The Subcellular Localization and Oligomerization Preferences of NME1/NME2 upon Radiation-Induced DNA Damage


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
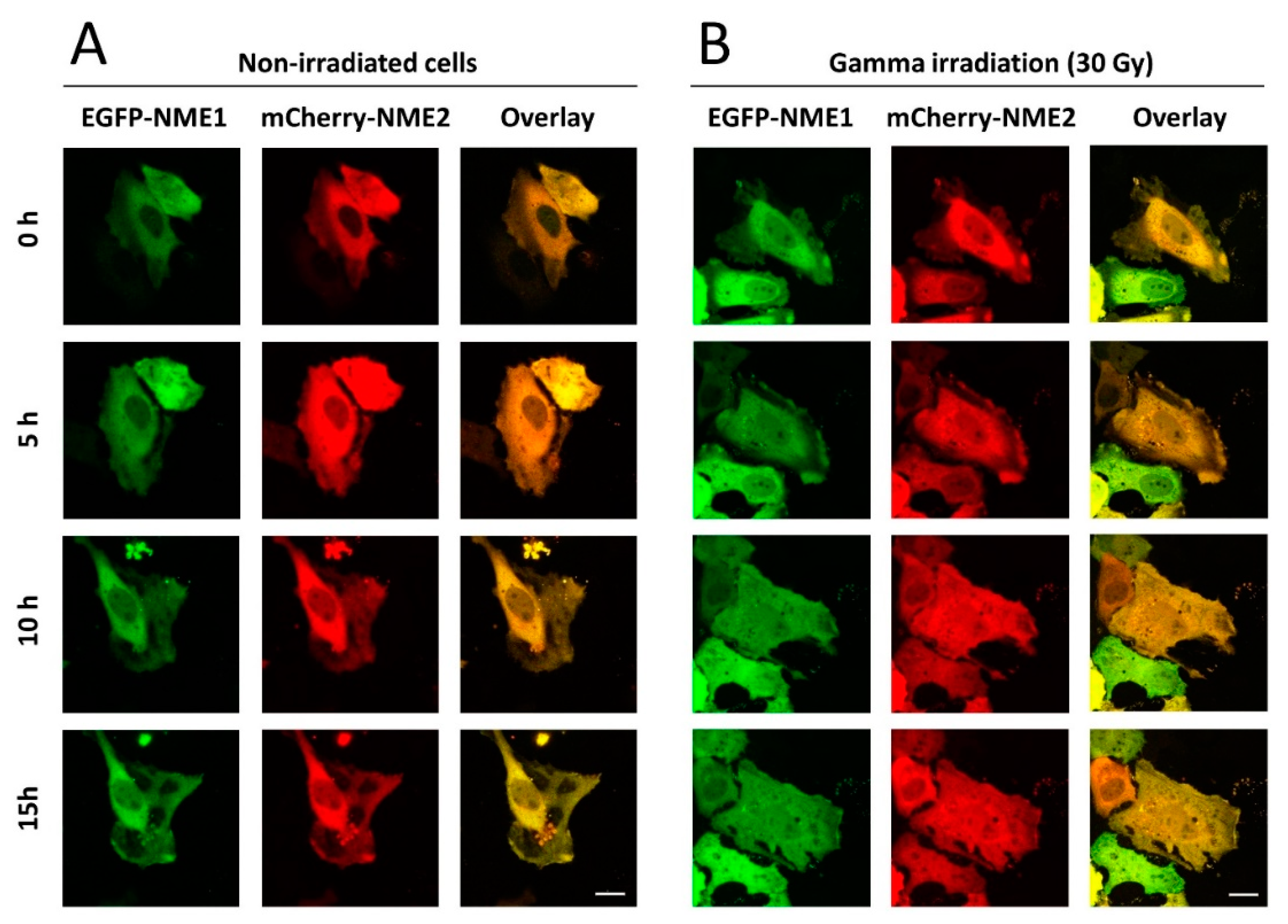
2.1. NME1 and NME2 Translocate to the Nucleus after Gamma Irradiation Treatment
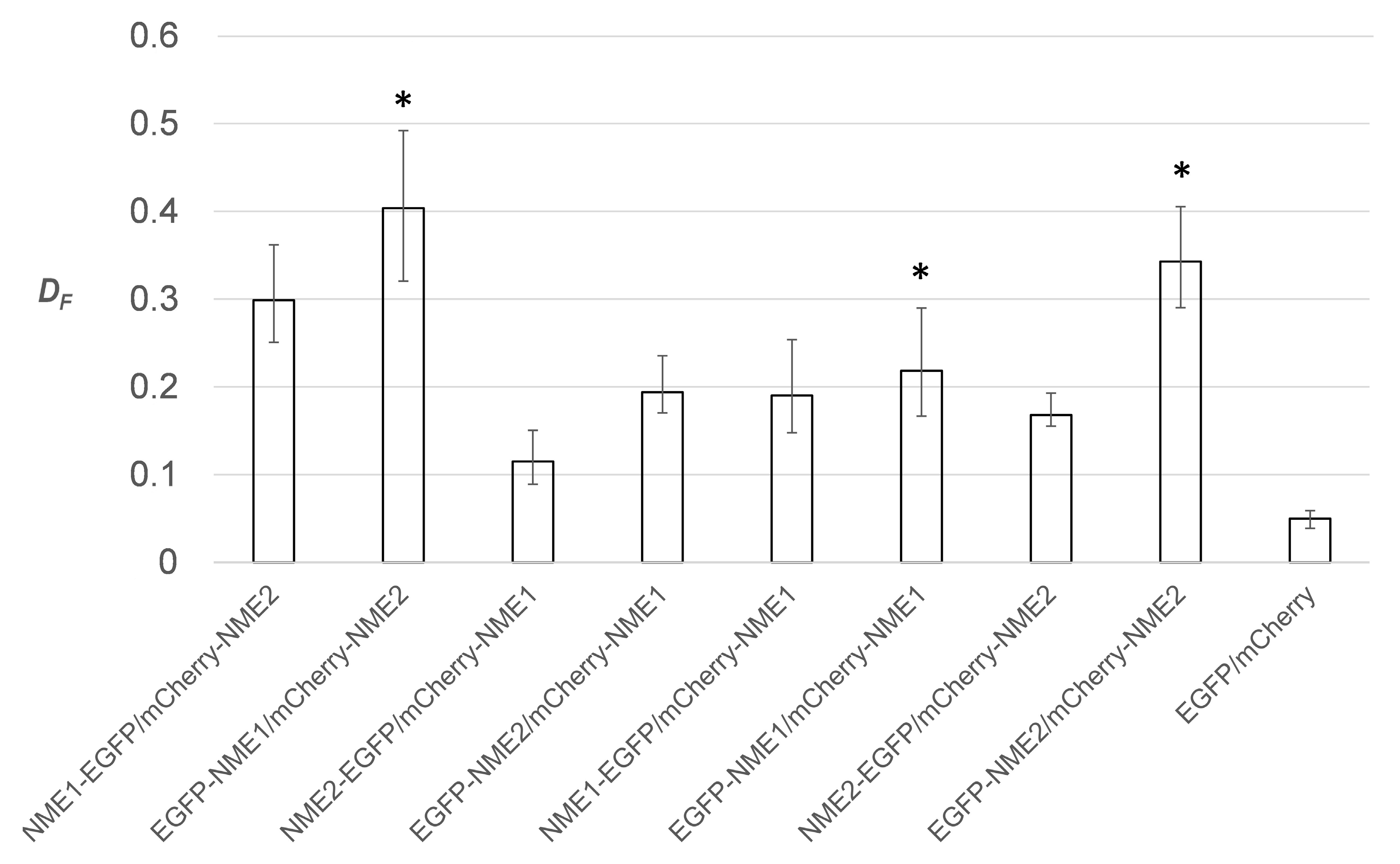
2.2. Choosing the Optimal FRET Reporter Orientation for Estimating Fractional FRET Population of Homomers and Heteromers
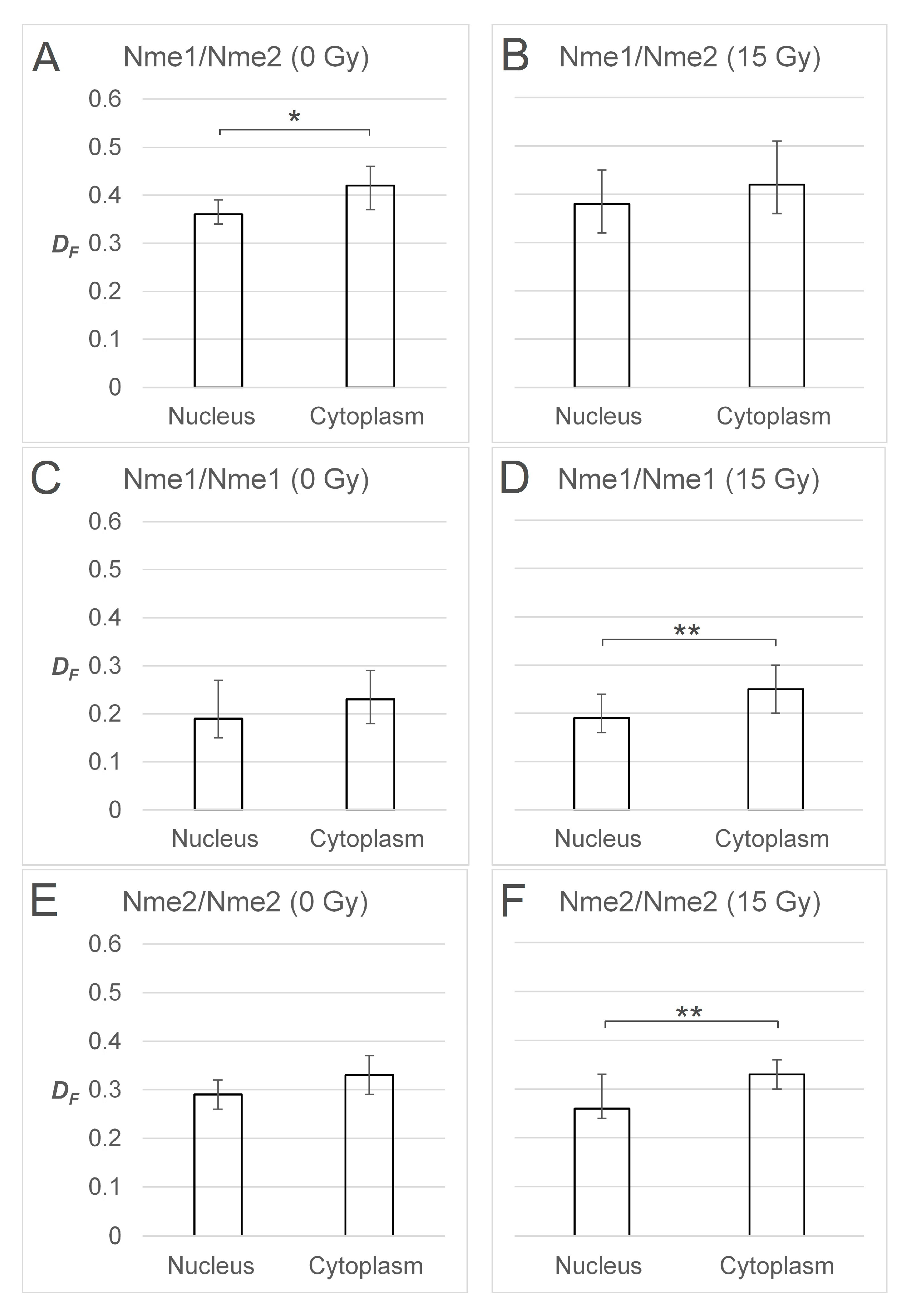
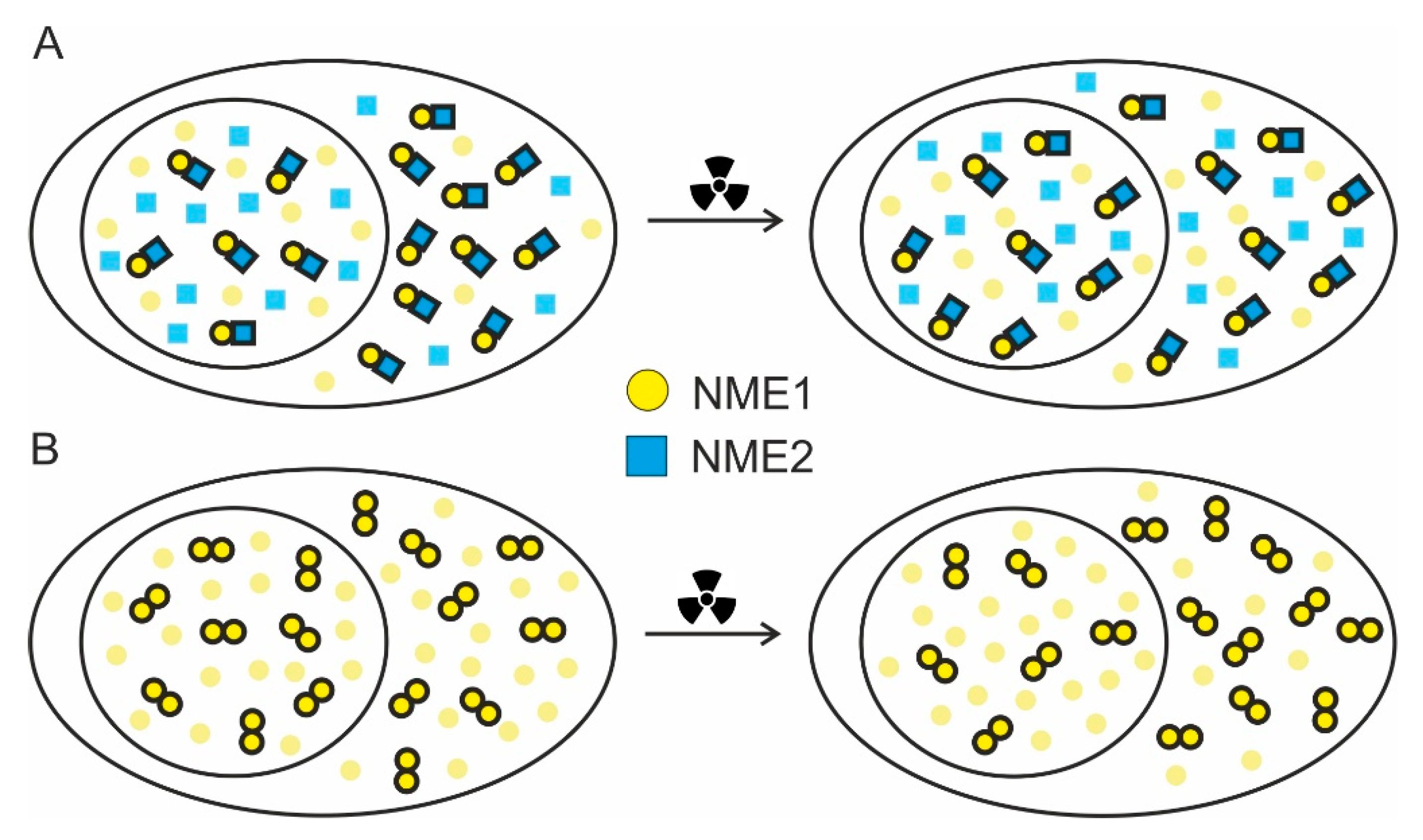
2.3. FRET/FLIM Experiments Indicate that DNA Damage Slightly Changes the Balance between the Oligomers in Cellular Compartments
3. Materials and Methods
3.1. Cell Lines
3.2. Plasmids
3.3. Transfection
3.4. DNA Damage
3.5. Co-immunoprecipitation
3.6. Western Blot Analysis
3.7. Confocal Imaging of Live and Fixed Cells
3.8. FRET-FLIM Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BER | Base Excision Repair |
| APE1 | Apurinic/Apyrimidinic Endonuclease 1 |
| NHEJ | Non-Homologous End Joining |
| CMV | Cytomegalovirus |
| DSBR | Double-Stranded Break Repair |
| EBNA 1–3C | Epstein–Barr Nuclear Antigen 1–3C |
| EGFP | Enhanced Green Fluorescent Protein |
| ERβ | Estrogen Receptor β |
| FLIM | Fluorescence-Lifetime Imaging Microscopy |
| FRET | Fluorescence Resonance Energy Transfer |
| ICAP1α | Integrin Cytoplasmic Domain-Associated Protein 1α |
| ER | Endoplasmic Reticulum |
| COP II | Coat Protein Complex II |
| GFAP | Glial Fibrillary Acidic Protein |
| ARF6 | ADP-Ribosylation Factor 6 |
| KSR | Kinase Suppressor of Ras |
| MDM2 | Mouse Double Minute 2 Homolog |
| MIF | Macrophage Migration Inhibitory Factor |
| NDP | Nucleoside Diphosphate |
| NDPK | Nucleoside Diphosphate Kinase |
| NER | Nucleotide Excision Repair |
| NES | Nuclear Export Signal |
| NLS | Nuclear Localization Signal |
| NTP | Nucleoside Triphosphate |
| PDGF | Platelet-Derived Growth Factor |
| STRAP | Serine/Threonine Kinase Receptor Associated Protein |
| TRF1 | Telomeric Repeat Binding Factor 1 |
| UVR | Ultraviolet Radiation |
| YNK1 | Yeast Homolog of NME1 |
References
- Agarwal, R.P.; Robison, B.; Parks, R.E. Nucleoside diphosphokinase from human erythrocytes. Methods Enzymol. 1978, 51, 376–386. [Google Scholar] [PubMed]
- Herak Bosnar, M.; Radić, M.; Ćetković, H. A young researcher’s guide to NME/Nm23/NDP Kinase. Period. Biol. 2018, 120, 3–9. [Google Scholar] [CrossRef]
- Lacombe, M.-L.; Milon, L.; Munier, A.; Mehus, J.G.; Lambeth, D.O. The Human Nm23/Nucleoside Diphosphate Kinases. J. Bioenerg. Biomembr. 2000, 32, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Gillesl, A.; Presecans, E.; Vonicas, A.; Lascusli, I.; Gilles, A.M.; Presecan, E.; Vonica, A.; Lascu, I. Nucleoside diphosphate kinase from human erythrocytes. Structural characterization of the two polypeptide chains responsible for heterogeneity of the hexameric enzyme. J. Biol. Chem. 1991, 266, 8784–8789. [Google Scholar]
- Massé, K.; Dabernat, S.; Bourbon, P.-M.; Larou, M.; Amrein, L.; Barraud, P.; Perel, Y.; Camara, M.; Landry, M.; Lacombe, M.-L.; et al. Characterization of the nm23-M2, nm23-M3 and nm23-M4 mouse genes: comparison with their human orthologs. Gene 2002, 296, 87–97. [Google Scholar] [CrossRef]
- Ćetković, H.; Perina, D.; Harcet, M.; Mikoč, A.; Herak Bosnar, M. Nme family of proteins—clues from simple animals. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 133–142. [Google Scholar] [CrossRef]
- Gonin, P.; Xu, Y.; Milon, L.; Dabernat, S.; Morr, M.; Kumar, R.; Lacombe, M.-L.; Janin, J.; Lascu, I. Catalytic Mechanism of Nucleoside Diphosphate Kinase Investigated Using Nucleotide Analogues, Viscosity Effects, and X-ray Crystallography. Biochemistry 1999, 38, 7265–7272. [Google Scholar] [CrossRef]
- Steeg, P.S.; Bevilacqua, G.; Kopper, L.; Thorgeirsson, U.P.; Talmadge, J.E.; Liotta, L.A.; Sobel, M.E. Evidence for a Novel Gene Associated With Low Tumor Metastatic Potential. JNCI J. Natl. Cancer Inst. 1988, 80, 200–204. [Google Scholar] [CrossRef]
- Hartsough, M.T.; Steeg, P.S. Nm23/nucleoside diphosphate kinase in human cancers. J. Bioenerg. Biomembr. 2000, 32, 301–308. [Google Scholar] [CrossRef]
- Li, Y.; Tong, Y.; Wong, Y.H. Regulatory functions of Nm23-H2 in tumorigenesis: insights from biochemical to clinical perspectives. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 243–256. [Google Scholar] [CrossRef]
- Cipollini, G.; Berti, A.; Fiore, L.; Rainaldi, G.; Basolo, F.; Merlo, G.; Bevilacqua, G.; Caligo, M.A. Down-regulation of the nm23.h1 gene inhibits cell proliferation. Int. J. cancer 1997, 73, 297–302. [Google Scholar] [CrossRef]
- Caligo, M.A.; Cipollini, G.; Berti, A.; Viacava, P.; Collecchi, P.; Bevilacqua, G. NM23 gene expression in human breast carcinomas: loss of correlation with cell proliferation in the advanced phase of tumor progression. Int. J. cancer 1997, 74, 102–111. [Google Scholar] [CrossRef]
- Herak Bosnar, M.; Dubravčić, K.; Bago, R.; Pavelić, J. Head and neck tumor cells exhibit altered proliferation upon overexpression of nm23 genes. Croat. Chem. Acta 2008, 81, 183–189. [Google Scholar]
- Choudhuri, T.; Murakami, M.; Kaul, R.; Sahu, S.K.; Mohanty, S.; Verma, S.C.; Kumar, P.; Robertson, E.S. Nm23-H1 can induce cell cycle arrest and apoptosis in B cells. Cancer Biol. Ther. 2010, 9, 1065–1078. [Google Scholar] [CrossRef]
- Kang, Y.; Lee, D.-C.; Han, J.; Yoon, S.; Won, M.; Yeom, J.-H.; Seong, M.-J.; Ko, J.-J.; Lee, K.-A.; Lee, K.; et al. NM23-H2 involves in negative regulation of Diva and Bcl2L10 in apoptosis signaling. Biochem. Biophys. Res. Commun. 2007, 359, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, D.; Lacombe, M.-L.; Paggi, M.G. nm23: Unraveling its biological function in cell differentiation. J. Cell. Physiol. 2000, 182, 144–149. [Google Scholar] [CrossRef]
- Bilitou, A.; Watson, J.; Gartner, A.; Ohnuma, S. The NM23 family in development. Mol. Cell. Biochem. 2009, 329, 17–33. [Google Scholar] [CrossRef]
- Smagghe, B.J.; Stewart, A.K.; Carter, M.G.; Shelton, L.M.; Bernier, K.J.; Hartman, E.J.; Calhoun, A.K.; Hatziioannou, V.M.; Lillacci, G.; Kirk, B.A.; et al. MUC1* ligand, NM23-H1, is a novel growth factor that maintains human stem cells in a more naïve state. PLoS One 2013, 8, e58601. [Google Scholar] [CrossRef]
- Lakso, M.; Steeg, P.S.; Westphal, H. Embryonic expression of nm23 during mouse organogenesis. Cell Growth Differ. 1992, 3, 873–879. [Google Scholar]
- Murakami, M.; Lan, K.; Subramanian, C.; Robertson, E.S. Epstein-Barr virus nuclear antigen 1 interacts with Nm23-H1 in lymphoblastoid cell lines and inhibits its ability to suppress cell migration. J. Virol. 2005, 79, 1559–1568. [Google Scholar] [CrossRef]
- Fournier, H.-N.; Dupé-Manet, S.; Bouvard, D.; Lacombe, M.-L.; Marie, C.; Block, M.R.; Albiges-Rizo, C. Integrin cytoplasmic domain-associated protein 1alpha (ICAP-1alpha ) interacts directly with the metastasis suppressor nm23-H2, and both proteins are targeted to newly formed cell adhesion sites upon integrin engagement. J. Biol. Chem. 2002, 277, 20895–20902. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.S.; Rikhy, R.; Rao, S.; Shivalkar, M.; Mosko, M.; Narayanan, R.; Etter, P.; Estes, P.S.; Ramaswami, M. Nucleoside diphosphate kinase, a source of GTP, is required for dynamin-dependent synaptic vesicle recycling. Neuron 2001, 30, 197–210. [Google Scholar] [CrossRef]
- Palacios, F.; Schweitzer, J.K.; Boshans, R.L.; D’Souza-Schorey, C. ARF6-GTP recruits Nm23-H1 to facilitate dynamin-mediated endocytosis during adherens junctions disassembly. Nat. Cell Biol. 2002, 4, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Boissan, M.; Montagnac, G.; Shen, Q.; Griparic, L.; Guitton, J.; Romao, M.; Sauvonnet, N.; Lagache, T.; Lascu, I.; Raposo, G.; et al. Membrane trafficking. Nucleoside diphosphate kinases fuel dynamin superfamily proteins with GTP for membrane remodeling. Science 2014, 344, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.D.; Vu, N.D. Histidine to aspartate phosphotransferase activity of nm23 proteins: phosphorylation of aldolase C on Asp-319. Biochem. J. 2000, 346, 623–630. [Google Scholar] [CrossRef]
- Wagner, P.D.; Vu, N.-D. Phosphorylation of ATP-Citrate Lyase by Nucleoside Diphosphate Kinase. J. Biol. Chem. 1995, 270, 21758–21764. [Google Scholar] [CrossRef]
- Hartsough, M.T.; Morrison, D.K.; Salerno, M.; Palmieri, D.; Ouatas, T.; Mair, M.; Patrick, J.; Steeg, P.S. Nm23-H1 Metastasis Suppressor Phosphorylation of Kinase Suppressor of Ras via a Histidine Protein Kinase Pathway. J. Biol. Chem. 2002, 277, 32389–32399. [Google Scholar] [CrossRef]
- Cuello, F.; Schulze, R.A.; Heemeyer, F.; Meyer, H.E.; Lutz, S.; Jakobs, K.H.; Niroomand, F.; Wieland, T. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Complex formation of NDPK B with Gbeta gamma dimers and phosphorylation of His-266 IN Gbeta. J. Biol. Chem. 2003, 278, 7220–7226. [Google Scholar] [CrossRef]
- Srivastava, S.; Li, Z.; Ko, K.; Choudhury, P.; Albaqumi, M.; Johnson, A.K.; Yan, Y.; Backer, J.M.; Unutmaz, D.; Coetzee, W.A.; et al. Histidine Phosphorylation of the Potassium Channel KCa3.1 by Nucleoside Diphosphate Kinase B Is Required for Activation of KCa3.1 and CD4 T Cells. Mol. Cell 2006, 24, 665–675. [Google Scholar] [CrossRef]
- Berberich, S.J.; Postel, E.H. PuF/NM23-H2/NDPK-B transactivates a human c-myc promoter-CAT gene via a functional nuclease hypersensitive element. Oncogene 1995, 10, 2343–2347. [Google Scholar]
- Michelotti, E.F.; Sanford, S.; Freije, J.M.P.; MacDonald, N.J.; Steeg, P.S.; Levens, D. Nm23/PuF Does Not Directly Stimulate Transcription through the CT Element in Vivo. J. Biol. Chem. 1997, 272, 22526–22530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; McCorkle, J.R.; Novak, M.; Yang, M.; Kaetzel, D.M. Metastasis suppressor function of NM23-H1 requires its 3′-5′ exonuclease activity. Int. J. Cancer 2011, 128, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Beresford, P.J.; Oh, D.Y.; Zhang, D.; Lieberman, J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 2003, 112, 659–672. [Google Scholar] [CrossRef]
- Vlatković, N.; Chang, S.-H.; Boyd, M.T. Janus-faces of NME–oncoprotein interactions. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Reymond, A.; Volorio, S.; Merla, G.; Al-Maghtheh, M.; Zuffardi, O.; Bulfone, A.; Ballabio, A.; Zollo, M. Evidence for interaction between human PRUNE and nm23-H1 NDPKinase. Oncogene 1999, 18, 7244–7252. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jung, H.; Seong, H.-A.; Ha, H. NM23-H1 tumor suppressor and its interacting partner STRAP activate p53 function. J. Biol. Chem. 2007, 282, 35293–35307. [Google Scholar] [CrossRef]
- Jung, H.; Seong, H.-A.; Ha, H. Direct interaction between NM23-H1 and macrophage migration inhibitory factor (MIF) is critical for alleviation of MIF-mediated suppression of p53 activity. J. Biol. Chem. 2008, 283, 32669–32679. [Google Scholar] [CrossRef]
- Lin, C.-H.; Dammai, V.; Adryan, B.; Hsu, T. Interaction between Nm23 and the tumor suppressor VHL. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 143–152. [Google Scholar] [CrossRef]
- Subramanian, C.; Cotter, M.A.; Robertson, E.S. Epstein-Barr virus nuclear protein EBNA-3C interacts with the human metastatic suppressor Nm23-H1: A molecular link to cancer metastasis. Nat. Med. 2001, 7, 350–355. [Google Scholar] [CrossRef]
- Filić, V.; Marinović, M.; Šoštar, M.; Weber, I. Modulation of small GTPase activity by NME proteins. Lab. Investig. 2018, 98, 589–601. [Google Scholar] [CrossRef]
- Polanski, R.; Maguire, M.; Nield, P.C.; Jenkins, R.E.; Park, B.K.; Krawczynska, K.; Devling, T.; Ray-Sinha, A.; Rubbi, C.P.; Vlatkovic, N.; et al. MDM2 interacts with NME2 (non-metastatic cells 2, protein) and suppresses the ability of NME2 to negatively regulate cell motility. Carcinogenesis 2011, 32, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, K.; Kawahara, M.; Masuda, M.; Satomi, Y.; Nishino, H. Association of Nucleoside Diphosphate Kinase nm23-H2 with Human Telomeres. Biochem. Biophys. Res. Commun. 1998, 243, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Urano, T.; Baba, H.; Furukawa, K.; Furukawa, K.; Shiku, H. Independent and differential expression of two isotypes of human Nm23: analysis of the promoter regions of the nm23-H1 and H2 genes. Oncogene 1996, 13, 1937–1943. [Google Scholar] [PubMed]
- Bosnar, M.H.; Bago, R.; Ćetković, H. Subcellular localization of Nm23/NDPK A and B isoforms: A reflection of their biological function? Mol. Cell. Biochem. 2009, 329, 63–71. [Google Scholar] [CrossRef]
- Pinon, V.P.-B.; Millot, G.; Munier, A.; Vassy, J.; Linares-Cruz, G.; Capeau, J.; Calvo, F.; Lacombe, M.-L. Cytoskeletal Association of the A and B Nucleoside Diphosphate Kinases of Interphasic But Not Mitotic Human Carcinoma Cell Lines: Specific Nuclear Localization of the B Subunit. Exp. Cell Res. 1999, 246, 355–367. [Google Scholar] [CrossRef]
- Bosnar, M.H.; De Gunzburg, J.; Bago, R.; Brečević, L.; Weber, I.; Pavelić, J. Subcellular localization of A and B Nm23/NDPK subunits. Exp. Cell Res. 2004, 298, 275–284. [Google Scholar] [CrossRef]
- Kraeft, S.-K.; Traincart, F.; Mesnildrey, S.; Bourdais, J.; Véron, M.; Chen, L.B. Nuclear Localization of Nucleoside Diphosphate Kinase Type B (nm23-H2) in Cultured Cells. Exp. Cell Res. 1996, 227, 63–69. [Google Scholar] [CrossRef]
- Roymans, D.; Willems, R.; Vissenberg, K.; De Jonghe, C.; Grobben, B.; Claes, P.; Lascu, I.; Van Bockstaele, D.; Verbelen, J.P.; Van Broeckhoven, C.; et al. Nucleoside diphosphate kinase β (Nm23-R1/NDPKβ) is associated with intermediate filaments and becomes upregulated upon cAMP-induced differentiation of rat C6 glioma. Exp. Cell Res. 2000, 261, 127–138. [Google Scholar] [CrossRef]
- Nickerson, J.A.; Wells, W.W. The microtubule-associated nucleoside diphosphate kinase. J. Biol. Chem. 1984, 259, 11297–11304. [Google Scholar]
- Lombardi, D.; Sacchi, A.; D’agostino, G.; Tibursi, G. The association of the Nm23-M1 protein and β-tubulin correlates with cell differentiation. Exp. Cell Res. 1995, 217, 267–271. [Google Scholar] [CrossRef]
- Gervasi, F.; D’Agnano, I.; Vossio, S.; Zupi, G.; Sacchi, A.; Lombardi, D. nm23 Influences proliferation and differentiation of PC12 cells in response to nerve growth factor. Cell Growth Differ. 1996, 7, 1689–1695. [Google Scholar] [PubMed]
- Biggs, J.; Hersperger, E.; Steeg, P.S.; Liotta, L.A.; Shearn, A. A Drosophila gene that is homologous to a mammalian gene associated with tumor metastasis codes for a nucleoside diphosphate kinase. Cell 1990, 63, 933–940. [Google Scholar] [CrossRef]
- Gallagher, B.C.; Parrott, K.A.; Szabo, G.; de, S.; Otero, A. Receptor activation regulates cortical, but not vesicular localization of NDP kinase. J. Cell Sci. 2003, 116, 3239–3250. [Google Scholar] [CrossRef] [PubMed]
- Melki, R.; Lascu, I.; Carlier, M.F.; Véron, M. Nucleoside diphosphate kinase does not directly interact with tubulin nor microtubules. Biochem. Biophys. Res. Commun. 1992, 187, 65–72. [Google Scholar] [CrossRef]
- Kapetanovich, L.; Baughman, C.; Lee, T.H. Nm23H2 facilitates coat protein complex II assembly and endoplasmic reticulum export in mammalian cells. Mol. Biol. Cell 2005, 16, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Sonnemann, J.; Mutzel, R. Cytosolic nucleoside diphosphate kinase associated with the translation apparatus may provide GTP for protein synthesis. Biochem. Biophys. Res. Commun. 1995, 209, 490–496. [Google Scholar] [CrossRef]
- Otero, A.D.S. Copurification of vimentin, energy metabolism enzymes, and a MER5 homolog with nucleoside diphosphate kinase: Identification of tissue-specific interactions. J. Biol. Chem. 1997, 272, 14690–14694. [Google Scholar] [CrossRef]
- Romani, P.; Papi, A.; Ignesti, M.; Soccolini, G.; Hsu, T.; Gargiulo, G.; Spisni, E.; Cavaliere, V. Dynamin controls extracellular level of Awd/Nme1 metastasis suppressor protein. Naunyn. Schmiedebergs. Arch. Pharmacol. 2016, 389, 1171–1182. [Google Scholar] [CrossRef]
- Kaetzel, D.M.; Zhang, Q.; Yang, M.; McCorkle, J.R.; Ma, D.; Craven, R.J. Potential roles of 3′-5′exonuclease activity of NM23-H1 in DNA repair and malignant progression. J. Bioenerg. Biomembr. 2006, 38, 163–167. [Google Scholar] [CrossRef]
- Rayner, K.; Chen, Y.X.; Hibbert, B.; White, D.; Miller, H.; Postel, E.H.; O’Brien, E.R. NM23-H2, an estrogen receptor β-associated protein, shows diminished expression with progression of atherosclerosis. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 2007, 292, R743–R750. [Google Scholar] [CrossRef]
- Yang, M.; Jarrett, S.G.; Craven, R.; Kaetzel, D.M. YNK1, the yeast homolog of human metastasis suppressor NM23, is required for repair of UV radiation- and etoposide-induced DNA damage. Mutat. Res. 2009, 660, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.M.; Yang, X.Q.; Wang, D.; Wang, G.; Yang, Z.Z.; Qing, Y.; Yang, Z.X.; Li, M.X.; Xiang, D.B. Nm23-H1 Protein Binds to APE1 at AP Sites and Stimulates AP Endonuclease Activity Following Ionizing Radiation of the Human Lung Cancer A549 Cells. Cell Biochem. Biophys. 2011, 61, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Novak, M.; Dabernat, S.; Daniel, J.-Y.; Mellon, I.; Zhang, Q.; Harris, N.; Ciesielski, M.J.; Fenstermaker, R.A.; Kovacic, D.; et al. Metastasis suppressor NM23-H1 promotes repair of UV-induced DNA damage and suppresses UV-induced melanomagenesis. Cancer Res. 2012, 72, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Xu, M.; Li, C.; Xiong, Y.; Yang, Y.; Kuang, X.; Wang, D.; Yang, X. Nm23-H1 is involved in the repair of ionizing radiation-induced DNA double-strand breaks in the A549 lung cancer cell line. BMC Cancer 2018, 18, 710. [Google Scholar] [CrossRef]
- Xue, R.; Peng, Y.; Han, B.; Li, X.; Chen, Y.; Pei, H. Metastasis suppressor NME1 promotes non-homologous end joining of DNA double-strand breaks. DNA Repair (Amst). 2019, 77, 27–35. [Google Scholar] [CrossRef]
- Potel, C.M.; Fasci, D.; Heck, A.J.R. Mix and match of the tumor metastasis suppressor Nm23 protein isoforms in vitro and in vivo. FEBS J. 2018, 285, 2856–2868. [Google Scholar] [CrossRef]
- Park, H.R.; Kim, S.-H.; Lee, S.Y.; Sung, J.M.; Park, A.R.; Kang, S.; Kim, B.G.; Choi, Y.P.; Kim, Y.-B.; Cho, N.H. Nuclear localization of Nm23-H1 in head and neck squamous cell carcinoma is associated with radiation resistance. Cancer 2011, 117, 1864–1873. [Google Scholar] [CrossRef]
- Qu, S.; Li, X.Y.; Liang, Z.G.; Li, L.; Huang, S.T.; Li, J.Q.; Li, D.R.; Zhu, X.D. Protein expression of nucleophosmin, annexin A3 and nm23-H1 correlates with human nasopharyngeal carcinoma radioresistance in vivo. Oncol. Lett. 2016, 12, 615–620. [Google Scholar] [CrossRef][Green Version]
- Lee, S.Y.; Park, H.R.; Cho, N.H.; Choi, Y.P.; Rha, S.Y.; Park, S.W.; Kim, S.H. Identifying genes related to radiation resistance in oral squamous cell carcinoma cell lines. Int. J. Oral Maxillofac. Surg. 2013, 42, 169–176. [Google Scholar] [CrossRef]
- Postel, E.H.; Abramczyk, B.M.; Levit, M.N.; Kyin, S. Catalysis of DNA cleavage and nucleoside triphosphate synthesis by NM23-H2/NDP kinase share an active site that implies a DNA repair function. Proc. Natl. Acad. Sci. USA 2000, 97, 14194–14199. [Google Scholar] [CrossRef]
- Newmeyer, D.D. The nuclear pore complex and nucleocytoplasmic transport. Curr. Opin. Cell Biol. 1993, 5, 395–407. [Google Scholar] [CrossRef]
- Schlattner, U.; Tokarska-Schlattner, M.; Epand, R.M.; Boissan, M.; Lacombe, M.-L.; Kagan, V.E. NME4/nucleoside diphosphate kinase D in cardiolipin signaling and mitophagy. Lab. Invest. 2018, 98, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Margineanu, A.; Chan, J.J.; Kelly, D.J.; Warren, S.C.; Flatters, D.; Kumar, S.; Katan, M.; Dunsby, C.W.; French, P.M.W. Screening for protein-protein interactions using Förster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Sci. Rep. 2016, 6, 28186. [Google Scholar] [CrossRef]
- Yasuda, R. Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Curr. Opin. Neurobiol. 2006, 16, 551–561. [Google Scholar] [CrossRef]
- Zhang, J.; Campbell, R.E.; Ting, A.Y.; Tsien, R.Y. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 2002, 3, 906–918. [Google Scholar] [CrossRef]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P.C. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012, 17, 4047–4132. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, X.; Yu, M.; Zhu, Y.; Xing, J.; Lin, J. Techniques for detecting protein-protein interactions in living cells: principles, limitations, and recent progress. Sci. China Life Sci. 2019, 62, 619–632. [Google Scholar] [CrossRef]
- Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A Guide to Fluorescent Protein FRET Pairs. Sensors (Basel) 2016, 16, 1488. [Google Scholar] [CrossRef]
- Mesnildrey, S.; Agou, F.; Karlsson, A.; Deville Bonne, D.; Véron, M. Coupling between catalysis and oligomeric structure in nucleoside diphosphate kinase. J. Biol. Chem. 1998, 273, 4436–4442. [Google Scholar] [CrossRef]
- Boshart, M.; Weber, F.; Jahn, G.; Dorsch-Hler, K.; Fleckenstein, B.; Schaffner, W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 1985, 41, 521–530. [Google Scholar] [CrossRef]
- Acsadi, G.; Jani, A.; Massie, B.; Simoneau, M.; Holland, P.; Blaschuk, K.; Karpati, G. A differential efficiency of adenovirus-mediated in vivo gene transfer into skeletal muscle cells of different maturity. Hum. Mol. Genet. 1994, 3, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, H.S.; Mickanin, C.; Buck, C. Adenovirus-mediated gene transfer during initial organogenesis in the mammalian embryo is promoter-dependent and tissue-specific. Gene Ther. 1997, 4, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.Y.; Zhang, L.; Clift, K.L.; Hulur, I.; Xiang, A.P.; Ren, B.Z.; Lahn, B.T. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS ONE 2010, 5, e10611. [Google Scholar] [CrossRef]
- Smith, R.L.; Traul, D.L.; Schaack, J.; Clayton, G.H.; Staley, K.J.; Wilcox, C.L. Characterization of Promoter Function and Cell-Type-Specific Expression from Viral Vectors in the Nervous System. J. Virol. 2000, 74, 11254–11261. [Google Scholar] [CrossRef]
- Johnson, L.G.; Pickles, R.J.; Boyles, S.E.; Morris, J.C.; Ye, H.; Zhou, Z.; Olsen, J.C.; Boucher, R.C. In vitro assessment of variables affecting the efficiency and efficacy of adenovirus-mediated gene transfer to cystic fibrosis airway epithelia. Hum. Gene Ther. 1996, 7, 51–59. [Google Scholar] [CrossRef]
- Yew, N.S.; Wysokenski, D.M.; Wang, K.X.; Ziegler, R.J.; Marshall, J.; Mcneilly, D.; Cherry, M.; Osburn, W.; Cheng, S.H. Optimization of plasmid vectors for high-level expression in lung epithelial cells. Hum. Gene Ther. 1997, 8, 575–584. [Google Scholar] [CrossRef]
- Bago, R.; Pavelić, J.; Vlahoviček, G.M.; Bosnar, M.H. Nm23-H1 promotes adhesion of CAL 27 cells in vitro. Mol. Carcinog. 2009, 48, 779–789. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radić, M.; Šoštar, M.; Weber, I.; Ćetković, H.; Slade, N.; Herak Bosnar, M. The Subcellular Localization and Oligomerization Preferences of NME1/NME2 upon Radiation-Induced DNA Damage. Int. J. Mol. Sci. 2020, 21, 2363. https://doi.org/10.3390/ijms21072363
Radić M, Šoštar M, Weber I, Ćetković H, Slade N, Herak Bosnar M. The Subcellular Localization and Oligomerization Preferences of NME1/NME2 upon Radiation-Induced DNA Damage. International Journal of Molecular Sciences. 2020; 21(7):2363. https://doi.org/10.3390/ijms21072363
Chicago/Turabian StyleRadić, Martina, Marko Šoštar, Igor Weber, Helena Ćetković, Neda Slade, and Maja Herak Bosnar. 2020. "The Subcellular Localization and Oligomerization Preferences of NME1/NME2 upon Radiation-Induced DNA Damage" International Journal of Molecular Sciences 21, no. 7: 2363. https://doi.org/10.3390/ijms21072363
APA StyleRadić, M., Šoštar, M., Weber, I., Ćetković, H., Slade, N., & Herak Bosnar, M. (2020). The Subcellular Localization and Oligomerization Preferences of NME1/NME2 upon Radiation-Induced DNA Damage. International Journal of Molecular Sciences, 21(7), 2363. https://doi.org/10.3390/ijms21072363