Soluble Alpha-Klotho Alleviates Cardiac Fibrosis without Altering Cardiomyocytes Renewal


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
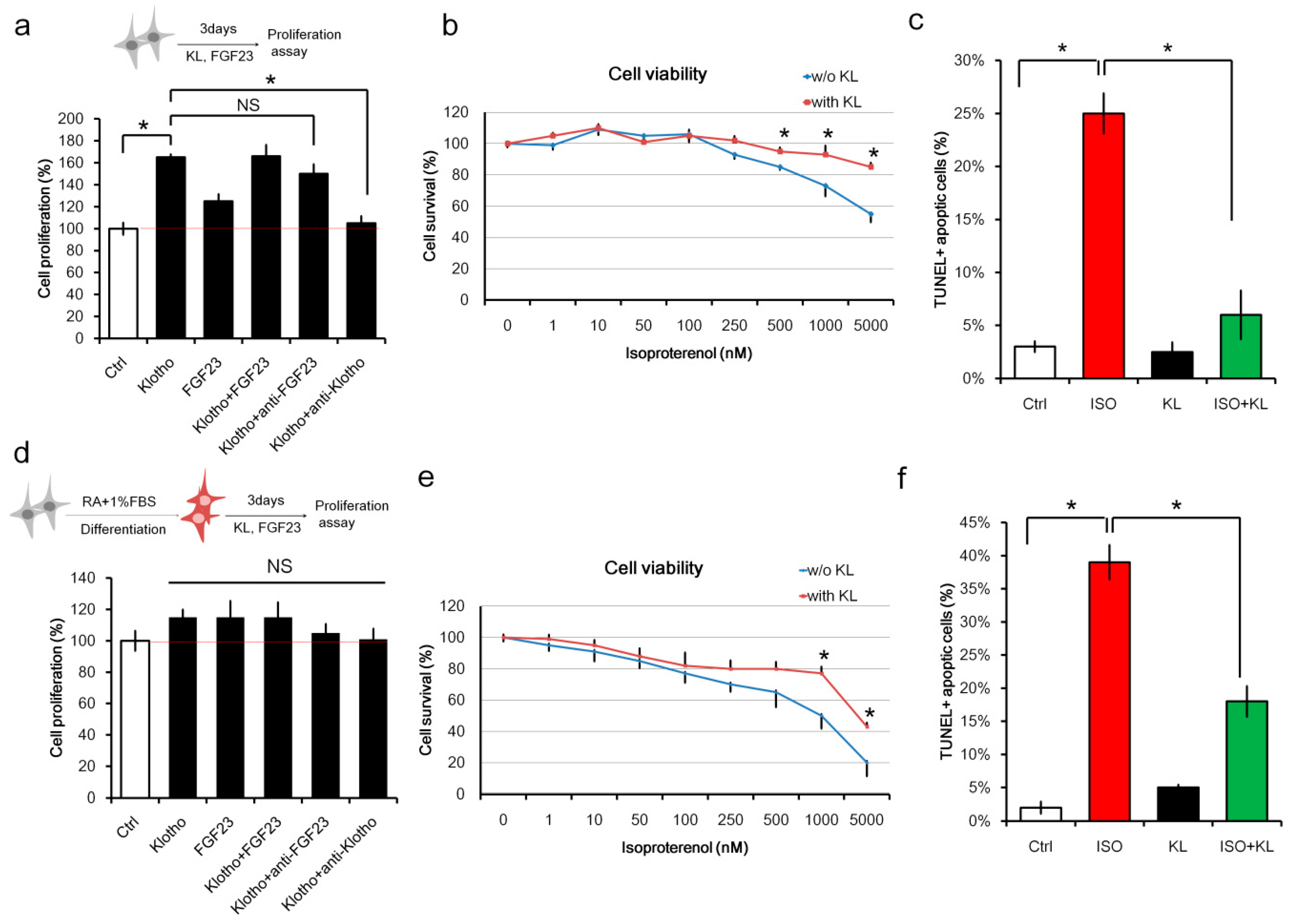
2.1. Klotho (KL) Attenuated Isoproterenol-Induced Cell Death in H9c2 Cardiomyocytes In Vitro
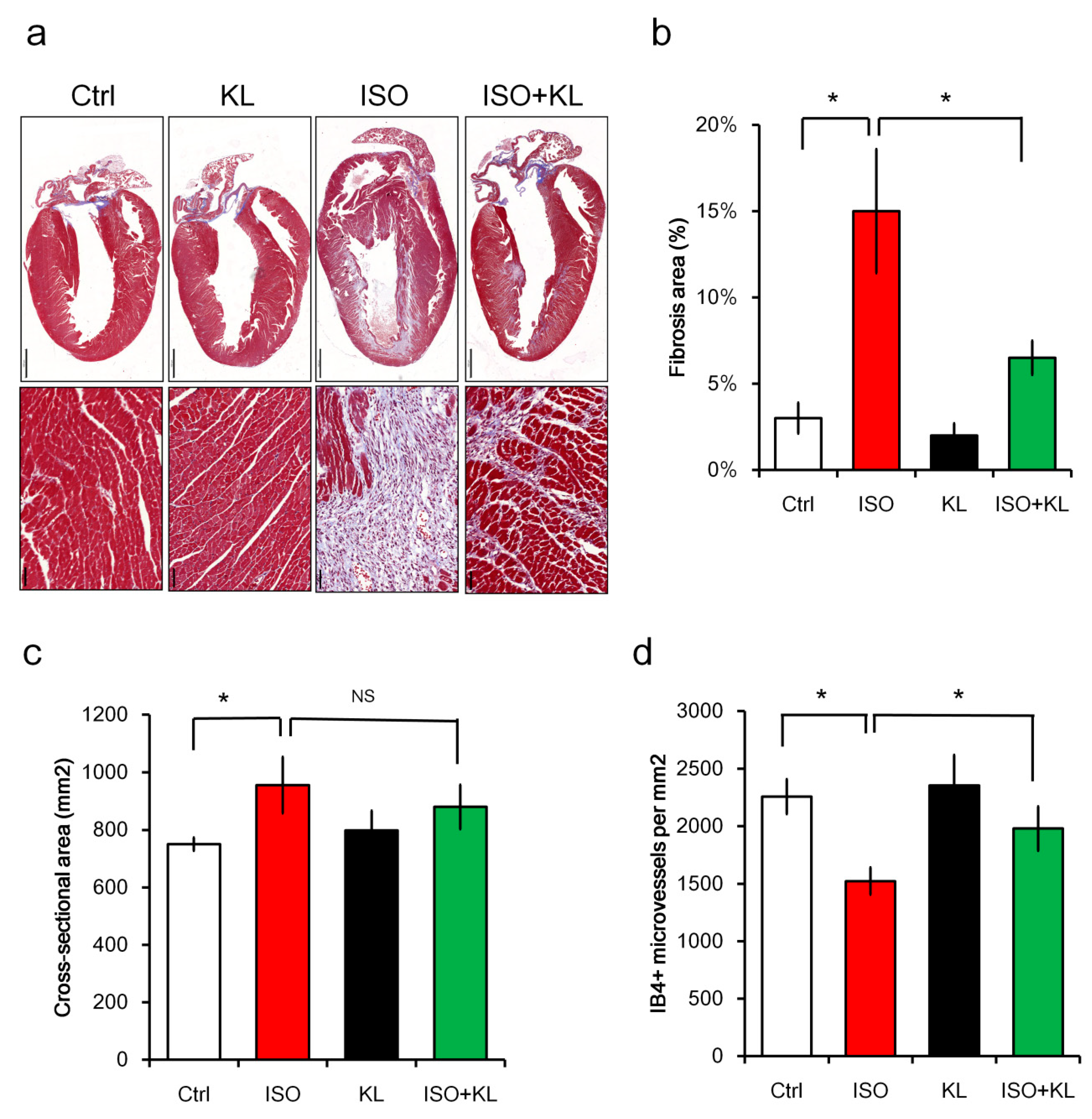
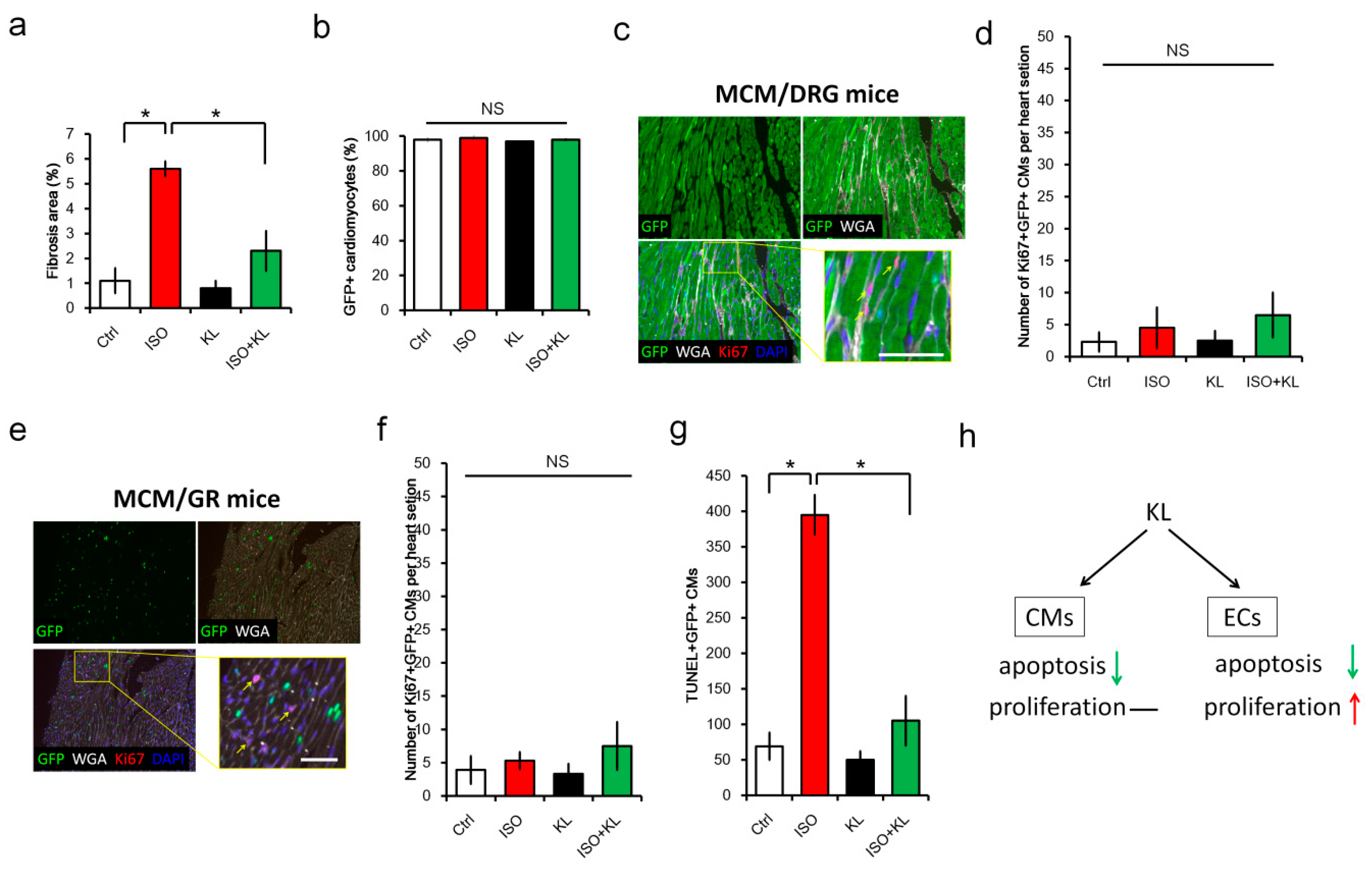
2.2. KL Inhibited Isoproterenol-Induced Cardiac Fibrosis and Cellular Apoptosis In Vivo
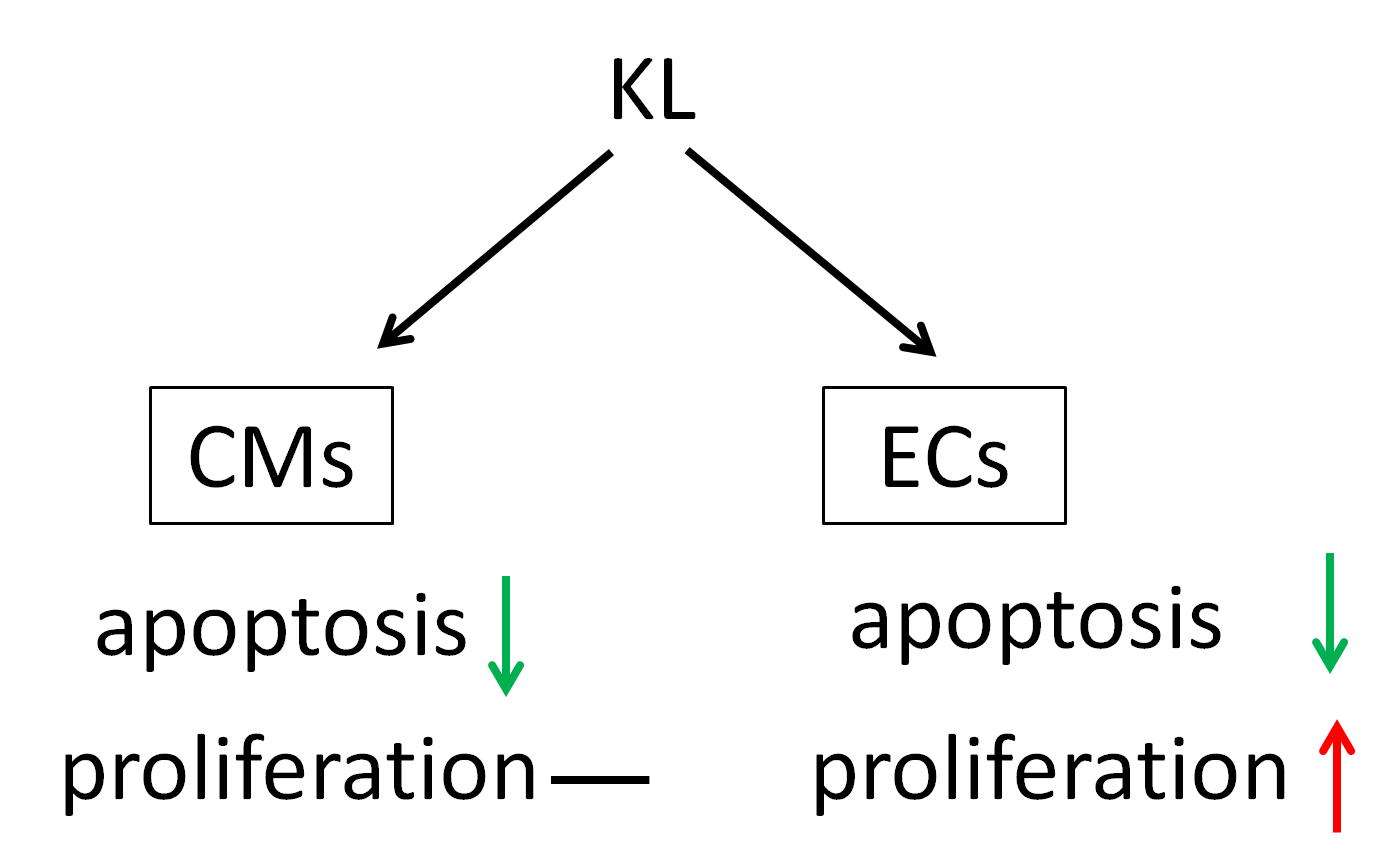
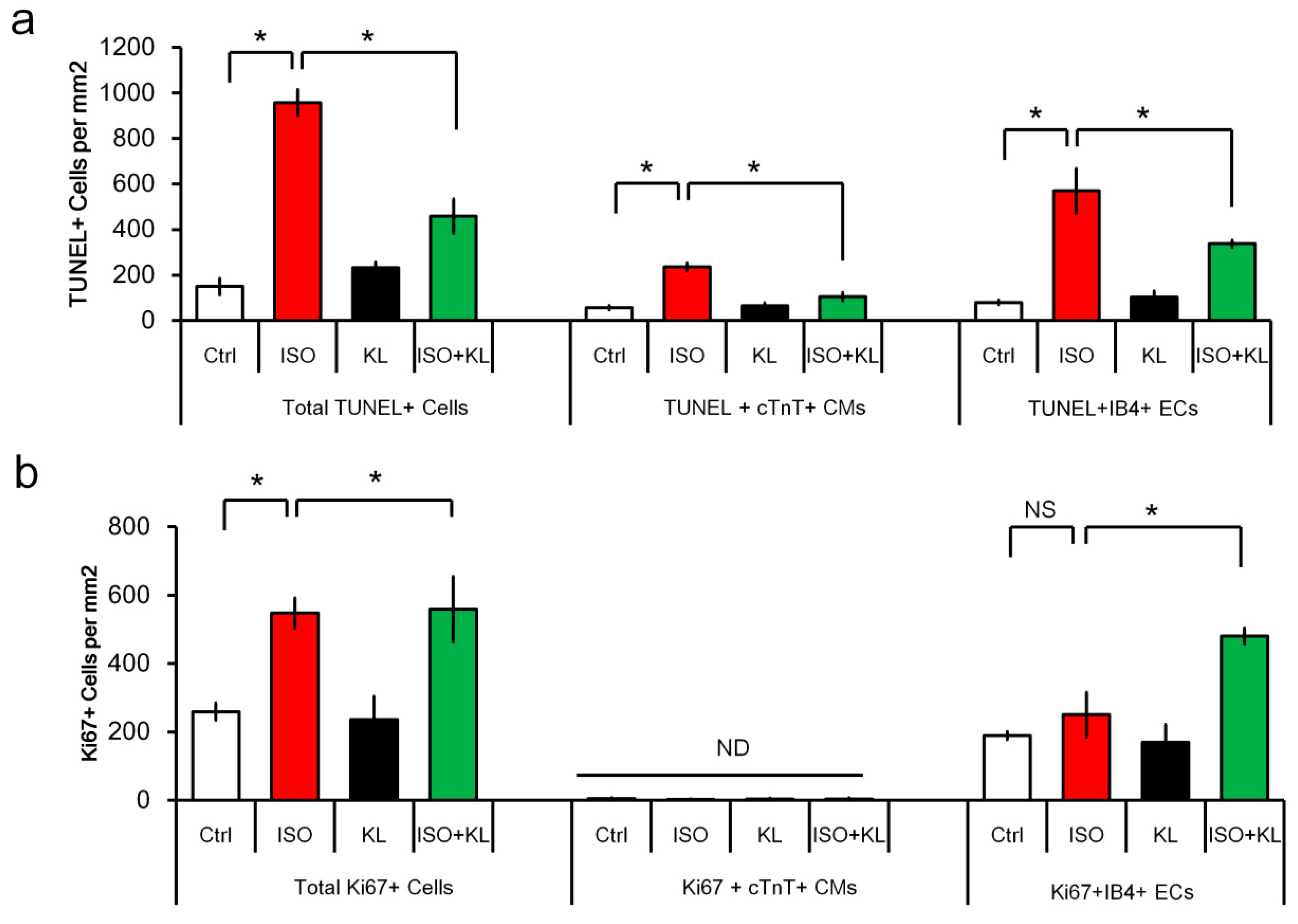
2.3. KL Attenuated Isoproterenol-Induced Apoptosis of Cardiomyocytes and Endothelial Cells
2.4. KL Increased the Number of Proliferating Endothelial Cells but Not Cardiomyocytes
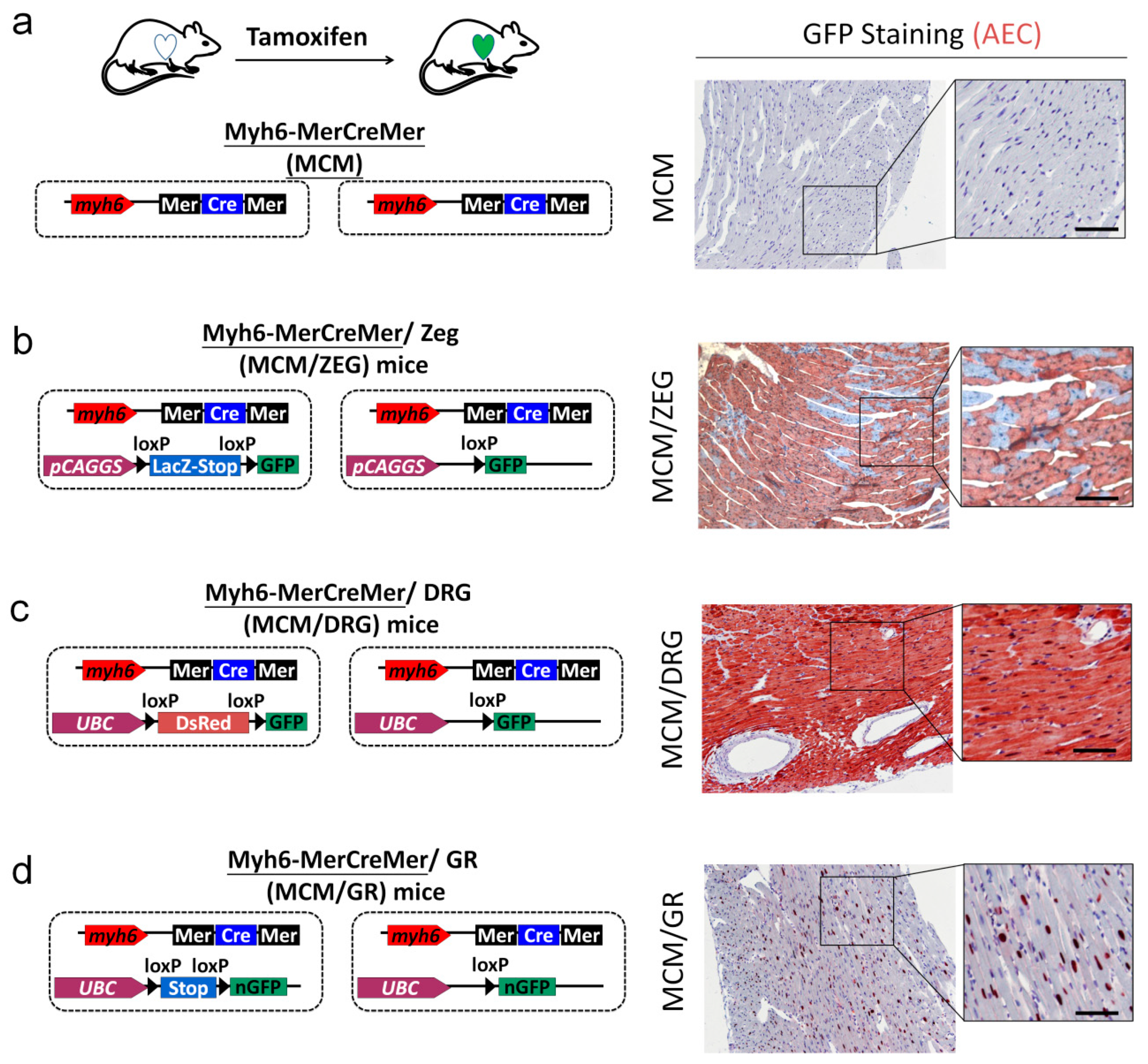
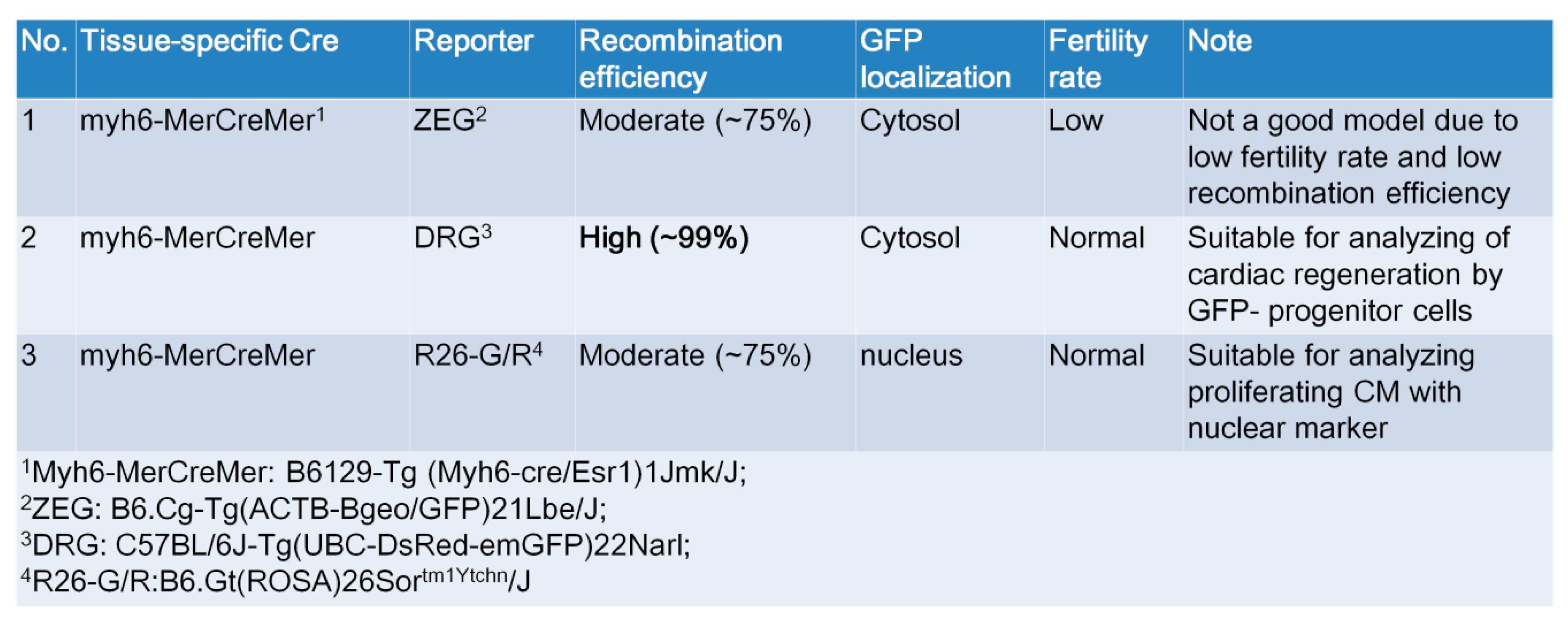
2.5. Generation of myh6-MerCreMer/GFP Reporter Mice for Studying Cardiomyocte Renewal
2.6. KL Ameliorated Isoproterenol-Induced Cardiac Fibrosis, Whereas Its Did Not Affect Cardiac Renewal in Adult Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Tamoxifen Pulse
4.3. Isoproterenol-Induced Cardiac Injury
4.4. Immunofluorescence Staining
4.5. Cell Viability Assay
4.6. Statistical Analysis
Funding
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Borchardt, T.; Braun, T. Cardiovascular regeneration in non-mammalian model systems: What are the differences between newts and man? Thromb. Haemost. 2007, 98, 311–318. [Google Scholar]
- Poss, K.D. Getting to the heart of regeneration in zebrafish. Semin. Cell Dev. Biol. 2007, 18, 36–45. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnable-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Malliaras, K.; Terrovitis, J. Cardiomyocyte proliferation vs progenitor cells in myocardial regeneration: The debate continues. Glob. Cardiol. Sci. Pract. 2013, 2013, 303–315. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Jopling, C.; Sleep, E.; Raya, M.; Marti, M.; Raya, A.; Belmonte, J.C.I. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010, 464, 606–611. [Google Scholar] [CrossRef]
- Bersell, K.; Arab, S.; Haring, B.; Kuhn, B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009, 138, 257–270. [Google Scholar] [CrossRef]
- Anversa, P.; Leri, A.; Kajstura, J. Cardiac regeneration. J. Am. Coll. Cardiol. 2006, 47, 1769–1776. [Google Scholar] [CrossRef]
- Ellison, G.M.; Vicinanza, C.; Smith, A.J.; Aquila, I.; Leone, A.; Waring, C.D.; Henning, B.J.; Stirparo, G.G.; Papait, R.; Scarfo, M.; et al. Adult c-kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 2013, 154, 827–842. [Google Scholar] [CrossRef]
- Hoch, M.; Fischer, P.; Stapel, B.; Missol-Kolka, E.; Sekkali, B.; Scherr, M.; Favret, F.; Braun, T.; Eder, M.; Schuster-Gossler, K.; et al. Erythropoietin preserves the endothelial differentiation capacity of cardiac progenitor cells and reduces heart failure during anticancer therapies. Cell Stem Cell 2011, 9, 131–143. [Google Scholar] [CrossRef]
- Laugwitz, K.L.; Moretti, A.; Lam, J.; Gruber, P.; Chen, Y.; Woodard, S.; Lin, L.Z.; Cai, C.L.; Lu, M.M.; Reth, M.; et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 2005, 433, 647–653. [Google Scholar] [CrossRef]
- Oh, H.; Bradfute, S.B.; Gallardo, T.D.; Nakamura, T.; Gaussin, V.; Mishina, Y.; Pocius, J.; Michael, L.H.; Behringer, R.R.; Garry, D.J.; et al. Cardiac progenitor cells from adult myocardium: Homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. USA 2003, 100, 12313–12318. [Google Scholar] [CrossRef]
- Pfister, O.; Mouquet, F.; Jain, M.; Summer, R.; Helmes, M.; Fine, A.; Colucci, W.S.; Liao, R. CD31- but Not CD31+ cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ. Res. 2005, 97, 52–61. [Google Scholar] [CrossRef]
- Rota, M.; Kajstura, J.; Hosoda, T.; Bearzi, C.; Vitale, S.; Esposito, G.; Iaffaldano, G.; Padin-Iruegas, M.E.; Gonzalez, A.; Rizzi, R.; et al. Bone marrow cells adopt the cardiomyogenic fate in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 17783–17788. [Google Scholar] [CrossRef]
- Smart, N.; Bollini, S.; Dube, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef]
- Smith, R.R.; Barile, L.; Cho, H.C.; Leppo, M.K.; Hare, J.M.; Messina, E.; Giacomello, A.; Abraham, M.R.; Marban, E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 2007, 115, 896–908. [Google Scholar] [CrossRef]
- Hsieh, P.C.; Segers, V.F.; Davis, M.E.; MacGillivray, C.; Gannon, J.; Molkentin, J.D.; Robbins, J.; Lee, R.T. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat. Med. 2007, 13, 970–974. [Google Scholar] [CrossRef]
- Loffredo, F.S.; Steinhauser, M.L.; Gannon, J.; Lee, R.T. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell 2011, 8, 389–398. [Google Scholar] [CrossRef]
- Malliaras, K.; Zhang, Y.; Seinfeld, J.; Galang, G.; Tseliou, E.; Cheng, K.; Sun, B.; Aminzadeh, M.; Marban, E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol. Med. 2013, 5, 191–209. [Google Scholar] [CrossRef]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Ginard, B.; Ellison, G.M.; Torella, D. The cardiac stem cell compartment is indispensable for myocardial cell homeostasis, repair and regeneration in the adult. Stem Cell Res. 2014, 13, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Senyo, S.E.; Lee, R.T.; Kuhn, B. Cardiac regeneration based on mechanisms of cardiomyocyte proliferation and differentiation. Stem Cell Res. 2014, 13, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Koudstaal, S.; Jansen Of Lorkeers, S.J.; Gaetani, R.; Gho, J.M.; van Slochteren, F.J.; Sluijter, J.P.; Doevendans, P.A.; Ellison, G.M.; Chamuleau, S.A. Concise review: Heart regeneration and the role of cardiac stem cells. Stem Cells Transl. Med. 2013, 2, 434–443. [Google Scholar] [CrossRef]
- Van Berlo, J.H.; Molkentin, J.D. An emerging consensus on cardiac regeneration. Nat. Med. 2014, 20, 1386–1393. [Google Scholar] [CrossRef]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef]
- Hsueh, Y.C.; Wu, J.M.; Yu, C.K.; Wu, K.K.; Hsieh, P.C. Prostaglandin E(2) promotes post-infarction cardiomyocyte replenishment by endogenous stem cells. EMBO Mol. Med. 2014, 6, 496–503. [Google Scholar] [CrossRef]
- Sohal, D.S.; Nghiem, M.; Crackower, M.A.; Witt, S.A.; Kimball, T.R.; Tymitz, K.M.; Penninger, J.M.; Molkentin, J.D. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 2001, 89, 20–25. [Google Scholar] [CrossRef]
- Novak, A.; Guo, C.; Yang, W.; Nagy, A.; Lobe, C.G. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 2000, 28, 147–155. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef]
- Chen, Y.T.; Tsai, M.S.; Yang, T.L.; Ku, A.T.; Huang, K.H.; Huang, C.Y.; Chou, F.J.; Fan, H.H.; Hong, J.B.; Yen, S.T.; et al. R26R-GR: A Cre-activable dual fluorescent protein reporter mouse. PLoS ONE 2012, 7, e46171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, W.Y.; Hong, J.; Gannon, J.; Kakkar, R.; Lee, R.T. Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc. Natl. Acad. Sci. USA 2015, 112, 7249–7254. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Chang, Y.J.; Su, C.H.; Tsai, T.H.; Chen, S.D.; Hsing, C.H.; Yang, J.L. Upregulation of Interleukin-33 in obstructive renal injury. Biochem. Biophys. Res. Commun. 2016, 473, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Branco, A.F.; Pereira, S.P.; Gonzalez, S.; Gusev, O.; Rizvanov, A.A.; Oliveira, P.J. Gene Expression Profiling of H9c2 Myoblast Differentiation towards a Cardiac-Like Phenotype. PLoS ONE 2015, 10, e0129303. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Xie, J.; Yoon, J.; An, S.W.; Kuro-o, M.; Huang, C.L. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. J. Am. Soc. Nephrol. JASN 2015, 26, 1150–1160. [Google Scholar] [CrossRef]
- Lim, K.; Halim, A.; Lu, T.S.; Ashworth, A.; Chong, I. Klotho: A Major Shareholder in Vascular Aging Enterprises. Int. J. Mol. Sci. 2019, 20, 4637. [Google Scholar] [CrossRef]
- Wright, J.D.; An, S.W.; Xie, J.; Lim, C.; Huang, C.L. Soluble klotho regulates TRPC6 calcium signaling via lipid rafts, independent of the FGFR-FGF23 pathway. FASEB J. 2019, 33, 9182–9193. [Google Scholar] [CrossRef]
- Song, S.; Si, L.Y. Klotho ameliorated isoproterenol-induced pathological changes in cardiomyocytes via the regulation of oxidative stress. Life Sci. 2015, 135, 118–123. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-Y. Soluble Alpha-Klotho Alleviates Cardiac Fibrosis without Altering Cardiomyocytes Renewal. Int. J. Mol. Sci. 2020, 21, 2186. https://doi.org/10.3390/ijms21062186
Chen W-Y. Soluble Alpha-Klotho Alleviates Cardiac Fibrosis without Altering Cardiomyocytes Renewal. International Journal of Molecular Sciences. 2020; 21(6):2186. https://doi.org/10.3390/ijms21062186
Chicago/Turabian StyleChen, Wei-Yu. 2020. "Soluble Alpha-Klotho Alleviates Cardiac Fibrosis without Altering Cardiomyocytes Renewal" International Journal of Molecular Sciences 21, no. 6: 2186. https://doi.org/10.3390/ijms21062186
APA StyleChen, W.-Y. (2020). Soluble Alpha-Klotho Alleviates Cardiac Fibrosis without Altering Cardiomyocytes Renewal. International Journal of Molecular Sciences, 21(6), 2186. https://doi.org/10.3390/ijms21062186