Abstract
Alveolar epithelial type II cells (AT2) are a heterogeneous population that have critical secretory and regenerative roles in the alveolus to maintain lung homeostasis. However, impairment to their normal functional capacity and development of a pro-fibrotic phenotype has been demonstrated to contribute to the development of idiopathic pulmonary fibrosis (IPF). A number of factors contribute to AT2 death and dysfunction. As a mucosal surface, AT2 cells are exposed to environmental stresses that can have lasting effects that contribute to fibrogenesis. Genetical risks have also been identified that can cause AT2 impairment and the development of lung fibrosis. Furthermore, aging is a final factor that adds to the pathogenic changes in AT2 cells. Here, we will discuss the homeostatic role of AT2 cells and the studies that have recently defined the heterogeneity of this population of cells. Furthermore, we will review the mechanisms of AT2 death and dysfunction in the context of lung fibrosis.
1. Introduction
Idiopathic Pulmonary Fibrosis (IPF) is a chronic, progressive scarring of the lungs that causes significant healthcare burden due to high morbidity and mortality [1,2,3]. It is the most common form of idiopathic interstitial pneumonia and exhibits the most severe manifestations carrying the poorest clinical outcomes [4,5,6]. Recently, two anti-fibrotic medications, Pirfenidone and Nintedanib, have been demonstrated to slow the progression of the disease and are now FDA-approved treatments for IPF [7,8,9,10,11,12,13,14,15]. Despite the progress made after decades of investigation, there is considerable room for improvement in the treatment of this incurable disease.
Pathological fibrogenesis that occurs in IPF is a dynamic process involving complex interactions between epithelial cells, fibroblasts, immune cells (macrophages, T-cells), and endothelial cells [16,17]. Early theories that chronic inflammation and repetitive damage to the alveolar epithelium promotes fibrogenesis and scar formation in the pathogenesis of lung fibrosis [18] have been largely rejected due to the lack of ongoing inflammation in IPF and the general ineffectiveness of immunosuppressive medications in curbing disease progression. In contrast, a preponderance of more recent evidence suggests that the alveolar epithelium plays a central role. Indeed, honeycombed regions of the lungs have discontinuous epithelium adjacent to hyperplastic alveolar epithelial type II cells (AT2) [19]. Accordingly, the more contemporary paradigm is that chronic injury to distal lung tissue leads to either loss or altered function of epithelial stem cells (i.e., AT2 cells), that promote dysregulated repair and pathogenic activation of fibroblasts.
Both intrinsic (e.g., genetic, aging) and environmental factors have been linked to damage of AT2 cells and contribute to the development of lung fibrosis. Tobacco smoking and viral infections have been associated with IPF [4,20]. IPF is also an age-related disease with a median age of diagnosis of 66 years old [21]. As such, several factors that accumulate with age have been found to contribute to AT2 dysfunction in IPF [22]. However, the most direct evidence supporting a role for AT2 cell dysfunction in the initiation and progression of IPF has come from the characterization of gene defects observed among patients with familial forms of the disease. Two general categories of gene mutations are observed; one category includes genes involved in the regulation of stem cell longevity, the other includes genes whose products contribute to specialized secretory functions of AT2 cells [23].
Altogether, the evidence supports a model in which AT2 injury/dysfunction serves as an early initiating event in IPF that leads to fibroproliferation and progressive loss of lung function [24]. AT2 loss can limit the ability for the repair of the damaged alveolus. Additionally, AT2 cells have maladaptive effects in the IPF lung that can drive fibrosis (Figure 1). Herein, we will review the homeostatic role of the AT2 cell and evidence for both AT2 depletion and dysfunction as contributors to IPF.
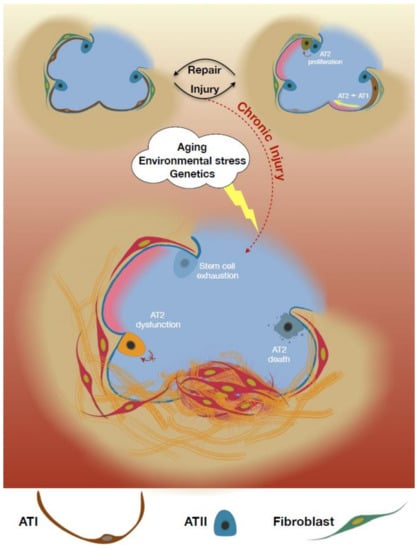
Figure 1.
Alveolar epithelial type II cells (AT2) functions in lung fibrosis. An overview schematic illustration described functional roles of AT2 cells to maintain lung homeostasis during injuries and their profibrotic phenotypic changes that promote lung fibrosis upon the presence of risk factors e.g., aging, environmental stress, etc.
2. Alveolar Epithelial Type II Cells (AT2) of the Normal Mammalian Lung
The epithelium lining airspaces of the mammalian lung is maintained by regional stem and progenitor cells that are responsible for replacement of functionally specialized cell types in each compartment during homeostasis and repair [25,26]. In the alveolus, AT2 cells serve as the predominant epithelial progenitor. Lineage tracing experiments in mice show that AT2 cells defined by their expression of surfactant protein C (Sftpc/SFTPC) are capable of long-term self-renewal and multipotent differentiation to yield alveolar type I (AT1) cells; two characteristics that suggest either the AT2 population as a whole or a subset of AT2 cells, represent adult tissue stem cells [27]. The observation in this study that a subset of Sftpc-lineage positive AT2 cells exhibit greater in vivo clonogenic potential than bulk AT2 cells provides evidence that “stemness” may be a property of a subset of AT2 cells, and by inference, that AT2 cells are functionally heterogeneous. What is not clear from this work is whether AT2 cell heterogeneity is the result of differences in intrinsic potential versus microenvironmental regulation. Recent work defining a Wnt-responsive subset of AT2 cells raises the potential that a Wnt signaling microenvironment may modulate “stemness” of AT2 cells [28,29]; an appealing concept but currently not confirmed by lineage-tracing experiments. However, these data do support the notion that AT2 cells represent a heterogeneous population that includes an abundant pool of facultative progenitors and a more stem-like subset that contributes to homeostatic replacement of specialized alveolar epithelial cells.
Other epithelial stem or progenitor cells have been proposed to contribute to alveolar epithelial renewal and replacement of AT2 cells. So-called bronchio-alveolar stem cells (BASCs) were initially proposed as a multipotent “stem” cell in mouse terminal bronchioles based upon their co-expression of Sftpc and Scgb1a1, contribution to repair after chemical injury and expansion following induction of activating mutations of the K-ras oncogene [30]. Evidence supporting the multipotency and “stemness” of BASCs includes recent studies involving use of dual lineage-tracing strategies demonstrating that Sftpc/Scgb1a1 dual lineage-labeled cells have potential to contribute to repair following injury to either airways or alveoli [31,32]. However, multipotency of BASCs has currently only been convincingly demonstrated in clonal culture experiments [33], with in vivo confirmation of multipotency confounded by heterogeneity within the lineage-labeled population [32]. Other candidate progenitors for replacement of AT2 cells and the alveolar epithelium include a rare subpopulation of AT1 cells [34] and basal cells that expand to repopulate damaged alveoli following influenza virus infection [35].
3. AT2 Depletion in Idiopathic Pulmonary Fibrosis (IPF)
A prevailing concept is that AT2 depletion potentially through repetitive microinjuries is the underlying cause of lung fibrosis [5]. Indeed, targeted deletion of AT2 cells is sufficient to induce a fibrotic response in the lungs, but it is not sustained [36,37]. Additional data supporting this idea of stem cell exhaustion is that the number of AT2 cells are diminished in IPF lungs [38,39]. However, the depletion of this vital progenitor cell is not only a function of the distorted lung architecture but may also be a precursor to fibrosis [39].
Apoptotic Death of AT2 Cells
Mechanisms of AT2 depletion particularly in the context of early events leading to IPF are not fully elucidated. Clearly, the lung epithelium is under constant stresses as a number of studies have identified increased levels of cells undergoing apoptosis in the lungs from IPF patients, which is not seen in non-fibrotic lungs [40,41,42,43]. In fact, TGFβ, a potent pro-fibrotic cytokine, has been demonstrated to mediate its fibroproliferative effects by induction of AT2 apoptosis [44,45]. Apoptosis is a form of programmed cell death that is required as a physiological process for tissue homeostasis but can also be activated in pathological situations [46]. This regulated cell death pathway is tightly controlled at multiple levels, but if the stimulus exceeds a critical threshold, a prescribed pattern of events mediated by caspase cleavage of cellular contents to induce cell death.
In IPF lungs, the Fas-FasL pathway has several components that are upregulated in the alveolar epithelium indicating a preexisting elevation in the signals that induce apoptosis [42,47]. Similar findings were demonstrated in murine models of fibrosis [48]. Global inhibition of apoptosis with captopril or z-VAD also blunts lung fibrosis [49,50]. Additionally, blockade of Fas signaling through various pharmacologic or genetic methods attenuates whereas Fas stimulation augments lung fibrosis in bleomycin-injured mice [51,52]. Many cells that succumb to Fas-mediated death must undergo an amplification loop (type II pathway) that intensifies the death signal by crossing over to the mitochondria through the cleave of BID, a B-cell lymphoma protein-2 (BCL2) family member [53]. Further supporting the importance of the Fas-FasL pathway and apoptosis in general as an important early initiator of lung fibrosis, mice genetically deficient in Bid are protected from bleomycin-induced lung fibrosis [45].
4. ER Stress Contributes to AT2 Apoptosis
In addition to receptor-mediated activation of apoptosis, an intrinsic apoptotic pathway can also induce programmed cell death [46]. Various intracellular stresses and damage signals activate pro-apoptotic BCL-2 proteins that permeabilize the mitochondria to release mediators (e.g., cytochrome C) that create a nidus for apoptosome formation and activation of executioner caspases that cause apoptotic death [54].
Endoplasmic reticulum (ER) stress is an important initiator of AT2 apoptosis via the intrinsic pathway that has been linked to IPF [55]. The ER is an important cellular organelle that facilitates the folding and trafficking of proteins to ensure the quality control of proteins required for cellular homeostasis. In situations that overwhelm the protein folding capacity of the ER, the unfolded protein response (UPR) is activated with the aim to restore the physiological activity of the ER. Three transmembrane sensors, specifically inositol-requiring enzyme 1α (IRE1α), pancreatic endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6), control the UPR [56]. When the UPR is prolonged or severe in nature, proteostasis is lost, and cells become dysfunctional and undergo apoptotic death [55].
Several studies have identified UPR activation in AT2 cells during lung fibrosis [57,58,59]. Immunohistochemical evaluation of lungs from sporadic and familial IPF demonstrated AT2 co-localization of various ER stress markers and activated caspase-3 [57,58]. Interestingly, AT2 activation of the UPR can be found in histologically normal appearing regions of IPF lungs suggesting ER stress precedes the development of fibrosis. Mice injured with bleomycin also demonstrate ER stress in AT2 cells [59]. More importantly, tunicamycin, an activator of ER stress, augments lung fibrosis [59,60], and specific activation of ER stress in AT2 cells can lead to spontaneous lung fibrosis in transgenic mice [61,62].
Several genetic variants that confer an inherited susceptibility to lung fibrosis can induce ER stress. In particular, pathologic variants of surfactant proteins, which are produced by AT2 cells, have been found to induce ER stress and augment lung fibrosis [23]. In experimental models with transgenic mice that conditionally overexpressed the SFTPC mutation (SFTPCL188Q) in AT2 cells, ER stress increased after the induction of the mutant SFTPCL188Q expression [59]. Although these transgenic mice did not develop spontaneous pulmonary fibrosis, they were more susceptible to bleomycin exposure and the resultant injury-induced fibrosis. In contrast, a different SFTPC mutation (SFTPCC121G) demonstrated exaggerated ER stress, AT2 apoptosis, and development of spontaneous lung fibrosis after induction of expression in AT2 cells [61].
With aging, the lungs also become more susceptible to ER stress [55]. GRP78 is a chaperone protein that is a central regulator of ER stress by repressing IRE1α, PERK, and ATF6, the three arms that initiate the UPR. A recent study by Borok et al. demonstrated that AT2 expression of GRP78 decreases in both aged mice and in IPF lungs [63]. AT2-specific deletion of Grp78 induced ER stress, apoptosis and lung fibrosis with an age-dependence on the severity of the effect.
A number of environmental factors that are associated with the development of IPF has also been found to induce ER stress. Smoking, which induces ER stress within the lung epithelium, is associated with an increased risk factor for acquiring IPF [64,65]. Several herpesvirus proteins were identified in AT2 cells that concomitantly showed evidence of UPR activation [20,58]. A recent study also found evidence for increased herpesvirus infections in at-risk subjects for developing interstitial lung disease [66]. Fibrosis can be augmented in mice infected with γherpesvirus [67,68,69,70]. Moreover, the viral susceptibility appears be age-related with increased AT2 ER stress and apoptosis, as well as augmented lung fibrosis in old compared to young mice infected with murine γherpesvirus [60,71].
5. Mitochondrial Dysfunction Causes AT2 Death
AT2 cells have the highest number of mitochondria in the lungs due to their high metabolic demands, especially during lung injury and repair [72]. Disturbance and interference of AT2 mitochondrial biogenesis, functions, and homeostasis is a known profibrotic signal [73,74,75]. The imbalance of mitochondrial dynamic due to impaired mitophagy and accumulation of mtDNA damage or irregularities of protein homeostasis leads to ER stress and program cell death of AT2 cells [74,76]. An accumulation of higher dysmorphic mitochondria (i.e., damaged mitochondria) and ER stress proteins in AT2 cells in IPF compared to control lungs suggests the significance of mitochondria dysfunction in the pathogenesis of lung fibrosis [60,77].
Mitochondrial homeostasis is regulated by integrated pathways primarily for biogenesis and recycling/disposal through mitophagy [78]. Mitofusin 1 and 2 are GTPases required for mitochondrial fusion, and AT2 deletion of either gene augmented lung fibrosis after bleomycin injury [79]. Moreover, combined AT2 deletion of both mitofusin 1 and 2 lead to spontaneous lung fibrosis. Transgenic mice expressing mitochondrial-targeted catalase (MCAT) are also protected from asbestos- and bleomycin-induced lung fibrosis mediated through the inhibition of AT2 cell mtDNA damage and apoptosis [80]. Corroborating with these data, the deficiency of sirtuin 3, a NAD-dependent deacetylase that prevents mtDNA damage, in AT2 cells promoted lung fibrosis by inducing cells apoptosis [81].
PTEN-induced putative kinase 1 (PINK1), a mitochondrial factor that facilitates mitophagy, is depleted during ER stress, with aging, and within fibrotic lungs [60,82,83]. AT2 cells in Pink1 deficient mice are morphologically similar to AT2 cells in IPF lung, [60]. Furthermore, bleomycin-induced lung fibrosis is augmented in Pink1 deficient mice and associated with less mitophagy, accumulation of dysmorphic mitochondria, and increased ER stress and AT2 apoptosis [60,77]. Activating transcription factor 3 (Aft3), a Pink1 transcription repressor, has higher expression in fibrotic and aged lungs, suggesting this factor may account for the concomitant depletion of Pink1 in similar conditions [60,82]. Accordingly, conditional AT2 deletion of Aft3 protects mice from developing lung fibrosis [82]. Further demonstrating the importance of mitophagy in mediating IPF, a recent finding demonstrated a dysfunctional thyroid axis in IPF, and thyroid hormone administration increased Pink1 levels, attenuated AT2 apoptosis, and reduced lung fibrosis via a Pink1-dependent mechanism [83].
A conflicting finding recently emerged where Pgam5 deficient mice had decreased mitophagy that improved mitochondrial homeostasis, which had a protective effect during bleomycin-induced lung fibrosis [84]. Because these findings were in global Pgam5 deficient mice, one possible explanation for the contradictory results is that mitophagy may potentially have profibrotic roles in non-AT2 compartments. Nevertheless, the majority of the evidence suggests mitophagy has a protective effect in AT2 cells and in preventing lung fibrosis [60,77,82,83].
6. AT2 Dysfunction in IPF
Persistent injury to the AT2 compartment not only causes a depletion of these facultative progenitor cells but can also cause irreversible alterations the capacity of these vital cells in carrying out their reparative functions. Indeed, hypertrophic and hyperplastic AT2 cells are found in the fibroblastic foci and have impaired renewal capacity [85]. Furthermore, dysfunctional AT2 cells in the fibrotic lung also produce pro-fibrotic factors that contribute to fibrogenesis [24]. Altogether, AT2 cells are not just simply depleted in the fibrotic lung as collateral damage to ongoing injury, but in addition, these cells have acquired a dysfunctional phenotype that places it as a central driver of fibrosis [37] as depicted in Figure 2.
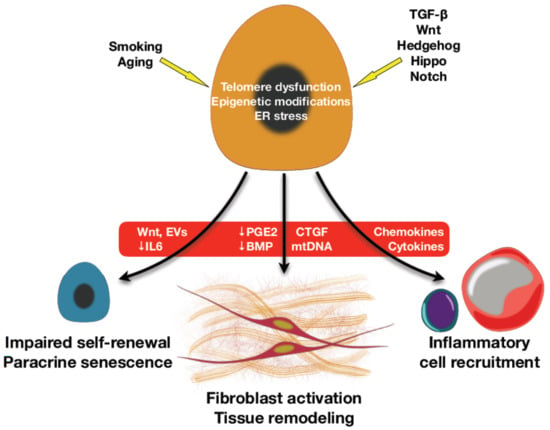
Figure 2.
Mechanisms and regulatory signaling of alveolar type II epithelial cells (AT2) dysfunction in lung fibrosis. In lung fibrosis, environmental or intrinsic factors and several signaling regulatory pathways e.g., TGF-β, Wnt, Hedgehog, etc. stimulate AT2 senescence to release senescence-associated secretory phenotype (SASP) and other mediators that can directly activate fibroblasts and tissue remodeling. The indirect effects of SASP include inflammatory cell recruitment and AT2 self-renewal exhaustion through a paracrine effect.
AT2 plasticity has been best demonstrated by the ability of TGFβ to alter their cellular phenotype [86,87,88]. Although epithelial cells do not directly contribute to the mesenchymal population in IPF [89], what has become evident is that the AT2 cells develop a fundamentally altered state in the IPF lung and acquire a distinctly pro-fibrotic phenotype that promotes expansion of the mesenchymal compartment with myofibroblast activation and matrix deposition [90]. In one respect, the inability of AT2 cells to carry out proper repair of the injured alveolus can lead to scar formation [85]. In fact, AT2 cells play a central role in the activation of TGFβ, which may be self-perpetuating through the rising tension within a fibrotic lung [91]. Additionally, AT2 cells have diminished capacity for transdifferentiation into AT1 cells in the IPF lungs [5]. As such, both ER stress and telomere dysfunction, both of which have the potential to activate a program of cellular senescence, have been found to impair differentiation by stem cells [92,93,94,95,96]. Emerging evidence also demonstrates that induction of AT2 senescence in itself is sufficient to promote lung fibrosis [37].
7. Impaired AT2 Self-Renewal in IPF
Cellular senescence, one of the signature characterizations of the aging process, plays an important role in the pathogenesis of lung fibroproliferative disorders [97,98,99]. Fibrotic regions of lung tissue from IPF patients show both regional depletion of AT2 cells and the presence of AT2 cells with a senescent-like phenotype [37,38]. As such, AT2 senescence may have a limited reparative capacity that contributes to the fibroproliferation [22,100]. In support of this concept, senolytics show some potential as anti-fibrotics in animal models [101,102,103]. Moreover, ER stress may not only contribute to IPF through UPR-mediated AT2 death but can also impair the renewal capacity of AT2 cells by inducing cellular senescence [63]. This effect appears to be ubiquitous as the regenerative capacity of multiple types of stem cells is impaired with ER stress [92].
A major contributor to cellular senescence in lung fibrosis is telomere dysfunction [93,104]. Telomeres protect the ends of chromosomes from replicative loss through providing a mechanism for telomerase-dependent repeat expansion, thus maintaining the proliferative potential of stem and progenitor cells [105]. Shortened telomere length has been found in a subset of patients with IPF and correlates with poor survival [66,106,107]. Smoking, a risk factor for IPF, also causes a dose-dependent shortening of telomeres [108]. Similarly, telomere attrition also occurs with aging, another risk for the development of IPF [104].
A number of variants in genes that regulate telomere function has been found in IPF and could be contributing to AT2 senescence and impairment in their renewal capacity [23]. Mutations in TERC and TERT, telomerase reverse transcriptase family genes that regulate telomere length and functions, are linked to IPF [107,109,110,111,112]. A null TERT allele conferred a dominant transmission of disease in a familial form of pulmonary fibrosis (FIP) [109]. Risk variants of regulator of telomere elongation helicase 1 (RTEL1), a DNA helicase necessary for telomere stability, is also linked to FIP [113,114,115]. Subsequent studies have also found TERT and RTEL1 mutations to be enriched in patients with IPF [116]. Moreover, a number of other genetic variants in DKC1 [117], PARN [115,116], NAF1 [118], and TINF2 [119,120] have been found in FIP and associated with short telomeres. Various animal models have also demonstrated the impact of telomeres in lung fibrosis. Mice with AT2-specific Tert deficiency did not develop spontaneous lung fibrosis but upon bleomycin injury had more AT2 senescence and more lung fibrosis [94]. In contrast, conditional AT2 deletion of telomere repeat-binding factor 1 (TRF1), a telomere shelterin protein, caused mice to develop spontaneous lung fibrosis due to an impairment of telomere integrity [95,121].
Several non-cell autonomous factors also contribute to AT2 health and self-renewal capacity [122]. Indeed, the extracellular matrix (ECM) also has a role in the maintenance of AT2 cells [122], and these cell-matrix interactions recently were revealed to be dysfunctional in IPF, diminishing the renewal capacity of these stem cells [39]. Hyaluronan (HA) is a glycosaminoglycan that has increased abundance in the IPF ECM and can promote fibroblast invasiveness to mediate progressive lung fibrosis [123,124]. Interestingly, HA is expressed on the cell surface of normal AT2 cells and promotes stem cell renewal in a Toll-like receptor 4-dependent manner through the release of IL-6 [39]. AT2 cells also have a deficiency in HA signaling during lung fibrosis, leading to IL-6-dependent reductions in their renewal capacity [39]. Furthermore, targeted deletion of hyaluronan synthase 2 in murine AT2 cells leads to loss of progenitor cell functions and increased susceptibility to bleomycin-induced fibrosis [39].
8. Dysregulated Signaling Pathways Causing Intrinsic AT2 Dysfunction in IPF
Advances in single genomics have provided unprecedented opportunities to reveal changes in cellular states during normal development, tissue homeostasis, and disease. As applied to IPF patient samples, these studies have demonstrated reactivation of a series of key developmental pathways (e.g., Wnt, Hippo, Hedgehog, Notch) and the induction of altered cell states (senescence, apoptosis), that provide insights into mechanisms of altered AT2 function [38,125,126,127]. The Wnt pathway is a particularly prominent signal that is reactivated in fibrotic lungs [125,126,127,128,129,130]. Although Wnt signaling is necessary for maintaining AT2 self-renewal in development [28,29,131], the aged lung has a maladaptive response where Wnt induces cellular senescence [132,133]. As such, inhibition of Wnt signaling improves lung fibrosis in bleomycin-challenged mice [134,135,136]. One of the key AT2 functions is to transdifferentiate into AT1 cells. Because of the essential AT1 role in gas exchange, AT2 self-renewal is an important feature of to provide an endless reparative population that is ready to respond to any injury that damages the alveolar surface [137]. Wnt signaling is a developmental pathway that is activated in adult AT2 cells to drive AT1 transdifferentiation [138]. However, Wnt signaling is largely established as a nefarious event in IPF [128,129,130]. One possible way to reconcile these divergent observations is that the activated Wnt pathways are a sign of ongoing repair and regeneration of damaged alveoli [139]. Another more likely explanation is that homeostatic signals become maladapted in pathological situations with Wnt signaling causing damaging effects, such as induction of AT2 senescence [132,133].
Hippo signaling, which primarily mediates its signaling via Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), is an evolutionarily conserved pathway that crosses over with Wnt signaling [140,141]. TAZ is expressed by embryonic epithelium and is necessary for branching morphogenesis and alveolarization by promoting AT1 differentiation [142,143,144,145,146,147]. YAP/TAZ activity is an important transducer of extracellular cues that has important effects in fibrotic lung diseases [148]. In particular, fibroblasts use this mechanosensing pathway to promote a fibrotic phenotype in the fibrotic lung [149,150,151]. A prediction would be that the epithelium may also alter their phenotype in the IPF as a result of the increased stiffness within their microenvironment. Indeed, Hippo signaling is dysregulated in AT2 cells in the IPF lung to alter cell shape, proliferation, and migration as a possible contributor to lung fibrosis [152].
Notch is another developmental control pathway that regulates cell differentiation and fate through mechanisms involving lateral inhibition [153]. Upon ligand binding, Notch receptors (Notch 1 through 4) are cleaved by γ-secretase, which releases the Notch intracellular domain (NICD) that translocates to the nucleus and induces target gene (e.g., HES, HEY) transcription [154]. Although Notch has distinct roles in distal lung development, AT2 cells also require Notch signaling to coordinate crosstalk with myofibroblasts for proper alveologenesis [155,156]. Reactivation of Notch signaling has been found to be profibrotic in skin, kidney, and cardiac fibrosis [157], and Notch pathways are re-activated in AT2 cells within the IPF lungs [125,158]. Interestingly, alveolar differentiation is inhibited with persistent Notch signaling, which led to a failure to regenerate the damaged alveolus and development of honeycombed cysts that are akin to the pathology found in IPF [159]. Furthermore, Notch activation in AT2 cells induces, whereas Notch inhibition attenuates lung fibrosis [158].
9. Altered AT2-Fibroblast Signaling in IPF
AT2-fibroblast interactions are tightly coordinated in lung development, and this unit works in conjunction after injury to repair a damaged alveolus [29,155,156,160]. This epithelial-mesenchymal crosstalk is clearly disrupted in IPF such that AT2 cells acquire a profibrotic phenotype to aberrantly secrete profibrotic mediators as paracrine factors that stimulate and activate fibroblasts [24]. In fact, TGFβ is predominantly produced by the epithelium where the integrins needed for activation of the latent form is primarily expressed, and abrogation of TGFβ signaling within the lung epithelium can attenuate lung fibrosis [91,161,162,163,164]. CTGF is produced by the lung epithelium in response to TGFβ signaling and plays a vital profibrotic role by stimulating collagen deposition by fibroblasts [165,166,167]. AT2 cells have increased expression of CTGF in bleomycin-induced lung fibrosis, whereas blockade can abrogate fibrosis [167,168,169]. Hippo signaling (via YAP) also induces activation of target genes such as CTGF and could partially explain how the reactivation of this developmental pathway in IPF contributes to the disease [152]. Release of mtDNA from damaged AT2 cells may also contribute to fibroblast activation in IPF [76].
Hedgehog signaling is a key regulator of the epithelial-mesenchymal interactions during development and fibrosis [127,160,170]. Hedgehog is regulated by the patched surface receptors and upon ligand binding releases smoothened to activate the Gli-family transcription factors [171]. This pathway is active in the alveolar epithelium during alveolarization, after injury, and in tumors as a means to crosstalk with mesenchymal cells and coordinate their activity [152,170]. In the adult lung, hedgehog signaling controls the epithelial-mesenchymal unit by maintaining fibroblast quiescence at homeostasis and during resolution after injury [160]. Pathological reactivation of hedgehog signaling in the alveolar compartment occurs in IPF and in bleomycin-induced lung fibrosis, and pharmacologic and genetic blockade of the hedgehog epithelial-fibroblast crosstalk can attenuate experimental lung fibrosis [127,172,173,174,175,176,177,178]. In contrast, it was recently demonstrated that mice deficient in Gli1 were not protected from lung fibrosis suggesting either Gli2 or non-canonical hedgehog signaling may be regulating pathological effects in lung fibrosis [179].
The epithelium has defined properties that appear to keep the mesenchymal compartment in check at homeostasis [160,180,181,182,183]. Prostaglandin E2 (PGE2) levels are suppressed in IPF and have been identified as an important epithelial factor that suppresses fibroblast proliferation and activation [183,184,185]. Recently, bone morphogenic protein (BMP) was also identified to mediate a similar inhibitory effect on fibroblasts [186]. Thus, AT2 cells not only promote fibrosis through release of profibrotic mediators, but the loss of negative reinforcement factors such as PGE2 and BMP can contribute to fibroproliferation.
10. The Senescence-associated Secretory Phenotype Has Pro-fibrotic Effects
AT2 senescence plays a prominent role in lung fibrosis through the acquisition of a senescence-associated secretory phenotype (SASP) and is a feature of senescent cells that result in the release of a myriad of factors with inflammatory and fibrogenic properties [187]. AT2 cells within the lungs of IPF patients and bleomycin-injured mice gain features consistent with a SASP [37,38,93,125,126]. Indeed, a number of secreted inflammatory and profibrotic factors are released from AT2 cells within the fibrotic lung that can have autocrine and paracrine actions within the fibrotic microenvironment to promote lung fibrosis [24].
Several genetic variants have been found to be associated with lung fibrosis and could be mediating fibrosis through the development of the SASP in AT2 cells. Telomere-mediated senescence causes the SASP [93], and it is plausible that the various telomerase mutations associated with lung fibrosis mediate their pro-fibrotic effects by inducing a SASP in AT2 cells [107,109,110,111,112]. Hermansky–Pudlak syndrome (HPS) is an autosomal recessive disease that causes oculocutaneous albinism, bleeding diathesis, and lung fibrosis [188]. AT2 produced excessive monocyte chemotactic protein-1 (MCP-1, also known as C-C motif ligand 2 (CCL2)) that in turn promoted macrophage recruitments and TGFβ production leading to lung fibrosis [189]. This finding is consistent with the acquisition of a SASP. A SFTPC genetic variant associated with IPF (SFPTCI73T) also induces AT2 secretion of several chemokines, which are consistent with a SASP, to recruit Ly6ChiCD64- monocytes [62]. Although not evaluated, this constellation of findings suggests a SASP may also be regulating the development of the spontaneous fibrosis found in this murine model.
Interestingly, a “bystander effect” is found with cellular senescence in which paracrine factors can induce neighboring cells to undergo cellular senescence [190,191,192]. Notch signaling can also promote the transfer of the senescent phenotype, which makes one posit if Notch activation in IPF may be perpetuating cell senescence in neighboring cells [193,194,195]. Extracellular vesicles (EVs) are released from cells and carry a number of factors including proteins, nucleic acids, and lipids as cargo that can be delivered to distal cells as a means of intercellular communication [196]. EVs not only change quantity and quality with cell senescence but can also play a role in causing senescence through a variety of cargo [197]. Recent evidence demonstrates increased EV production in IPF lungs could potentially be produced by senescent cells; Moreover, these EVs carry cargo such as interferon-induced transmembrane protein 3 to promote paracrine senescence [198]. EV quantify increases in the IPF lungs, and EVs isolated from fibrotic lungs can augment fibroproliferation in bleomycin-injured mice [126,199]. Wnt5a, which can induce senescence, is found on EVs from IPF lungs [132,199]. Furthermore, several microRNA cargos within EVs from IPF lungs have been found to regulate cellular senescence [126]. With the ability of EVs to carry a number of cargos that can functionally regulate distal cell phenotype, they are poised to facilitate cellular dysfunction in lung fibrosis and understanding how EVs mediate the pathological effects of the SASP could lead to interesting targets for therapeutic intervention.
11. Epigenetic Changes in the AT2 Promote Lung Fibrosis
The accumulation of environmental and age-related stresses over time can permanently reshape the cellular response through epigenetic changes [200]. Epigenetics refers to hereditable changes in gene expression that occur in the absence of DNA sequence alterations. Specifically, DNA methylation, histone modifications, chromatin high-order structure and remodeling, and noncoding RNAs regulate genetic accessibility to the transcriptional machinery and post-transcriptional regulation of protein translation [201]. Distinctly different patterns of DNA methylation are found in IPF patients, and multiple histone modifications have been associated with alterations in key pro-fibrotic pathways [200,202,203,204].
IPF tends to occur in older adults, and aging is an inherent risk in accumulating epigenetic changes [24,205,206,207]. IPF is more prevalent in smokers [4], and several lines of evidence have demonstrated tobacco smoke to induce long-lasting changes in gene expression, which are largely attributed to epigenetic modification of the lung epithelium [208,209,210,211,212]. Moreover, differential patterns in DNA methylation patterns can be determined by gender, which may also contribute to the increased incidence of IPF in males [213,214,215,216]. Several lines of evidence support the ability of epigenetic modulators to alter the course of lung fibrosis. Changes to the methylation state and Histone deacetylase (HDAC) inhibition by pharmacological treatment or with genetically-modified mice can blunt lung fibrosis [37,217,218,219,220,221,222]. Several noncoding RNAs have also been demonstrated to regulate lung fibrosis [200,223].
Noncoding RNAs operate through suppress of protein translation, and although without controversy, is generally considered epigenetic modifiers [201]. Moreover, multiple lines of evidence support a crossover of traditional mechanisms of epigenetic modification with microRNA expression. For example, HDAC3 controls expression of the miR-17-92 family to regulate TGFβ expression, which controls alveolar sacculation [224]. The miR-17-92 family has decreased expression in IPF and after bleomycin-induced fibrosis, whereas augmented expression after 5′-aza-2′-deoxycytidine treatment attenuated lung fibrosis [217]. The Dlk-Dio3 domain is an imprinted region that contains clusters of miRNAs and genes [225]. Methylation of the paternal intergenic germline-derived differentially methylated region (IG-DMR) represses miRNA expression from this allele such that only the miRNAs on the maternally inherited allele are transcribed [226]. Expression changes of miRNAs within the Dlk-Dio3 regions alters WNT signaling and has been associated with lung fibrosis [227,228]. Similar to the miR-17-92 family, HDAC3 controls the expression of the microRNAs within the Dlk-Dio3 region [224]. Moreover, microRNAs in the Dlk-Dio3 region have decreased expression in fibrotic conditions and can target TGFβ expression and signaling [180,224]. In particular, miR-323a-3p, which is located in the Dlk-Dio3 region, has been demonstrated to attenuate lung fibrosis in bleomycin-injured mice [180].
12. Conclusions
AT2 cells as a driver of IPF is an established concept supported by an abundance of evidence that demonstrates their loss and dysfunction to have a central role in fibroproliferation. Recent delineation of AT2 subsets has improved the understanding of the way these cells participate in lung development and repair after injury. What remains to be determined is how this cellular heterogeneity may have differential AT2 responses to the various environmental exposures, genetics, and age-associated changes that contribute to injury and culminate over time in a dysfunctional phenotype that shifts from reparative programs to pathogenic fibrogenesis.
Funding
This research was funded by the National Institute of Health (NIH)/National Center for Advancing Translational Sciences UCLA-CTSI-KL2-UL1TR001881 (T.P.), R01 HL13707 (P.C.), T32 HL134637 (P.W.N., C.Y.), P01 HL108793 (B.R.S., P.W.N.), California Institute of Regenerative Medicine LA1-06915 (B.R.S.), and the Parker B Francis Foundation Fellowship (C.Y.).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| IPF | Idiopathic Pulmonary Fibrosis |
| FDA | U.S. Food and Drug Administration |
| U.S. | The United States of America |
| AT2 | Alveolar Epithelial Type II Cells |
References
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.P.; McKeever, T.M.; Fogarty, A.W.; Navaratnam, V.; Hubbard, R.B. Increasing Global Mortality from Idiopathic Pulmonary Fibrosis in the Twenty-First Century. Ann. ATS 2014, 11, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef] [PubMed]
- Robbie, H.; Daccord, C.; Chua, F.; Devaraj, A. Evaluating disease severity in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2017, 26, 170051. [Google Scholar] [CrossRef]
- Karimi-Shah, B.A.; Chowdhury, B.A. Forced vital capacity in idiopathic pulmonary fibrosis—FDA review of pirfenidone and nintedanib. N. Engl. J. Med. 2015, 372, 1189–1191. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Du Bois, R.M.; Fagan, E.A.; Fishman, R.S.; Glaspole, I.; Glassberg, M.K.; Lancaster, L.; et al. Pirfenidone for idiopathic pulmonary fibrosis: Analysis of pooled data from three multinational phase 3 trials. Eur. Respir. J. 2016, 47, 243–253. [Google Scholar] [CrossRef]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Azuma, A.; Nukiwa, T.; Tsuboi, E.; Suga, M.; Abe, S.; Nakata, K.; Taguchi, Y.; Nagai, S.; Itoh, H.; Ohi, M.; et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Corte, T.; Bonella, F.; Crestani, B.; Demedts, M.G.; Richeldi, L.; Coeck, C.; Pelling, K.; Quaresma, M.; Lasky, J.A. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Costabel, U.; Inoue, Y.; Richeldi, L.; Collard, H.R.; Tschoepe, I.; Stowasser, S.; Azuma, A. Efficacy of Nintedanib in Idiopathic Pulmonary Fibrosis across Prespecified Subgroups in INPULSIS. Am. J. Respir. Crit. Care Med. 2016, 193, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Noble, P.W. Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am. J. Physiol. Cell Physiol. 2014, 306, C987–C996. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. Molecular mechanisms of pulmonary fibrosis. Front. Biosci. 2002, 7, d1743–d1761. [Google Scholar] [CrossRef]
- Tang, Y.W.; Johnson, J.E.; Browning, P.J.; Cruz-Gervis, R.A.; Davis, A.; Graham, B.S.; Brigham, K.L.; Oates, J.A., Jr.; Loyd, J.E.; Stecenko, A.A. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J. Clin. Microbiol. 2003, 41, 2633–2640. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Meiners, S.; Eickelberg, O.; Konigshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.K. Insights from human genetic studies of lung and organ fibrosis. J. Clin. Investig. 2018, 128, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell Signal. 2019, 66, 109482. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.L.; Barkauskas, C.E.; Chapman, H.A.; Epstein, J.A.; Jain, R.; Hsia, C.C.; Niklason, L.; Calle, E.; Le, A.; Randell, S.H.; et al. Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 2014, 15, 123–138. [Google Scholar] [CrossRef]
- Rackley, C.R.; Stripp, B.R. Building and maintaining the epithelium of the lung. J. Clin. Investig. 2012, 122, 2724–2730. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef]
- Salwig, I.; Spitznagel, B.; Vazquez-Armendariz, A.I.; Khalooghi, K.; Guenther, S.; Herold, S.; Szibor, M.; Braun, T. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, K.; Cui, G.; Huang, X.; Yao, S.; Guo, W.; Qin, Z.; Li, Y.; Yang, R.; Pu, W.; et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat. Genet. 2019, 51, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.H.; Ryeom, S.; Kim, C.F. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Barkauskas, C.E.; Takeda, N.; Bowie, E.J.; Aghajanian, H.; Wang, Q.; Padmanabhan, A.; Manderfield, L.J.; Gupta, M.; Li, D.; et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat. Commun. 2015, 6, 6727. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.H.; Mendez, M.; Choi, K.; Subbotina, N.; Courey, A.; Cunningham, A.; Dave, A.; Engelhardt, J.F.; Liu, X.; White, E.S.; et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 181, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Soukiasian, H.J.; David, G.; Weigt, S.S.; Belperio, J.A.; et al. Senescence of alveolar stem cells drives progressive pulmonary fibrosis. Cell Stem Cell 2019, 59, 00545. [Google Scholar] [CrossRef]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhang, Y.; Xie, T.; Liu, N.; Chen, H.; Geng, Y.; Kurkciyan, A.; Mena, J.M.; Stripp, B.R.; Jiang, D.; et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 2016, 22, 1285–1293. [Google Scholar] [CrossRef]
- Uhal, B.D.; Joshi, I.; Hughes, W.F.; Ramos, C.; Pardo, A.; Selman, M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am. J. Physiol. 1998, 275, L1192–L1199. [Google Scholar] [CrossRef]
- Barbas-Filho, J.V.; Ferreira, M.A.; Sesso, A.; Kairalla, R.A.; Carvalho, C.R.; Capelozzi, V.L. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J. Clin. Pathol. 2001, 54, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Maeyama, T.; Kuwano, K.; Kawasaki, M.; Kunitake, R.; Hagimoto, N.; Matsuba, T.; Yoshimi, M.; Inoshima, I.; Yoshida, K.; Hara, N. Upregulation of Fas-signalling molecules in lung epithelial cells from patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2001, 17, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Plataki, M.; Koutsopoulos, A.V.; Darivianaki, K.; Delides, G.; Siafakas, N.M.; Bouros, D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest 2005, 127, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Cho, S.J.; Kang, M.J.; Chapoval, S.P.; Lee, P.J.; Noble, P.W.; Yehualaeshet, T.; Lu, B.; Flavell, R.A.; Milbrandt, J.; et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J. Exp. Med. 2004, 200, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Budinger, G.R.; Mutlu, G.M.; Eisenbart, J.; Fuller, A.C.; Bellmeyer, A.A.; Baker, C.M.; Wilson, M.; Ridge, K.; Barrett, T.A.; Lee, V.Y.; et al. Proapoptotic Bid is required for pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4604–4609. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kuwano, K.; Miyazaki, H.; Hagimoto, N.; Kawasaki, M.; Fujita, M.; Kunitake, R.; Kaneko, Y.; Hara, N. The involvement of Fas-Fas ligand pathway in fibrosing lung diseases. Am. J. Respir. Cell Mol. Biol. 1999, 20, 53–60. [Google Scholar] [CrossRef]
- Hagimoto, N.; Kuwano, K.; Nomoto, Y.; Kunitake, R.; Hara, N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 1997, 16, 91–101. [Google Scholar] [CrossRef]
- Wang, R.; Ibarra-Sunga, O.; Verlinski, L.; Pick, R.; Uhal, B.D. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L143–L151. [Google Scholar] [CrossRef]
- Kuwano, K.; Kunitake, R.; Maeyama, T.; Hagimoto, N.; Kawasaki, M.; Matsuba, T.; Yoshimi, M.; Inoshima, I.; Yoshida, K.; Hara, N. Attenuation of bleomycin-induced pneumopathy in mice by a caspase inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L316–L325. [Google Scholar] [CrossRef]
- Kuwano, K.; Hagimoto, N.; Kawasaki, M.; Yatomi, T.; Nakamura, N.; Nagata, S.; Suda, T.; Kunitake, R.; Maeyama, T.; Miyazaki, H.; et al. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J. Clin. Investig. 1999, 104, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Hagimoto, N.; Kuwano, K.; Miyazaki, H.; Kunitake, R.; Fujita, M.; Kawasaki, M.; Kaneko, Y.; Hara, N. Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of Fas antigen. Am. J. Respir. Cell Mol. Biol. 1997, 17, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Schutze, S.; Tchikov, V.; Schneider-Brachert, W. Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Investig. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef]
- Lawson, W.E.; Cheng, D.S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef]
- Katzen, J.; Wagner, B.D.; Venosa, A.; Kopp, M.; Tomer, Y.; Russo, S.J.; Headen, A.C.; Basil, M.C.; Stark, J.M.; Mulugeta, S.; et al. An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Nureki, S.I.; Tomer, Y.; Venosa, A.; Katzen, J.; Russo, S.J.; Jamil, S.; Barrett, M.; Nguyen, V.; Kopp, M.; Mulugeta, S.; et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Investig. 2018, 128, 4008–4024. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Horie, M.; Flodby, P.; Wang, H.; Liu, Y.; Ganesh, S.; Ryan Firth, A.L.; Minoo, P.; Li, C.; Beers, M.F.; et al. Grp78 Loss in Epithelial Progenitors Reveals an Age-Linked Role for ER Stress in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019. [Google Scholar] [CrossRef]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef]
- Jorgensen, E.; Stinson, A.; Shan, L.; Yang, J.; Gietl, D.; Albino, A.P. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 2008, 8, 229. [Google Scholar] [CrossRef]
- Kropski, J.A.; Pritchett, J.M.; Zoz, D.F.; Crossno, P.F.; Markin, C.; Garnett, E.T.; Degryse, A.L.; Mitchell, D.B.; Polosukhin, V.V.; Rickman, O.B.; et al. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 417–426. [Google Scholar] [CrossRef]
- McMillan, T.R.; Moore, B.B.; Weinberg, J.B.; Vannella, K.M.; Fields, W.B.; Christensen, P.J.; Van Dyk, L.F.; Toews, G.B. Exacerbation of established pulmonary fibrosis in a murine model by gammaherpesvirus. Am. J. Respir. Crit. Care Med. 2008, 177, 771–780. [Google Scholar] [CrossRef]
- Vannella, K.M.; Luckhardt, T.R.; Wilke, C.A.; Van Dyk, L.F.; Toews, G.B.; Moore, B.B. Latent herpesvirus infection augments experimental pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 181, 465–477. [Google Scholar] [CrossRef]
- Mora, A.L.; Woods, C.R.; Garcia, A.; Xu, J.; Rojas, M.; Speck, S.H.; Roman, J.; Brigham, K.L.; Stecenko, A.A. Lung infection with gamma-herpesvirus induces progressive pulmonary fibrosis in Th2-biased mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L711–L721. [Google Scholar] [CrossRef]
- Lok, S.S.; Haider, Y.; Howell, D.; Stewart, J.P.; Hasleton, P.S.; Egan, J.J. Murine gammaherpes virus as a cofactor in the development of pulmonary fibrosis in bleomycin resistant mice. Eur. Respir. J. 2002, 20, 1228–1232. [Google Scholar] [CrossRef]
- Torres-Gonzalez, E.; Bueno, M.; Tanaka, A.; Krug, L.T.; Cheng, D.S.; Polosukhin, V.V.; Sorescu, D.; Lawson, W.E.; Blackwell, T.S.; Rojas, M.; et al. Role of endoplasmic reticulum stress in age-related susceptibility to lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 46, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Suliman, H.B. Mitochondrial Dysfunction in Lung Pathogenesis. Annu. Rev. Physiol. 2017, 79, 495–515. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bernard, K.; Thannickal, V.J. Mitochondrial Dysfunction in Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S383–S388. [Google Scholar] [CrossRef]
- Bueno, M.; Zank, D.; Buendia-Roldan, I.; Fiedler, K.; Mays, B.G.; Alvarez, D.; Sembrat, J.; Kimball, B.; Bullock, J.K.; Martin, J.L.; et al. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PLoS ONE 2019, 14, e0218003. [Google Scholar] [CrossRef]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef]
- Chung, K.-P.; Hsu, C.-L.; Fan, L.-C.; Huang, Z.; Bhatia, D.; Chen, Y.-J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390. [Google Scholar] [CrossRef]
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Morales-Nebreda, L.; Cheng, Y.; Hogan, E.; Yeldandi, A.; Chi, M.; Piseaux, R.; Ridge, K.; et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic. Biol. Med. 2016, 101, 482–490. [Google Scholar] [CrossRef]
- Jablonski, R.P.; Kim, S.-J.; Cheresh, P.; Williams, D.B.; Morales-Nebreda, L.; Cheng, Y.; Yeldandi, A.; Bhorade, S.; Pardo, A.; Selman, M.; et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. 2017, 31, 2520–2532. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; St Croix, C.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; De Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ganzleben, I.; He, G.W.; Gunther, C.; Prigge, E.S.; Richter, K.; Rieker, R.J.; Mougiakakos, D.; Neurath, M.F.; Becker, C. PGAM5 is a key driver of mitochondrial dysfunction in experimental lung fibrosis. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Kulkarni, T.; De Andrade, J.; Zhou, Y.; Luckhardt, T.; Thannickal, V.J. Alveolar epithelial disintegrity in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L185–L191. [Google Scholar] [CrossRef]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef]
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 213–224. [Google Scholar] [CrossRef]
- Kim, Y.; Kugler, M.C.; Wei, Y.; Kim, K.K.; Li, X.; Brumwell, A.N.; Chapman, H.A. Integrin alpha3beta1-dependent beta-catenin phosphorylation links epithelial Smad signaling to cell contacts. J. Cell Biol. 2009, 184, 309–322. [Google Scholar] [CrossRef]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef]
- Borok, Z.; Whitsett, J.A.; Bitterman, P.B.; Thannickal, V.J.; Kotton, D.N.; Reynolds, S.D.; Krasnow, M.A.; Bianchi, D.W.; Morrisey, E.E.; Hogan, B.L.; et al. Cell plasticity in lung injury and repair: Report from an NHLBI workshop, April 19–20, 2010. Proc. Am. Thorac. Soc. 2011, 8, 215–222. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2019, 180, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cheung, H.H.; Tu, J.; Miu, K.K.; Chan, W.Y. New insights into the unfolded protein response in stem cells. Oncotarget 2016, 7, 54010–54027. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Gonzalez De Los Santos, F.; Zhao, Y.; Wu, Z.; Rinke, A.E.; Kim, K.K.; Phan, S.H. Telomerase reverse transcriptase ameliorates lung fibrosis by protecting alveolar epithelial cells against senescence. J. Biol. Chem. 2019, 294, 8861–8871. [Google Scholar] [CrossRef]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef]
- Povedano, J.M.; Martinez, P.; Serrano, R.; Tejera, A.; Gomez-Lopez, G.; Bobadilla, M.; Flores, J.M.; Bosch, F.; Blasco, M.A. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Thannickal, V.J. The Aging Lung and Idiopathic Pulmonary Fibrosis. Am. J. Med. Sci. 2019, 357, 384–389. [Google Scholar] [CrossRef]
- Selman, M.; Buendia-Roldan, I.; Pardo, A. Aging and Pulmonary Fibrosis. Rev. Investig. Clin. 2016, 68, 75–83. [Google Scholar]
- Thannickal, V.J. Mechanistic links between aging and lung fibrosis. Biogerontology 2013, 14, 609–615. [Google Scholar] [CrossRef]
- Chilosi, M.; Carloni, A.; Rossi, A.; Poletti, V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl. Res. 2013, 162, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Arish, N.; Petukhov, D.; Wallach-Dayan, S.B. The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Snetselaar, R.; Van Batenburg, A.A.; Van Oosterhout, M.F.M.; Kazemier, K.M.; Roothaan, S.M.; Peeters, T.; Van der Vis, J.J.; Goldschmeding, R.; Grutters, J.C.; Van Moorsel, C.H.M. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS ONE 2017, 12, e0189467. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef]
- Morla, M.; Busquets, X.; Pons, J.; Sauleda, J.; MacNee, W.; Agusti, A.G. Telomere shortening in smokers with and without COPD. Eur. Respir. J. 2006, 27, 525–528. [Google Scholar] [CrossRef]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Diaz de Leon, A.; Cronkhite, J.T.; Katzenstein, A.L.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE 2010, 5, e10680. [Google Scholar] [CrossRef] [PubMed]
- Kannengiesser, C.; Borie, R.; Menard, C.; Reocreux, M.; Nitschke, P.; Gazal, S.; Mal, H.; Taille, C.; Cadranel, J.; Nunes, H.; et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur. Respir. J. 2015, 46, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Cogan, J.D.; Kropski, J.A.; Zhao, M.; Mitchell, D.B.; Rives, L.; Markin, C.; Garnett, E.T.; Montgomery, K.H.; Mason, W.R.; McKean, D.F.; et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2015, 191, 646–655. [Google Scholar] [CrossRef]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef]
- Petrovski, S.; Todd, J.L.; Durheim, M.T.; Wang, Q.; Chien, J.W.; Kelly, F.L.; Frankel, C.; Mebane, C.M.; Ren, Z.; Bridgers, J.; et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 82–93. [Google Scholar] [CrossRef]
- Kropski, J.A.; Mitchell, D.B.; Markin, C.; Polosukhin, V.V.; Choi, L.; Johnson, J.E.; Lawson, W.E.; Phillips, J.A., 3rd; Cogan, J.D.; Blackwell, T.S.; et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest 2014, 146, e1–e7. [Google Scholar] [CrossRef]
- Stanley, S.E.; Gable, D.L.; Wagner, C.L.; Carlile, T.M.; Hanumanthu, V.S.; Podlevsky, J.D.; Khalil, S.E.; DeZern, A.E.; Rojas-Duran, M.F.; Applegate, C.D.; et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci. Transl. Med. 2016, 8, 351ra107. [Google Scholar] [CrossRef]
- Alder, J.K.; Stanley, S.E.; Wagner, C.L.; Hamilton, M.; Hanumanthu, V.S.; Armanios, M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest 2015, 147, 1361–1368. [Google Scholar] [CrossRef]
- Hoffman, T.W.; Van der Vis, J.J.; Van Oosterhout, M.F.; Van Es, H.W.; Van Kessel, D.A.; Grutters, J.C.; Van Moorsel, C.H. TINF2 Gene Mutation in a Patient with Pulmonary Fibrosis. Case Rep. Pulmonol. 2016, 2016, 1310862. [Google Scholar] [CrossRef]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Adamson, I.Y.; King, G.M.; Young, L. Influence of extracellular matrix and collagen components on alveolar type 2 cell morphology and function. In Vitro Cell Dev. Biol. 1989, 25, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Bjermer, L.; Lundgren, R.; Hallgren, R. Hyaluronan and type III procollagen peptide concentrations in bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. Thorax 1989, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Habiel, D.M.; Ge, L.; Bora, S.A.; Brauer, R.; Evans, C.M.; Xie, T.; Alonso-Valenteen, F.; Medina-Kauwe, L.K.; et al. Syndecan-1 promotes lung fibrosis by regulating epithelial reprogramming through extracellular vesicles. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Raredon, M.S.B.; Adams, T.S.; Suhail, Y.; Schupp, J.C.; Poli, S.; Neumark, N.; Leiby, K.L.; Greaney, A.M.; Yuan, Y.; Horien, C.; et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 2019, 5, eaaw3851. [Google Scholar] [CrossRef]
- Konigshoff, M.; Kramer, M.; Balsara, N.; Wilhelm, J.; Amarie, O.V.; Jahn, A.; Rose, F.; Fink, L.; Seeger, W.; Schaefer, L.; et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Investig. 2009, 119, 772–787. [Google Scholar] [CrossRef]
- Konigshoff, M.; Balsara, N.; Pfaff, E.M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Zamo, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Frank, D.B.; Peng, T.; Zepp, J.A.; Snitow, M.; Vincent, T.L.; Penkala, I.J.; Cui, Z.; Herriges, M.J.; Morley, M.P.; Zhou, S.; et al. Emergence of a Wave of Wnt Signaling that Regulates Lung Alveologenesis by Controlling Epithelial Self-Renewal and Differentiation. Cell Rep. 2016, 17, 2312–2325. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, T.; Csongei, V.; Feller, D.; Ernszt, D.; Smuk, G.; Sarosi, V.; Jakab, L.; Kvell, K.; Bartis, D.; Pongracz, J.E. Alteration in the Wnt microenvironment directly regulates molecular events leading to pulmonary senescence. Aging Cell 2014, 13, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.P.; Herazo-Maya, J.D.; Sennello, J.A.; Flozak, A.S.; Russell, S.; Mutlu, G.M.; Budinger, G.R.; DasGupta, R.; Varga, J.; Kaminski, N.; et al. Wnt coreceptor Lrp5 is a driver of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 185–195. [Google Scholar] [CrossRef]
- Henderson, W.R., Jr.; Chi, E.Y.; Ye, X.; Nguyen, C.; Tien, Y.T.; Zhou, B.; Borok, Z.; Knight, D.A.; Kahn, M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14309–14314. [Google Scholar] [CrossRef]
- Ulsamer, A.; Wei, Y.; Kim, K.K.; Tan, K.; Wheeler, S.; Xi, Y.; Thies, R.S.; Chapman, H.A. Axin pathway activity regulates in vivo pY654-beta-catenin accumulation and pulmonary fibrosis. J. Biol. Chem. 2012, 287, 5164–5172. [Google Scholar] [CrossRef]
- Olajuyin, A.M.; Zhang, X.; Ji, H.L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. 2019, 5, 63. [Google Scholar] [CrossRef]
- Mutze, K.; Vierkotten, S.; Milosevic, J.; Eickelberg, O.; Konigshoff, M. Enolase 1 (ENO1) and protein disulfide-isomerase associated 3 (PDIA3) regulate Wnt/beta-catenin-driven trans-differentiation of murine alveolar epithelial cells. Dis. Model. Mech. 2015, 8, 877–890. [Google Scholar] [CrossRef]
- Konigshoff, M.; Eickelberg, O. WNT signaling in lung disease: A failure or a regeneration signal? Am. J. Respir. Cell Mol. Biol. 2010, 42, 21–31. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Azzolin, L.; Zanconato, F.; Bresolin, S.; Forcato, M.; Basso, G.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Role of TAZ as mediator of Wnt signaling. Cell 2012, 151, 1443–1456. [Google Scholar] [CrossRef] [PubMed]
- Mitani, A.; Nagase, T.; Fukuchi, K.; Aburatani, H.; Makita, R.; Kurihara, H. Transcriptional coactivator with PDZ-binding motif is essential for normal alveolarization in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Whitsett, J.A.; Di Palma, T.; Hong, J.H.; Yaffe, M.B.; Zannini, M. TAZ interacts with TTF-1 and regulates expression of surfactant protein-C. J. Biol. Chem. 2004, 279, 17384–17390. [Google Scholar] [CrossRef] [PubMed]
- Makita, R.; Uchijima, Y.; Nishiyama, K.; Amano, T.; Chen, Q.; Takeuchi, T.; Mitani, A.; Nagase, T.; Yatomi, Y.; Aburatani, H.; et al. Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am. J. Physiol. Ren. Physiol. 2008, 294, F542–F553. [Google Scholar] [CrossRef]
- Lin, C.; Yao, E.; Zhang, K.; Jiang, X.; Croll, S.; Thompson-Peer, K.; Chuang, P.T. YAP is essential for mechanical force production and epithelial cell proliferation during lung branching morphogenesis. Elife 2017, 6. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef]
- Sun, T.; Huang, Z.; Zhang, H.; Posner, C.; Jia, G.; Ramalingam, T.R.; Xu, M.; Brightbill, H.; Egen, J.G.; Dey, A.; et al. TAZ is required for lung alveolar epithelial cell differentiation after injury. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Ligresti, G.; Hilscher, M.B.; Shah, V.H. Mechanosensing and fibrosis. J. Clin. Investig. 2018, 128, 74–84. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A. Keeping fibroblasts in suspense: TAZ-mediated signaling activates a context-dependent profibrotic phenotype. Focus on “TAZ activation drives fibroblast spheroid growth, expression of profibrotic paracrine signals, and context-dependent ECM gene expression”. Am. J. Physiol. Cell Physiol. 2017, 312, C274–C276. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Mikami, Y.; Urushiyama, H.; Horie, M.; Matsuzaki, H.; Takeshima, H.; Makita, K.; Miyashita, N.; Mitani, A.; et al. TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci. Rep. 2017, 7, 42595. [Google Scholar] [CrossRef]
- Gokey, J.J.; Sridharan, A.; Xu, Y.; Green, J.; Carraro, G.; Stripp, B.R.; Perl, A.-K.T.; Whitsett, J.A. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight 2018, 3, e98738. [Google Scholar] [CrossRef]
- Hussain, M.; Xu, C.; Ahmad, M.; Yang, Y.; Lu, M.; Wu, X.; Tang, L.; Wu, X. Notch Signaling: Linking Embryonic Lung Development and Asthmatic Airway Remodeling. Mol. Pharmacol. 2017, 92, 676–693. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Tsao, P.N.; Matsuoka, C.; Wei, S.C.; Sato, A.; Sato, S.; Hasegawa, K.; Chen, H.K.; Ling, T.Y.; Mori, M.; Cardoso, W.V.; et al. Epithelial Notch signaling regulates lung alveolar morphogenesis and airway epithelial integrity. Proc. Natl. Acad. Sci. USA 2016, 113, 8242–8247. [Google Scholar] [CrossRef]
- Xu, K.; Nieuwenhuis, E.; Cohen, B.L.; Wang, W.; Canty, A.J.; Danska, J.S.; Coultas, L.; Rossant, J.; Wu, M.Y.; Piscione, T.D.; et al. Lunatic Fringe-mediated Notch signaling is required for lung alveogenesis. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L45–L56. [Google Scholar] [CrossRef]
- Kavian, N.; Servettaz, A.; Weill, B.; Batteux, F. New insights into the mechanism of notch signalling in fibrosis. Open Rheumatol. J. 2012, 6, 96–102. [Google Scholar] [CrossRef]
- Wasnick, R.M.; Korfei, M.; Piskulak, K.; Henneke, I.; Wilhelm, J.; Mahavadi, P.; Von der Beck, D.; Koch, M.; Shalashova, I.; Klymenko, O.; et al. Restored alveolar epithelial differentiation and reversed human lung fibrosis upon Notch inhibition. BioRxiv 2019, 580498. [Google Scholar] [CrossRef]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef]
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.S.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 2015, 526, 578–582. [Google Scholar] [CrossRef]
- Li, M.; Krishnaveni, M.S.; Li, C.; Zhou, B.; Xing, Y.; Banfalvi, A.; Li, A.; Lombardi, V.; Akbari, O.; Borok, Z.; et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Investig. 2011, 121, 277–287. [Google Scholar] [CrossRef]
- Coker, R.K.; Laurent, G.J.; Jeffery, P.K.; Du Bois, R.M.; Black, C.M.; McAnulty, R.J. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax 2001, 56, 549–556. [Google Scholar] [CrossRef]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Bonniaud, P.; Martin, G.; Margetts, P.J.; Ask, K.; Robertson, J.; Gauldie, J.; Kolb, M. Connective tissue growth factor is crucial to inducing a profibrotic environment in “fibrosis-resistant” BALB/c mouse lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 510–516. [Google Scholar] [CrossRef]
- Bonniaud, P.; Margetts, P.J.; Kolb, M.; Haberberger, T.; Kelly, M.; Robertson, J.; Gauldie, J. Adenoviral gene transfer of connective tissue growth factor in the lung induces transient fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 770–778. [Google Scholar] [CrossRef]
- Watts, K.L.; Spiteri, M.A. Connective tissue growth factor expression and induction by transforming growth factor-beta is abrogated by simvastatin via a Rho signaling mechanism. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L1323–L1332. [Google Scholar] [CrossRef]
- Lasky, J.A.; Ortiz, L.A.; Tonthat, B.; Hoyle, G.W.; Corti, M.; Athas, G.; Lungarella, G.; Brody, A.; Friedman, M. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am. J. Physiol. 1998, 275, L365–L371. [Google Scholar] [CrossRef]
- Pan, L.H.; Yamauchi, K.; Uzuki, M.; Nakanishi, T.; Takigawa, M.; Inoue, H.; Sawai, T. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur. Respir. J. 2001, 17, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cassandras, M.; Peng, T. The Role of Hedgehog Signaling in Adult Lung Regeneration and Maintenance. J. Dev. Biol. 2019, 7. [Google Scholar] [CrossRef]
- Kugler, M.C.; Joyner, A.L.; Loomis, C.A.; Munger, J.S. Sonic hedgehog signaling in the lung. From development to disease. Am. J. Respir. Cell Mol. Biol. 2015, 52, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Liu, J.; Wu, Z.; Liu, T.; Ullenbruch, M.R.; Ding, L.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Reemergence of hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Bolanos, A.L.; Milla, C.M.; Lira, J.C.; Ramirez, R.; Checa, M.; Barrera, L.; Garcia-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M.; et al. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L978–L990. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.A.; Hoyne, G.F.; Ahmad, S.A.; Jarman, E.; Wallace, W.A.; Harrison, D.J.; Haslett, C.; Lamb, J.R.; Howie, S.E. Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. J. Pathol. 2003, 199, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Coon, D.R.; Roberts, D.J.; Loscertales, M.; Kradin, R. Differential epithelial expression of SHH and FOXF1 in usual and nonspecific interstitial pneumonia. Exp. Mol. Pathol. 2006, 80, 119–123. [Google Scholar] [CrossRef]
- Fitch, P.M.; Howie, S.E.; Wallace, W.A. Oxidative damage and TGF-beta differentially induce lung epithelial cell sonic hedgehog and tenascin-C expression: Implications for the regulation of lung remodelling in idiopathic interstitial lung disease. Int. J. Exp. Pathol. 2011, 92, 8–17. [Google Scholar] [CrossRef]
- Moshai, E.F.; Wemeau-Stervinou, L.; Cigna, N.; Brayer, S.; Somme, J.M.; Crestani, B.; Mailleux, A.A. Targeting the hedgehog-glioma-associated oncogene homolog pathway inhibits bleomycin-induced lung fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2014, 51, 11–25. [Google Scholar] [CrossRef]
- Liu, L.; Kugler, M.C.; Loomis, C.A.; Samdani, R.; Zhao, Z.; Chen, G.J.; Brandt, J.P.; Brownell, I.; Joyner, A.L.; Rom, W.N.; et al. Hedgehog signaling in neonatal and adult lung. Am. J. Respir. Cell Mol. Biol. 2013, 48, 703–710. [Google Scholar] [CrossRef]
- Kugler, M.C.; Yie, T.A.; Cai, Y.; Berger, J.Z.; Loomis, C.A.; Munger, J.S. The Hedgehog target Gli1 is not required for bleomycin-induced lung fibrosis. Exp. Lung Res. 2019, 45, 22–29. [Google Scholar] [CrossRef]
- Ge, L.; Habiel, D.M.; Hansbro, P.M.; Kim, R.Y.; Gharib, S.A.; Edelman, J.D.; Konigshoff, M.; Parimon, T.; Brauer, R.; Huang, Y.; et al. miR-323a-3p regulates lung fibrosis by targeting multiple profibrotic pathways. JCI Insight 2016, 1, e90301. [Google Scholar] [CrossRef]
- Pan, T.; Mason, R.J.; Westcott, J.Y.; Shannon, J.M. Rat alveolar type II cells inhibit lung fibroblast proliferation in vitro. Am. J. Respir. Cell Mol. Biol. 2001, 25, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Portnoy, J.; Pan, T.; Dinarello, C.A.; Shannon, J.M.; Westcott, J.Y.; Zhang, L.; Mason, R.J. Alveolar type II cells inhibit fibroblast proliferation: Role of IL-1alpha. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L307–L316. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Peters-Golden, M.; Christensen, P.J.; Lama, V.; Kuziel, W.A.; Paine, R., 3rd; Toews, G.B. Alveolar epithelial cell inhibition of fibroblast proliferation is regulated by MCP-1/CCR2 and mediated by PGE2. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L342–L349. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Gillissen, A.; Buhl, R.; Hoyt, R.F.; Hubbard, R.C.; Ozaki, T.; Rennard, S.I.; Crystal, R.G. Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am. Rev. Respir. Dis. 1991, 144, 1080–1084. [Google Scholar] [CrossRef]
- Lama, V.; Moore, B.B.; Christensen, P.; Toews, G.B.; Peters-Golden, M. Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am. J. Respir. Cell Mol. Biol. 2002, 27, 752–758. [Google Scholar] [CrossRef]
- Tan, Q.; Ma, X.Y.; Liu, W.; Meridew, J.A.; Jones, D.L.; Haak, A.J.; Sicard, D.; Ligresti, G.; Tschumperlin, D.J. Nascent Lung Organoids Reveal Epithelium- and Bone Morphogenetic Protein-mediated Suppression of Fibroblast Activation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 607–619. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Gahl, W.A.; Brantly, M.; Kaiser-Kupfer, M.I.; Iwata, F.; Hazelwood, S.; Shotelersuk, V.; Duffy, L.F.; Kuehl, E.M.; Troendle, J.; Bernardini, I. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome). N. Engl. J. Med. 1998, 338, 1258–1264. [Google Scholar] [CrossRef]
- Young, L.R.; Gulleman, P.M.; Short, C.W.; Tanjore, H.; Sherrill, T.; Qi, A.; McBride, A.P.; Zaynagetdinov, R.; Benjamin, J.T.; Lawson, W.E.; et al. Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky-Pudlak syndrome. JCI Insight 2016, 1, e88947. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; Von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFbeta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging (Albany N.Y.) 2012, 4, 932–951. [Google Scholar] [CrossRef]
- Hoare, M.; Narita, M. Notch and Senescence. Adv. Exp. Med. Biol. 2018, 1066, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Narita, M. NOTCH and the 2 SASPs of senescence. Cell Cycle 2017, 16, 239–240. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17. [Google Scholar] [CrossRef]
- Borghesan, M.; Fafian-Labora, J.; Eleftheriadou, O.; Carpintero-Fernandez, P.; Paez-Ribes, M.; Vizcay-Barrena, G.; Swisa, A.; Kolodkin-Gal, D.; Ximenez-Embun, P.; Lowe, R.; et al. Small Extracellular Vesicles Are Key Regulators of Non-cell Autonomous Intercellular Communication in Senescence via the Interferon Protein IFITM3. Cell Rep. 2019, 27, 3956–3971. [Google Scholar] [CrossRef]
- Martin-Medina, A.; Lehmann, M.; Burgy, O.; Hermann, S.; Baarsma, H.A.; Wagner, D.E.; De Santis, M.M.; Ciolek, F.; Hofer, T.P.; Frankenberger, M.; et al. Increased Extracellular Vesicles Mediate WNT-5A Signaling in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018. [Google Scholar] [CrossRef]
- Yang, I.V.; Schwartz, D.A. Epigenetics of idiopathic pulmonary fibrosis. Transl. Res. 2015, 165, 48–60. [Google Scholar] [CrossRef]
- Chen, Z.; Li, S.; Subramaniam, S.; Shyy, J.Y.; Chien, S. Epigenetic Regulation: A New Frontier for Biomedical Engineers. Annu. Rev. Biomed. Eng. 2017, 19, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.R.; Zhang, Y.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Li, N.; Ferreira, H.J.; Moran, S.; Pisano, D.G.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.A.; Vachani, A.; Whitney, D.; Elashoff, M.; Porta Smith, K.; Ferguson, J.S.; Parsons, E.; Mitra, N.; Brody, J.; Lenburg, M.E.; et al. A Bronchial Genomic Classifier for the Diagnostic Evaluation of Lung Cancer. N. Engl. J. Med. 2015, 373, 243–251. [Google Scholar] [CrossRef]
- Steiling, K.; Kadar, A.Y.; Bergerat, A.; Flanigon, J.; Sridhar, S.; Shah, V.; Ahmad, Q.R.; Brody, J.S.; Lenburg, M.E.; Steffen, M.; et al. Comparison of proteomic and transcriptomic profiles in the bronchial airway epithelium of current and never smokers. PLoS ONE 2009, 4, e5043. [Google Scholar] [CrossRef]
- Sridhar, S.; Schembri, F.; Zeskind, J.; Shah, V.; Gustafson, A.M.; Steiling, K.; Liu, G.; Dumas, Y.M.; Zhang, X.; Brody, J.S.; et al. Smoking-induced gene expression changes in the bronchial airway are reflected in nasal and buccal epithelium. BMC Genom. 2008, 9, 259. [Google Scholar] [CrossRef]
- Shah, V.; Sridhar, S.; Beane, J.; Brody, J.S.; Spira, A. SIEGE: Smoking Induced Epithelial Gene Expression Database. Nucleic Acids Res. 2005, 33, D573–D579. [Google Scholar] [CrossRef] [PubMed]
- Buro-Auriemma, L.J.; Salit, J.; Hackett, N.R.; Walters, M.S.; Strulovici-Barel, Y.; Staudt, M.R.; Fuller, J.; Mahmoud, M.; Stevenson, C.S.; Hilton, H.; et al. Cigarette smoking induces small airway epithelial epigenetic changes with corresponding modulation of gene expression. Hum. Mol. Genet. 2013, 22, 4726–4738. [Google Scholar] [CrossRef] [PubMed]
- Cotton, A.M.; Lam, L.; Affleck, J.G.; Wilson, I.M.; Penaherrera, M.S.; McFadden, D.E.; Kobor, M.S.; Lam, W.L.; Robinson, W.P.; Brown, C.J. Chromosome-wide DNA methylation analysis predicts human tissue-specific X inactivation. Hum. Genet. 2011, 130, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.P.; Derks, E.M.; Weisenberger, D.J.; Strengman, E.; Janson, E.; Sommer, I.E.; Kahn, R.S.; Ophoff, R.A. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS ONE 2009, 4, e6767. [Google Scholar] [CrossRef] [PubMed]
- Sarter, B.; Long, T.I.; Tsong, W.H.; Koh, W.P.; Yu, M.C.; Laird, P.W. Sex differential in methylation patterns of selected genes in Singapore Chinese. Hum. Genet. 2005, 117, 402–403. [Google Scholar] [CrossRef]
- El-Maarri, O.; Becker, T.; Junen, J.; Manzoor, S.S.; Diaz-Lacava, A.; Schwaab, R.; Wienker, T.; Oldenburg, J. Gender specific differences in levels of DNA methylation at selected loci from human total blood: A tendency toward higher methylation levels in males. Hum. Genet. 2007, 122, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Dakhlallah, D.; Batte, K.; Wang, Y.; Cantemir-Stone, C.Z.; Yan, P.; Nuovo, G.; Mikhail, A.; Hitchcock, C.L.; Wright, V.P.; Nana-Sinkam, S.P.; et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 187, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Rubio, K.; Singh, I.; Dobersch, S.; Sarvari, P.; Gunther, S.; Cordero, J.; Mehta, A.; Wujak, L.; Cabrera-Fuentes, H.; Chao, C.M.; et al. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 2229. [Google Scholar] [CrossRef] [PubMed]
- Korfei, M.; Skwarna, S.; Henneke, I.; MacKenzie, B.; Klymenko, O.; Saito, S.; Ruppert, C.; Von der Beck, D.; Mahavadi, P.; Klepetko, W.; et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 2015, 70, 1022–1032. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Hagood, J.S.; Liu, H.; Zhang, W.; Ambalavanan, N.; Thannickal, V.J. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur. Respir. J. 2014, 43, 1448–1458. [Google Scholar] [CrossRef]
- Conforti, F.; Davies, E.R.; Calderwood, C.J.; Thatcher, T.H.; Jones, M.G.; Smart, D.E.; Mahajan, S.; Alzetani, A.; Havelock, T.; Maher, T.M.; et al. The histone deacetylase inhibitor, romidepsin, as a potential treatment for pulmonary fibrosis. Oncotarget 2017, 8, 48737–48754. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Peng, R.; Phillips, J.E.; Deguzman, J.; Ren, Y.; Apparsundaram, S.; Luo, Q.; Bauer, C.M.; Fuentes, M.E.; DeMartino, J.A.; et al. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am. J. Pathol. 2013, 183, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Xiong, Y.; Zhang, G.; Chang, J. MicroRNAs in idiopathic pulmonary fibrosis, new research progress and their pathophysiological implication. Exp. Lung Res. 2018, 44, 178–190. [Google Scholar] [CrossRef]
- Wang, Y.; Frank, D.B.; Morley, M.P.; Zhou, S.; Wang, X.; Lu, M.M.; Lazar, M.A.; Morrisey, E.E. HDAC3-Dependent Epigenetic Pathway Controls Lung Alveolar Epithelial Cell Remodeling and Spreading via miR-17-92 and TGF-beta Signaling Regulation. Dev. Cell 2016, 36, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008, 24, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.; Royo, H.; Bortolin, M.L.; Lin, S.P.; Ferguson-Smith, A.C.; Cavaille, J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004, 14, 1741–1748. [Google Scholar] [CrossRef]
- Snyder, C.M.; Rice, A.L.; Estrella, N.L.; Held, A.; Kandarian, S.C.; Naya, F.J. MEF2A regulates the Gtl2-Dio3 microRNA mega-cluster to modulate WNT signaling in skeletal muscle regeneration. Development 2013, 140, 31–42. [Google Scholar] [CrossRef]
- Milosevic, J.; Pandit, K.; Magister, M.; Rabinovich, E.; Ellwanger, D.C.; Yu, G.; Vuga, L.J.; Weksler, B.; Benos, P.V.; Gibson, K.F.; et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 879–887. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).