. Changes in Gene Expression Induced by Acute SERCA2a Overexpression
3. Discussion
4. Materials and Methods
4.1. Generation of Double KO-TG Mouse Models
4.2. Electrocardiographic Recordings
4.3. Cardiomyocyte Isolation and Confocal Ca Imaging
4.4. Western Blots
4.5. RNA Extraction, Library Construction and Sequencing
4.6. RNA-seq Read Processing, Transcript Assembly, and Differential Expression
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | atrial fibrillation |
| CAMKII | Ca2+/calmodulin-dependent protein kinase II |
| Ca | calcium |
| CASQ2 | calsequestrin 2 |
| CICR | calcium-induced calcium release |
| CPVT | catecholaminergic polymorphic ventricular tachycardia |
| DAD | delayed afterdepolarizations |
| DOX | doxycycline |
| HF | heart failure |
| KO | knock out |
| ISO | isoproterenol |
| PLB | phospholamban |
| RyR2 | ryanodine receptor 2 |
| SR | sarcoplasmic reticulum |
| SERCA | SR Ca ATPase |
| SCW | Spontaneous calcium waves |
| WT | wild type |
References
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Trimarco, B.; Iaccarino, G.; Santulli, G. New Insights in Cardiac Calcium Handling and Excitation-Contraction Coupling. Adv. Exp. Med. Biol. 2018, 1067, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Belevych, A.E.; Radwanski, P.B.; Carnes, C.A.; Gyorke, S. ‘Ryanopathy’: Causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc. Res. 2013, 98, 240–247. [Google Scholar] [CrossRef]
- Belevych, A.E.; Terentyev, D.; Terentyeva, R.; Ho, H.T.; Gyorke, I.; Bonilla, I.M.; Carnes, C.A.; Billman, G.E.; Gyorke, S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 2012, 110, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Gyorke, S. Molecular basis of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2009, 6, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, A.V.; Kalyanasundaram, A.; Lou, Q.; Hage, L.T.; Hansen, B.J.; Belevych, A.E.; Mohler, P.J.; Knollmann, B.C.; Periasamy, M.; Gyorke, S.; et al. Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex1. Eur. Heart J. 2015, 36, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Macquaide, N.; Tuan, H.T.; Hotta, J.; Sempels, W.; Lenaerts, I.; Holemans, P.; Hofkens, J.; Jafri, M.S.; Willems, R.; Sipido, K.R. Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc. Res. 2015, 108, 387–398. [Google Scholar] [CrossRef]
- Santulli, G.; Lewis, D.; des Georges, A.; Marks, A.R.; Frank, J. Ryanodine Receptor Structure and Function in Health and Disease. Subcell. Biochem. 2018, 87, 329–352. [Google Scholar] [CrossRef]
- Greiser, M.; Kerfant, B.G.; Williams, G.S.; Voigt, N.; Harks, E.; Dibb, K.M.; Giese, A.; Meszaros, J.; Verheule, S.; Ravens, U.; et al. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J. Clin. Investig. 2014, 124, 4759–4772. [Google Scholar] [CrossRef]
- Gyorke, S.; Terentyev, D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc. Res. 2008, 77, 245–255. [Google Scholar] [CrossRef]
- Liu, B.; Walton, S.D.; Ho, H.T.; Belevych, A.E.; Tikunova, S.B.; Bonilla, I.; Shettigar, V.; Knollmann, B.C.; Priori, S.G.; Volpe, P.; et al. Gene Transfer of Engineered Calmodulin Alleviates Ventricular Arrhythmias in a Calsequestrin-Associated Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. J. Am. Heart Assoc. 2018, 7, e00155. [Google Scholar] [CrossRef] [PubMed]
- Faggioni, M.; Savio-Galimberti, E.; Venkataraman, R.; Hwang, H.S.; Kannankeril, P.J.; Darbar, D.; Knollmann, B.C. Suppression of spontaneous ca elevations prevents atrial fibrillation in calsequestrin 2-null hearts. Circ. Arrhythm. Electrophysiol. 2014, 7, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Belevych, A.E.; Terentyev, D.; Terentyeva, R.; Nishijima, Y.; Sridhar, A.; Hamlin, R.L.; Carnes, C.A.; Gyorke, S. The relationship between arrhythmogenesis and impaired contractility in heart failure: Role of altered ryanodine receptor function. Cardiovasc. Res. 2011, 90, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Ann. Rev. Physiol. 2014, 76, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, A.; Lacombe, V.A.; Belevych, A.E.; Brunello, L.; Carnes, C.A.; Janssen, P.M.; Knollmann, B.C.; Periasamy, M.; Gyorke, S. Up-regulation of sarcoplasmic reticulum Ca(2+) uptake leads to cardiac hypertrophy, contractile dysfunction and early mortality in mice deficient in CASQ2. Cardiovasc. Res. 2013, 98, 297–306. [Google Scholar] [CrossRef]
- Zhang, T.; Guo, T.; Mishra, S.; Dalton, N.D.; Kranias, E.G.; Peterson, K.L.; Bers, D.M.; Brown, J.H. Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ. Res. 2010, 106, 354–362. [Google Scholar] [CrossRef]
- Bai, Y.; Jones, P.P.; Guo, J.; Zhong, X.; Clark, R.B.; Zhou, Q.; Wang, R.; Vallmitjana, A.; Benitez, R.; Hove-Madsen, L.; et al. Phospholamban knockout breaks arrhythmogenic Ca(2)(+) waves and suppresses catecholaminergic polymorphic ventricular tachycardia in mice. Circ. Res. 2013, 113, 517–526. [Google Scholar] [CrossRef]
- Valverde, C.A.; Mazzocchi, G.; Di Carlo, M.N.; Ciocci Pardo, A.; Salas, N.; Ragone, M.I.; Felice, J.I.; Cely-Ortiz, A.; Consolini, A.E.; Portiansky, E.; et al. Ablation of phospholamban rescues reperfusion arrhythmias but exacerbates myocardium infarction in hearts with Ca2+/calmodulin kinase II constitutive phosphorylation of ryanodine receptors. Cardiovasc. Res. 2019, 115, 556–569. [Google Scholar] [CrossRef]
- Byrne, M.J.; Power, J.M.; Preovolos, A.; Mariani, J.A.; Hajjar, R.J.; Kaye, D.M. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008, 15, 1550–1557. [Google Scholar] [CrossRef]
- Hammoudi, N.; Ishikawa, K.; Hajjar, R.J. Adeno-associated virus-mediated gene therapy in cardiovascular disease. Curr. Opin. Cardiol. 2015, 30, 228–234. [Google Scholar] [CrossRef]
- Kang, S.; Dahl, R.; Hsieh, W.; Shin, A.; Zsebo, K.M.; Buettner, C.; Hajjar, R.J.; Lebeche, D. Small Molecular Allosteric Activator of the Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA) Attenuates Diabetes and Metabolic Disorders. J. Biol. Chem. 2016, 291, 5185–5198. [Google Scholar] [CrossRef] [PubMed]
- Motloch, L.J.; Cacheux, M.; Ishikawa, K.; Xie, C.; Hu, J.; Aguero, J.; Fish, K.M.; Hajjar, R.J.; Akar, F.G. Primary Effect of SERCA 2a Gene Transfer on Conduction Reserve in Chronic Myocardial Infarction. J. Am. Heart Assoc. 2018, 7, e009598. [Google Scholar] [CrossRef] [PubMed]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Lou, Q.; Belevych, A.E.; Radwanski, P.B.; Liu, B.; Kalyanasundaram, A.; Knollmann, B.C.; Fedorov, V.V.; Gyorke, S. Alternating membrane potential/calcium interplay underlies repetitive focal activity in a genetic model of calcium-dependent atrial arrhythmias. J. Physiol. 2015, 593, 1443–1458. [Google Scholar] [CrossRef] [PubMed]
- Salvage, S.C.; King, J.H.; Chandrasekharan, K.H.; Jafferji, D.I.; Guzadhur, L.; Matthews, H.R.; Huang, C.L.; Fraser, J.A. Flecainide exerts paradoxical effects on sodium currents and atrial arrhythmia in murine RyR2-P2328S hearts. Acta Physiol. 2015, 214, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Liu, B.; Ho, H.T.; Brunello, L.; Unudurthi, S.D.; Lou, Q.; Belevych, A.E.; Qian, L.; Kim do, H.; Cho, C.; Janssen, P.M.; et al. Ablation of HRC alleviates cardiac arrhythmia and improves abnormal Ca handling in CASQ2 knockout mice prone to CPVT. Cardiovasc. Res. 2015, 108, 299–311. [Google Scholar] [CrossRef]
- Notari, M.; Hu, Y.; Sutendra, G.; Dedeic, Z.; Lu, M.; Dupays, L.; Yavari, A.; Carr, C.A.; Zhong, S.; Opel, A.; et al. iASPP, a previously unidentified regulator of desmosomes, prevents arrhythmogenic right ventricular cardiomyopathy (ARVC)-induced sudden death. Proc. Natl. Acad. Sci. USA 2015, 112, E973–E981. [Google Scholar] [CrossRef]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef]
- Charlet, B.N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.A.; Cooper, T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 2002, 10, 45–53. [Google Scholar] [CrossRef]
- Chen, J.Y.; Liou, Y.M.; Wu, H.D.; Lin, K.H.; Chang, K.C. Promoter polymorphism G-6A, which modulates angiotensinogen gene expression, is associated with non-familial sick sinus syndrome. PLoS ONE 2012, 7, e29951. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laforest, B.; Dai, W.; Tyan, L.; Lazarevic, S.; Shen, K.M.; Gadek, M.; Broman, M.T.; Weber, C.R.; Moskowitz, I.P. Atrial fibrillation risk loci interact to modulate Ca2+-dependent atrial rhythm homeostasis. J. Clin. Investig. 2019, 129, 4937–4950. [Google Scholar] [CrossRef] [PubMed]
- Canon, S.; Caballero, R.; Herraiz-Martinez, A.; Perez-Hernandez, M.; Lopez, B.; Atienza, F.; Jalife, J.; Hove-Madsen, L.; Delpon, E.; Bernad, A. miR-208b upregulation interferes with calcium handling in HL-1 atrial myocytes: Implications in human chronic atrial fibrillation. J. Mol. Cell. Cardiol. 2016, 99, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; Santulli, G.; Guerra, G.; Trotta, M.C.; Santamaria, M.; Sacra, C.; Testa, N.; Ducceschi, V.; Gatta, G.; Amico, M.; et al. Modulation of SERCA in Patients with Persistent Atrial Fibrillation Treated by Epicardial Thoracoscopic Ablation: The CAMAF Study. J. Clin. Med. 2020, 9, 544. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, H.; van Sloun, B.; Schonleitner, P.; van Zandvoort, M.; Antoons, G.; Heijman, J. The Subcellular Distribution of Ryanodine Receptors and L-Type Ca(2+) Channels Modulates Ca(2+)-Transient Properties and Spontaneous Ca(2+)-Release Events in Atrial Cardiomyocytes. Front. Physiol. 2018, 9, 1108. [Google Scholar] [CrossRef] [PubMed]
- Herraiz-Martinez, A.; Llach, A.; Tarifa, C.; Gandia, J.; Jimenez-Sabado, V.; Lozano-Velasco, E.; Serra, S.A.; Vallmitjana, A.; Vazquez Ruiz de Castroviejo, E.; Benitez, R.; et al. The 4q25 variant rs13143308T links risk of atrial fibrillation to defective calcium homoeostasis. Cardiovasc. Res. 2019, 115, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, G.; Sommese, L.; Palomeque, J.; Felice, J.I.; Di Carlo, M.N.; Fainstein, D.; Gonzalez, P.; Contreras, P.; Skapura, D.; McCauley, M.D.; et al. Phospholamban ablation rescues the enhanced propensity to arrhythmias of mice with CaMKII-constitutive phosphorylation of RyR2 at site S2814. J. Physiol. 2016, 594, 3005–3030. [Google Scholar] [CrossRef]
- Keizer, J.; Smith, G.D. Spark-to-wave transition: Saltatory transmission of calcium waves in cardiac myocytes. Biophys. Chem. 1998, 72, 87–100. [Google Scholar] [CrossRef]
- Keizer, J.; Smith, G.D.; Ponce-Dawson, S.; Pearson, J.E. Saltatory propagation of Ca2+ waves by Ca2+ sparks. Biophys. J. 1998, 75, 595–600. [Google Scholar] [CrossRef]
- Gyorke, I.; Hester, N.; Jones, L.R.; Gyorke, S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 2004, 86, 2121–2128. [Google Scholar] [CrossRef]
- Blatter, L.A.; Kockskamper, J.; Sheehan, K.A.; Zima, A.V.; Huser, J.; Lipsius, S.L. Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J. Physiol. 2003, 546, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Walden, A.P.; Dibb, K.M.; Trafford, A.W. Differences in intracellular calcium homeostasis between atrial and ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.T.; Klassen, T.L.; Goldman, A.M.; Marini, C.; Guerrini, R.; Noebels, J.L. Novel brain expression of ClC-1 chloride channels and enrichment of CLCN1 variants in epilepsy. Neurology 2013, 80, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.S.; Li, Y.G.; Chen, X.D.; Yu, J.F.; Wang, J.; Sun, J.; Lu, S.B.; Jin, L.; Wang, X.F. Angiotensinogen polymorphisms and acquired atrial fibrillation in Chinese. J. Electrocardiol. 2010, 43, 373–377. [Google Scholar] [CrossRef]
- Houser, S.R. Can novel therapies for arrhythmias caused by spontaneous sarcoplasmic reticulum Ca2+ release be developed using mouse models? Circ. Res. 2005, 96, 1031–1032. [Google Scholar] [CrossRef]
- Milani-Nejad, N.; Janssen, P.M. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharmacol. Ther. 2014, 141, 235–249. [Google Scholar] [CrossRef]
- Clauss, S.; Bleyer, C.; Schuttler, D.; Tomsits, P.; Renner, S.; Klymiuk, N.; Wakili, R.; Massberg, S.; Wolf, E.; Kaab, S. Animal models of arrhythmia: Classic electrophysiology to genetically modified large animals. Nat. Rev. Cardiol. 2019, 16, 457–475. [Google Scholar] [CrossRef]
- Suarez, J.; Gloss, B.; Belke, D.D.; Hu, Y.; Scott, B.; Dieterle, T.; Kim, Y.K.; Valencik, M.L.; McDonald, J.A.; Dillmann, W.H. Doxycycline inducible expression of SERCA2a improves calcium handling and reverts cardiac dysfunction in pressure overload-induced cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2164–H2172. [Google Scholar] [CrossRef]
- Liu, B.; Ho, H.T.; Velez-Cortes, F.; Lou, Q.; Valdivia, C.R.; Knollmann, B.C.; Valdivia, H.H.; Gyorke, S. Genetic ablation of ryanodine receptor 2 phosphorylation at Ser-2808 aggravates Ca(2+)-dependent cardiomyopathy by exacerbating diastolic Ca2+ release. J. Physiol. 2014, 592, 1957–1973. [Google Scholar] [CrossRef]
- Zhong, S.; Joung, J.G.; Zheng, Y.; Chen, Y.R.; Liu, B.; Shao, Y.; Xiang, J.Z.; Fei, Z.; Giovannoni, J.J. High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 2011, 940–949. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Down-Regulated | Gene | log2Fold Change | Adjusted p Value |
| Fos | −2.7 | 1.17 × 10−28 | |
| Vaultrc5 | −1.7 | 1.85 × 10−18 | |
| Atf3 | −2.1 | 2.20 × 10−12 | |
| Ppp1r13l | −1.7 | 2.24 × 10−12 | |
| Egr1 | −1.5 | 2.75 × 10−11 | |
| Grin2c | −1.9 | 5.97 × 10−11 | |
| Nr4a3 | −1.8 | 6.86 × 10−11 | |
| H2-T23 | −2 | 5.71 × 10−9 | |
| H2-Eb1 | 5 | 5.90 × 10−8 | |
| Rn45s | −2 | 5.47 × 10−7 | |
| 5730408K05Rik | −1.7 | 7.19 × 10−6 | |
| Ifltd1 | −2.6 | 5.70 × 10−5 | |
| Clcn1 | −1.5 | 0.004 | |
| Prr5 | −1.6 | 0.004 | |
| Rprl3 | −9.7 | 0.006 | |
| Mir3113 | −1.5 | 0.01 | |
| Nr4a1 | −1.9 | 0.028 | |
| Up-regulated | Gene | log2Fold Change | Adjusted p Value |
| Col4a6 | 4.5 | 3.61 × 10−29 | |
| Zdhhc2 | 1.6 | 1.17 × 10−28 | |
| Agt | 2.2 | 1.19 × 10−21 | |
| Rps15a-ps6 | 1.5 | 0.001 | |
| Selenbp2 | 2.4 | 0.004 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Lou, Q.; Smith, H.; Velez-Cortes, F.; Dillmann, W.H.; Knollmann, B.C.; Armoundas, A.A.; Györke, S. Conditional Up-Regulation of SERCA2a Exacerbates RyR2-Dependent Ventricular and Atrial Arrhythmias. Int. J. Mol. Sci. 2020, 21, 2535. https://doi.org/10.3390/ijms21072535
Liu B, Lou Q, Smith H, Velez-Cortes F, Dillmann WH, Knollmann BC, Armoundas AA, Györke S. Conditional Up-Regulation of SERCA2a Exacerbates RyR2-Dependent Ventricular and Atrial Arrhythmias. International Journal of Molecular Sciences. 2020; 21(7):2535. https://doi.org/10.3390/ijms21072535
Chicago/Turabian StyleLiu, Bin, Qing Lou, Heather Smith, Florencia Velez-Cortes, Wolfgang H. Dillmann, Björn C. Knollmann, Antonis A. Armoundas, and Sándor Györke. 2020. "Conditional Up-Regulation of SERCA2a Exacerbates RyR2-Dependent Ventricular and Atrial Arrhythmias" International Journal of Molecular Sciences 21, no. 7: 2535. https://doi.org/10.3390/ijms21072535
APA StyleLiu, B., Lou, Q., Smith, H., Velez-Cortes, F., Dillmann, W. H., Knollmann, B. C., Armoundas, A. A., & Györke, S. (2020). Conditional Up-Regulation of SERCA2a Exacerbates RyR2-Dependent Ventricular and Atrial Arrhythmias. International Journal of Molecular Sciences, 21(7), 2535. https://doi.org/10.3390/ijms21072535