Extracellular Vesicles and Tumor-Immune Escape: Biological Functions and Clinical Perspectives




Abstract
1. Introduction
2. Mechanisms of the TEVs-Mediated Immunoescape
2.1. Functional Activation
2.1.1. TEVs and Myeloid-Derived Suppressor Cells
2.1.2. TEVs and Regulatory T cells
2.2. Functional Inhibition
2.2.1. TEVs and Dendritic Cells
2.2.2. TEVs and Natural Killer Cells
2.2.3. TEVs and T lymphocyte Cells
2.3. Functional Polarization
TEVs and Macrophage Polarization
3. Immune Checkpoints and Cancer
3.1. The Immune Checkpoint Inhibitor PD-1
3.2. TEVs as the Carriers of PD-L1
3.3. TEVs as Modulators of PD-L1 Expression in Target Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EVs | Extracellular vesicles |
| TEVs | Tumor-derived extracellular vesicles |
| sEVs | Small extracellular vesicles |
| TME | Tumor microenvironment |
| Tregs | Regulatory T cells |
| MDSCs | Myeloid-derived suppressor cells |
| mRNAs | Messenger RNAs |
| miRNAs | microRNAs |
| lncRNAs | long non-coding RNAs |
| DCs | Dendritic cells |
| NK | Natural killer |
| APCs | Antigen-presenting cells |
| TAMs | Tumor-associated macrophages |
| CTLs | cytotoxic T lymphocytes |
| PD-1 | Programmed cell death 1 |
| PD-L1 | Programmed death-ligand 1 |
References
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Zhou, X.; Fang, M.; Li, H.; Su, P.; Tu, Y.; Zhang, L.; Zhou, F. Extracellular Vesicles in Cancer Immune Microenvironment and Cancer Immunotherapy. Adv. Sci. (Weinh) 2019, 6, 1901779. [Google Scholar] [CrossRef]
- Maji, S.; Chaudhary, P.; Akopova, I.; Nguyen, P.M.; Hare, R.J.; Gryczynski, I.; Vishwanatha, J.K. Exosomal Annexin II Promotes Angiogenesis and Breast Cancer Metastasis. Mol. Cancer Res. 2017, 15, 93–105. [Google Scholar] [CrossRef]
- Chiba, M.; Kubota, S.; Sato, K.; Monzen, S. Exosomes released from pancreatic cancer cells enhance angiogenic activities via dynamin-dependent endocytosis in endothelial cells in vitro. Sci. Rep. 2018, 8, 11972. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.L.; Hu, G.W.; Zhang, B.; Kuang, W.; Chen, Y.; Wu, L.; Xu, G.H. Glioma cells enhance angiogenesis and inhibit endothelial cell apoptosis through the release of exosomes that contain long non-coding RNA CCAT2. Oncol. Rep. 2017, 38, 785–798. [Google Scholar] [CrossRef]
- Emmanouilidi, A.; Paladin, D.; Greening, D.W.; Falasca, M. Oncogenic and Non-Malignant Pancreatic Exosome Cargo Reveal Distinct Expression of Oncogenic and Prognostic Factors Involved in Tumor Invasion and Metastasis. Proteomics 2019, 19, e1800158. [Google Scholar] [CrossRef]
- Li, M.; Lu, Y.; Xu, Y.; Wang, J.; Zhang, C.; Du, Y.; Wang, L.; Li, L.; Wang, B.; Shen, J.; et al. Horizontal transfer of exosomal CXCR4 promotes murine hepatocarcinoma cell migration, invasion and lymphangiogenesis. Gene 2018, 676, 101–109. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, C.; Zhan, Y.; Wang, H.; Jiang, X.; Li, W. Aberrant low expression of p85alpha in stromal fibroblasts promotes breast cancer cell metastasis through exosome-mediated paracrine Wnt10b. Oncogene 2017, 36, 4692–4705. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [PubMed]
- Menck, K.; Sonmezer, C.; Worst, T.S.; Schulz, M.; Dihazi, G.H.; Streit, F.; Erdmann, G.; Kling, S.; Boutros, M.; Binder, C.; et al. Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J. Extracell. Vesicles 2017, 6, 1378056. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Jin, F.; Bian, Z.; Li, L.; Liang, H.; Li, M.; Shi, L.; Pan, C.; Zhu, D.; et al. Pyruvate kinase type M2 promotes tumour cell exosome release via phosphorylating synaptosome-associated protein 23. Nat. Commun. 2017, 8, 14041. [Google Scholar] [CrossRef]
- Yang, L.; Peng, X.; Li, Y.; Zhang, X.; Ma, Y.; Wu, C.; Fan, Q.; Wei, S.; Li, H.; Liu, J. Long non-coding RNA HOTAIR promotes exosome secretion by regulating RAB35 and SNAP23 in hepatocellular carcinoma. Mol. Cancer 2019, 18, 78. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef]
- Cheng, W.C.; Liao, T.T.; Lin, C.C.; Yuan, L.E.; Lan, H.Y.; Lin, H.H.; Teng, H.W.; Chang, H.C.; Lin, C.H.; Yang, C.Y.; et al. RAB27B-activated secretion of stem-like tumor exosomes delivers the biomarker microRNA-146a-5p, which promotes tumorigenesis and associates with an immunosuppressive tumor microenvironment in colorectal cancer. Int. J. Cancer 2019, 145, 2209–2224. [Google Scholar] [CrossRef]
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Thery, C. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012, 72, 4920–4930. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.M.; Carneiro, F.; Machado, J.C.; Melo, S.A. Exosomes and Immune Response in Cancer: Friends or Foes? Front. Immunol. 2018, 9, 730. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Shen, H.; Li, Z.; Wang, T.; Wang, S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J. Hematol. Oncol. 2019, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef]
- Ren, W.; Zhang, X.; Li, W.; Feng, Q.; Feng, H.; Tong, Y.; Rong, H.; Wang, W.; Zhang, D.; Zhang, Z.; et al. Exosomal miRNA-107 induces myeloid-derived suppressor cell expansion in gastric cancer. Cancer Manag. Res. 2019, 11, 4023–4040. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Rayman, P.; Huang, P.; Grabowski, M.; Hambardzumyan, D.; Finke, J.H.; Vogelbaum, M.A. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J. Neurooncol. 2015, 122, 293–301. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, W.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Xue, H.; Li, G. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene 2018, 37, 4239–4259. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, W.; Wang, J.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Guo, Q.; et al. Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int. J. Cancer 2019, 144, 3111–3126. [Google Scholar] [CrossRef] [PubMed]
- Himes, B.T.; Peterson, T.E.; De Mooij, T.; Garcia, M.; Jung, M.Y.; Uhm, S.; Yan, D.; Tyson, J.; Li, H.J.L.; Parney, D.; et al. The role of extracellular vesicles and PD-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Elkord, E.; Alcantar-Orozco, E.M.; Dovedi, S.J.; Tran, D.Q.; Hawkins, R.E.; Gilham, D.E. T regulatory cells in cancer: Recent advances and therapeutic potential. Expert Opin. Biol. Ther. 2010, 10, 1573–1586. [Google Scholar] [CrossRef] [PubMed]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef]
- Muller, L.; Simms, P.; Hong, C.S.; Nishimura, M.I.; Jackson, E.K.; Watkins, S.C.; Whiteside, T.L. Human tumor-derived exosomes (TEX) regulate Treg functions via cell surface signaling rather than uptake mechanisms. Oncoimmunology 2017, 6, e1261243. [Google Scholar] [CrossRef]
- Wada, J.; Onishi, H.; Suzuki, H.; Yamasaki, A.; Nagai, S.; Morisaki, T.; Katano, M. Surface-bound TGF-beta1 on effusion-derived exosomes participates in maintenance of number and suppressive function of regulatory T-cells in malignant effusions. Anticancer Res. 2010, 30, 3747–3757. [Google Scholar]
- Yamada, N.; Kuranaga, Y.; Kumazaki, M.; Shinohara, H.; Taniguchi, K.; Akao, Y. Colorectal cancer cell-derived extracellular vesicles induce phenotypic alteration of T cells into tumor-growth supporting cells with transforming growth factor-beta1-mediated suppression. Oncotarget 2016, 7, 27033–27043. [Google Scholar] [CrossRef]
- Muller, L.; Mitsuhashi, M.; Simms, P.; Gooding, W.E.; Whiteside, T.L. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 2016, 6, 20254. [Google Scholar] [CrossRef]
- Ye, S.B.; Li, Z.L.; Luo, D.H.; Huang, B.J.; Chen, Y.S.; Zhang, X.S.; Cui, J.; Zeng, Y.X.; Li, J. Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal microRNAs in human nasopharyngeal carcinoma. Oncotarget 2014, 5, 5439–5452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Carbone, D.P. Tumor-host immune interactions and dendritic cell dysfunction. Adv. Cancer Res. 2004, 92, 13–27. [Google Scholar] [PubMed]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E.; et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef]
- Huang, S.H.; Li, Y.; Zhang, J.; Rong, J.; Ye, S. Epidermal growth factor receptor-containing exosomes induce tumor-specific regulatory T cells. Cancer Investig. 2013, 31, 330–335. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef]
- Lion, E.; Willemen, Y.; Berneman, Z.N.; Van Tendeloo, V.F.; Smits, E.L. Natural killer cell immune escape in acute myeloid leukemia. Leukemia 2012, 26, 2019–2026. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Schlosser, H.A.; Wang, Z.; Qin, J.; Li, J.; Popp, F.; Popp, M.C.; Alakus, H.; Chon, S.H.; Hansen, H.P.; et al. Tumor-Derived Extracellular Vesicles Inhibit Natural Killer Cell Function in Pancreatic Cancer. Cancers (Basel) 2019, 11, 874. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qin, X.; Zhu, X.; Chen, W.; Zhang, J. Oral cancer-derived exosomal NAP1 enhances cytotoxicity of natural killer cells via the IRF-3 pathway. Oral Oncol. 2018, 76, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef]
- Liu, Z.M.; Wang, Y.B.; Yuan, X.H. Exosomes from murine-derived GL26 cells promote glioblastoma tumor growth by reducing number and function of CD8+T cells. Asian Pac. J. Cancer Prev. 2013, 14, 309–314. [Google Scholar] [CrossRef]
- Maybruck, B.T.; Pfannenstiel, L.W.; Diaz-Montero, M.; Gastman, B.R. Tumor-derived exosomes induce CD8(+) T cell suppressors. J. Immunother. Cancer 2017, 5, 65. [Google Scholar] [CrossRef]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z.; et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 2019, 10, 3000. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Merelli, B.; Massi, D.; Cattaneo, L.; Mandala, M. Targeting the PD1/PD-L1 axis in melanoma: Biological rationale, clinical challenges and opportunities. Crit. Rev. Oncol. Hematol. 2014, 89, 140–165. [Google Scholar] [CrossRef] [PubMed]
- Theodoraki, M.N.; Yerneni, S.S.; Hoffmann, T.K.; Gooding, W.E.; Whiteside, T.L. Clinical Significance of PD-L1(+) Exosomes in Plasma of Head and Neck Cancer Patients. Clin. Cancer Res. 2018, 24, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.W.; Chan, L.C.; Wei, Y.; Hsu, J.M.; Xia, W.; Cha, J.H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Lubin, J.A.; Zhang, R.R.; Kuo, J.S. Extracellular Vesicles Containing PD-L1 Contribute to Immune Evasion in Glioblastoma. Neurosurgery 2018, 83, E98–E100. [Google Scholar] [CrossRef] [PubMed]
- Lux, A.; Kahlert, C.; Grutzmann, R.; Pilarsky, C. c-Met and PD-L1 on Circulating Exosomes as Diagnostic and Prognostic Markers for Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 3305. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weller, C.; Weber, R.; Groth, C.; Riester, Z.; Huser, L.; Sun, Q.; Nagibin, V.; Kirschning, C.; et al. Melanoma Extracellular Vesicles Generate Immunosuppressive Myeloid Cells by Upregulating PD-L1 via TLR4 Signaling. Cancer Res. 2019, 79, 4715–4728. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Kuranaga, Y.; Kumazaki, M.; Sugito, N.; Yoshikawa, Y.; Takai, T.; Taniguchi, K.; Ito, Y.; Akao, Y. Regulated Polarization of Tumor-Associated Macrophages by miR-145 via Colorectal Cancer-Derived Extracellular Vesicles. J. Immunol. 2017, 199, 1505–1515. [Google Scholar] [CrossRef]
- Wang, F.; Li, B.; Wei, Y.; Zhao, Y.; Wang, L.; Zhang, P.; Yang, J.; He, W.; Chen, H.; Jiao, Z.; et al. Tumor-derived exosomes induce PD1(+) macrophage population in human gastric cancer that promotes disease progression. Oncogenesis 2018, 7, 41. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef]
- Chen, F.; Wang, N.; Tan, H.Y.; Guo, W.; Zhang, C.; Feng, Y. The functional roles of exosomes-derived long non-coding RNA in human cancer. Cancer Biol. Ther. 2019, 20, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Conigliaro, A.; Costa, V.; Lo Dico, A.; Saieva, L.; Buccheri, S.; Dieli, F.; Manno, M.; Raccosta, S.; Mancone, C.; Tripodi, M.; et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol. Cancer 2015, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.X.; Liu, H.S.; Wang, F.W.; Xiong, L.; Zhou, C.; Hu, T.; He, X.W.; Wu, X.J.; Xie, D.; Wu, X.R.; et al. LncRNA RPPH1 promotes colorectal cancer metastasis by interacting with TUBB3 and by promoting exosomes-mediated macrophage M2 polarization. Cell Death Dis. 2019, 10, 829. [Google Scholar] [CrossRef]
- Boon, T.; Coulie, P.G.; Van den Eynde, B.J.; Van der Bruggen, P. Human T cell responses against melanoma. Annu. Rev. Immunol. 2006, 24, 175–208. [Google Scholar] [CrossRef]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J.; Mellman, I. Designing vaccines based on biology of human dendritic cell subsets. Immunity 2010, 33, 464–478. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Kooi, S.; Zhang, H.Z.; Patenia, R.; Edwards, C.L.; Platsoucas, C.D.; Freedman, R.S. HLA class I expression on human ovarian carcinoma cells correlates with T-cell infiltration in vivo and T-cell expansion in vitro in low concentrations of recombinant interleukin-2. Cell. Immunol. 1996, 174, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef]
- Brodská, B.; Otevřelová, P.; Šálek, C.; Fuchs, O.; Gašová, Z.; Kuželová, K. PD-1/PD-L1 as a prognostic factor in leukemia. J. Hematopathol. 2017, 10, 17–24. [Google Scholar]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Acuto, O.; Michel, F. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Schwartz, R.H. A cell culture model for T lymphocyte clonal anergy. Science 1990, 248, 1349–1356. [Google Scholar] [CrossRef]
- Schwartz, R.H. T cell anergy. Annu. Rev. Immunol. 2003, 21, 305–334. [Google Scholar] [CrossRef]
- Meyers, J.H.; Sabatos, C.A.; Chakravarti, S.; Kuchroo, V.K. The TIM gene family regulates autoimmune and allergic diseases. Trends Mol. Med. 2005, 11, 362–369. [Google Scholar] [CrossRef]
- Kuchroo, V.K.; Umetsu, D.T.; DeKruyff, R.H.; Freeman, G.J. The TIM gene family: Emerging roles in immunity and disease. Nat. Rev. Immunol. 2003, 3, 454–462. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar]
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin. Cancer Res. 2016, 22, 3057–3066. [Google Scholar] [CrossRef]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 2, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Bercovici, N.; Guerin, M.V.; Trautmann, A.; Donnadieu, E. The Remarkable Plasticity of Macrophages: A Chance to Fight Cancer. Front. Immunol. 2019, 10, 1563. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.S.; Datta, P.K. Targeting the Transforming Growth Factor-β Signaling Pathway in Human Cancer. Expert Opin. Investig. Drugs 2010, 19, 77–91. [Google Scholar] [CrossRef]
- Marti, L.C.; Pavon, L.; Severino, P.; Sibov, T.; Guilhen, D.; Moreira-Filho, C.A. Vascular endothelial growth factor-A enhances indoleamine 2,3-dioxygenase expression by dendritic cells and subsequently impacts lymphocyte proliferation. Memórias do Instituto Oswaldo Cruz 2014, 109, 70–79. [Google Scholar] [CrossRef]
- De Vries, J.E. Immunosuppressive and anti-inflammatory properties of interleukin 10. Ann. Med. 1995, 27, 537–541. [Google Scholar] [CrossRef]
- Lai, Y.S.; Wahyuningtyas, R.; Aui, S.P.; Chang, K.T. Autocrine VEGF signalling on M2 macrophages regulates PD-L1 expression for immunomodulation of T cells. J. Cell. Mol. Med. 2019, 23, 1257–1267. [Google Scholar]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000 Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315. [Google Scholar] [CrossRef]
- Minas, V.; Rolaki, A.; Kalantaridou, S.N.; Sidiropoulos, J.; Mitrou, S.; Petsas, G.; Jeschke, U.; Paraskevaidis, E.A.; Fountzilas, G.; Chrousos, G.P.; et al. Intratumoral CRH modulates immuno-escape of ovarian cancer cells through FasL regulation. Br. J. Cancer 2007, 97, 637–645. [Google Scholar] [CrossRef]
- Maher, S.; Toomey, D.; Condron, C.; Bouchier-Hayes, D. Activation-induced cell death: The controversial role of Fas and Fas ligand in immune privilege and tumour counterattack. Immunol. Cell Biol. 2002, 80, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Figueroa, D.J.; Fay, A.P.; Signoretti, S.; Liu, Y.; Gagnon, R.; Deen, K.; Carpenter, C.; Benson, P.; Ho, T.H.; et al. Correlation of PD-L1 Tumor Expression and Treatment Outcomes in Patients with Renal Cell Carcinoma Receiving Sunitinib or Pazopanib: ResultsfromCOMPARZ, aRandomizedControlled Trial. Clin Cancer Res 2014, 21, 1071–1077. [Google Scholar] [CrossRef]
- Geng, L.; Huang, D.; Liu, J.; Qian, Y.; Deng, J.; Li, D.; Hu, Z.; Zhang, J.; Jiang, G.; Zheng, S. B7-H1 up-regulated expression in human pancreatic carcinoma tissue associates with tumor progression. J. Cancer Res. Clin. Oncol. 2008, 134, 1021–1027. [Google Scholar] [CrossRef]
- Ghebeh, H.; Mohammed, S.; Al-Omair, A.; Qattan, A.; Lehe, C.; Al-Qudaihi, G.; Elkum, N.; Alshabanah, M.; Amer, S.B.; Tulbah, A.; et al. The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressedin breast cancerpatientswith infiltrating ductal carcinoma: Correlation with important high-risk prognostic factors. Neoplasia 2006, 8, 190–198. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Wu, C.; Zhu, Y.; Jiang, J.; Zhao, J.; Zhang, X.G.; Xu, L.N. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem. 2006, 108, 19–24. [Google Scholar] [CrossRef]
- Qing, Y.; Li, Q.; Ren, T.; Xia, W.; Peng, Y.; Liu, G.L.; Luo, H.; Yang, Y.X.; Dai, X.Y.; Zhou, S.F.; et al. Upregulation of PD-L1 and APE1 is associated with tumorigenesis and poor prognosis of gastric cancer. Drug Des. Dev. Ther. 2015, 9, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Yu, Z.; Xiang, R.; Li, C.; Wang, L.; Chen, S.; Li, Q.; Chen, M.; Wang, L. Correlation between infiltration of FOXP3+ regulatory T cells and expression of B7-H1 in the tumor tissues of gastric cancer. Exp. Mol. Pathol. 2014, 96, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Takayama, K.M.K.; Takamori, S.; Kage, M.; Hoshino, T. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmallcell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.Y.; Huang, J.; Chen, Y.; Chen, C.; Zhang, X.G. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med. Oncol. 2011, 28, 682–688. [Google Scholar] [CrossRef]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Wang, H.B.; Yao, H.; Li, C.S.; Liang, L.X.; Zhang, Y.; Chen, Y.X.; Fang, J.Y.; Xu, J. Rise of PD-L1 expression during metastasis of colorectal cancer: Implications for immunotherapy. J. Dig. Dis. 2017, 18, 574–581. [Google Scholar] [CrossRef]
- Lu, C.; Paschall, A.V.; Shi, H.; Savage, N.; Waller, J.L.; Sabbatini, M.E.; Oberlies, N.H.; Pearce, C.; Liu, K. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J. Natl. Cancer Inst. 2016. [Google Scholar] [CrossRef]
- Xie, F.; Xu, M.; Lu, J.; Mao, L.; Wang, S. The role of exosomal PD-L1 in tumor progression and immunotherapy. Mol. Cancer 2019, 18, 146. [Google Scholar] [CrossRef]
- Li, C.; Zhi, C.; Liang, W.; Wang, X.; Chen, X.; Lv, T.; Shen, Q.; Song, Y.; Lin, D.; Liu, H. Clinical significance of PD-L1 expression in serum-derived exosomes in NSCLC patients. J. Transl. Med. 2019, 17, 355. [Google Scholar] [CrossRef]
- Theodoraki, M.N.; Yerneni, S.; Gooding, W.E.; Ohr, J.; Clump, D.A.; Bauman, J.E.; Ferris, R.L.; Whiteside, T.L. Circulating exosomes measure responses to therapy in head and neck cancer patients treated with cetuximab, ipilimumab, and IMRT. Oncoimmunology 2019, 8, 1593805. [Google Scholar] [CrossRef]
- Muller, L.; Muller-Haegele, S.; Mitsuhashi, M.; Gooding, W.; Okada, H.; Whiteside, T.L. Exosomes isolated from plasma of glioma patients enrolled in a vaccination trial reflect antitumor immune activity and might predict survival. Oncoimmunology 2015, 4, e1008347. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Eljaszewicz, A.; Jankowski, M.; Gackowska, L.; Helmin-Basa, A.; Wiese, M.; Kubiszewska, I.; Kaszewski, W.; Michalkiewicz, J.; Zegarski, W. Clinical immunologyGastric cancer increase the percentage of intermediate (CD14++CD16+) and nonclassical (CD14+CD16+) monocytes. Cent. Eur. J. Immunol. 2012, 37, 355–361. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Health-related quality-of-life results for pembrolizumab versus chemotherapy in advanced, PD-L1-positive NSCLC (KEYNOTE-024): A multicentre, international, randomised, open-label phase 3 trial. Lancet Oncol. 2017, 18, 1600–1609. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Ellerton, J.; Infante, J.R.; Agrawal, M.; Gordon, M.; Aljumaily, R.; Britten, C.D.; Dirix, L.; Lee, K.W.; Taylor, M.; et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): Pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol. 2018, 19, 51–64. [Google Scholar] [CrossRef]
- Powles, T.; O’Donnell, P.H.; Massard, C.; Arkenau, H.T.; Friedlander, T.W.; Hoimes, C.J.; Lee, J.L.; Ong, M.; Sridhar, S.S.; Vogelzang, N.J.; et al. Efficacy and Safety of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma: Updated Results From a Phase 1/2 Open-label Study. JAMA Oncol. 2017, 3, e172411. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Chung, H.C.; Ros, W.; Delord, J.P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.J.; Weber, J.S.; et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Shukuya, T.; Carbone, D.P. Predictive Markers for the Efficacy of Anti-PD-1/PD-L1 Antibodies in Lung Cancer. J. Thorac. Oncol. 2016, 11, 976–988. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhang, M.; Li, J.; Young, K.H. PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Front. Immunol. 2017, 8, 1597. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Sung, Y.H.; Pack, C.G.; Jung, M.K.; Han, B.; et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2, eaah5509. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Mandal, G.; Roy Chowdhury, S.; Purohit, S.; Payne, K.K.; Anadon, C.; Gupta, A.; Swanson, P.; Yu, X.; Conejo-Garcia, J.R.; et al. Exosomes Produced by Mesenchymal Stem Cells Drive Differentiation of Myeloid Cells into Immunosuppressive M2-Polarized Macrophages in Breast Cancer. J. Immunol. 2019, 203, 3447–3460. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
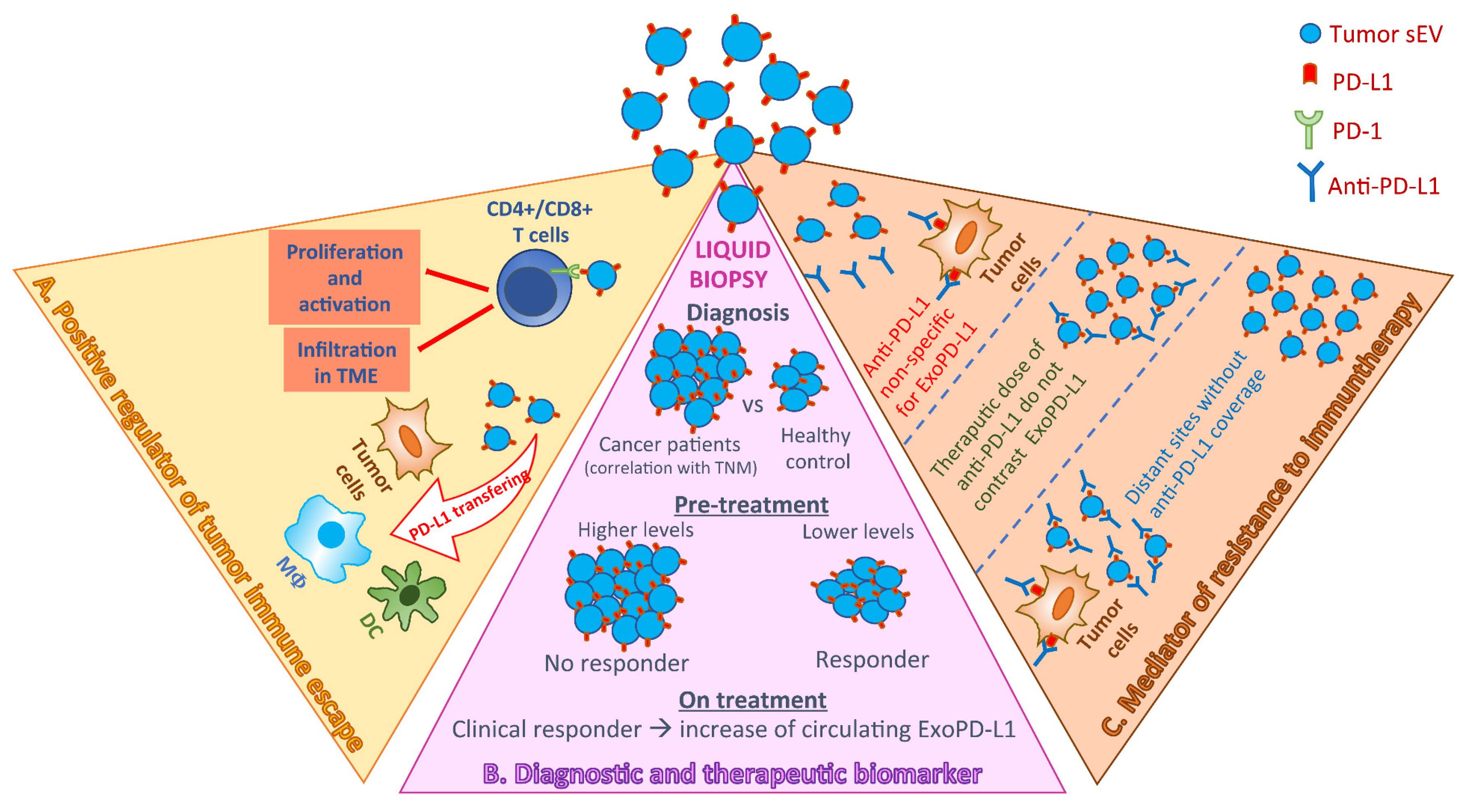{kind=link}
| Tumor Models | TEV Cargo | Target Cells | Biological Effects | References |
|---|---|---|---|---|
| Functional Activation | ||||
| Breast cancer | PGE2 and TGF-β | MDSCs | Tumor growth promotion by inducing the release of IL-6, Cox2, and VEGF | [25] |
| Breast, colon cancer and lymphoma | Hsp72 | MDSCs | Immunosuppressive capacity promotion by activating STAT3 pathway | [26] |
| Gastric cancer | miR-107 | MDSCs | Cell expansion and activation by targeting DICER1 and PTEN | [28] |
| Glioma | miR-10a and miR-21 | MDSCs | Cell functions activation by targeting RAR-related orphan receptor alpha (RORA) and PTEN | [30] |
| Glioma | miR-29a and miR-92a | MDSCs | Increased proliferation and immunosoppressive function promotion | [31] |
| Ovarian cancer | TGF-β1 and IL-10 | Treg | Increased immunosuppressive capacity by expressing IL-10, TGF-β1, CTLA-4, granzyme B | [36] |
| Colorectal cancer | TGF-β1 | Treg | Up-regulation of Treg-related genes by activating SMAD pathway | [39] |
| Functional Inhibition | ||||
| Lung cancer | EGFR | DCs | Tolerogenic induction | [47] |
| Mesothelioma | NKG2D ligands and TGF-β | NK and CD8+ | Cell exhaustion through NKG2D activating receptors down-regulation | [48] |
| Pancreatic cancer | TGF-β, nectin-2 and PVR | NK | Cell functions impairment by down-regulating NKG2D, CD107a, TNF-α, and INF-γ expression | [52] |
| Oral cancer | NAP-1 | NK | Increased cell toxicity by activating the interferon regulatory factor 3 (IRF-3) pathway | [53] |
| Head and neck cancer, melanoma | FasL and MHC class I molecules | CD8+ | Cell expansion inhibition and cell death induction | [56] |
| Head and neck cancer | Galectin-1 | CD8+ | Cell dysfunction induction | [58] |
| Ovarian carcinoma | Arginase I | CD4+ and CD8+ | Cell proliferation inhibition | [59] |
| Gliobastoma | PD-L1 | CD4+ and CD8+ | Cell activation and proliferation inhibition | [63] |
| Breast cancer | PD-L1 | CD4+ and CD8+ | Cell activation inhibition | [64] |
| Functional Polarization | ||||
| Colorectal cancer | miR-145 | macrophages | Tumor growth promotion through M2-like macrophage polarization induction | [72] |
| Melanoma, skin, lung cancer | CSF-1, CCL2, FTH, FTL, TGFβ | macrophages | Tumor growth promotion through M2-like macrophage polarization induction | [74] |
| Colorectal cancer | lncRNA RPPH1 | macrophages | Tumor growth promotion through M2-like macrophage polarization induction | [77] |
| Glioblastoma | STAT3 pathway activator | monocytes | Tumor growth promotion through M2-like macrophage polarization induction | [69] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raimondo, S.; Pucci, M.; Alessandro, R.; Fontana, S. Extracellular Vesicles and Tumor-Immune Escape: Biological Functions and Clinical Perspectives. Int. J. Mol. Sci. 2020, 21, 2286. https://doi.org/10.3390/ijms21072286
Raimondo S, Pucci M, Alessandro R, Fontana S. Extracellular Vesicles and Tumor-Immune Escape: Biological Functions and Clinical Perspectives. International Journal of Molecular Sciences. 2020; 21(7):2286. https://doi.org/10.3390/ijms21072286
Chicago/Turabian StyleRaimondo, Stefania, Marzia Pucci, Riccardo Alessandro, and Simona Fontana. 2020. "Extracellular Vesicles and Tumor-Immune Escape: Biological Functions and Clinical Perspectives" International Journal of Molecular Sciences 21, no. 7: 2286. https://doi.org/10.3390/ijms21072286
APA StyleRaimondo, S., Pucci, M., Alessandro, R., & Fontana, S. (2020). Extracellular Vesicles and Tumor-Immune Escape: Biological Functions and Clinical Perspectives. International Journal of Molecular Sciences, 21(7), 2286. https://doi.org/10.3390/ijms21072286