Computational Study on the Effect of Inactivating/Activating Mutations on the Inhibition of MEK1 by Trametinib


Abstract
1. Introduction
2. Results and Discussion
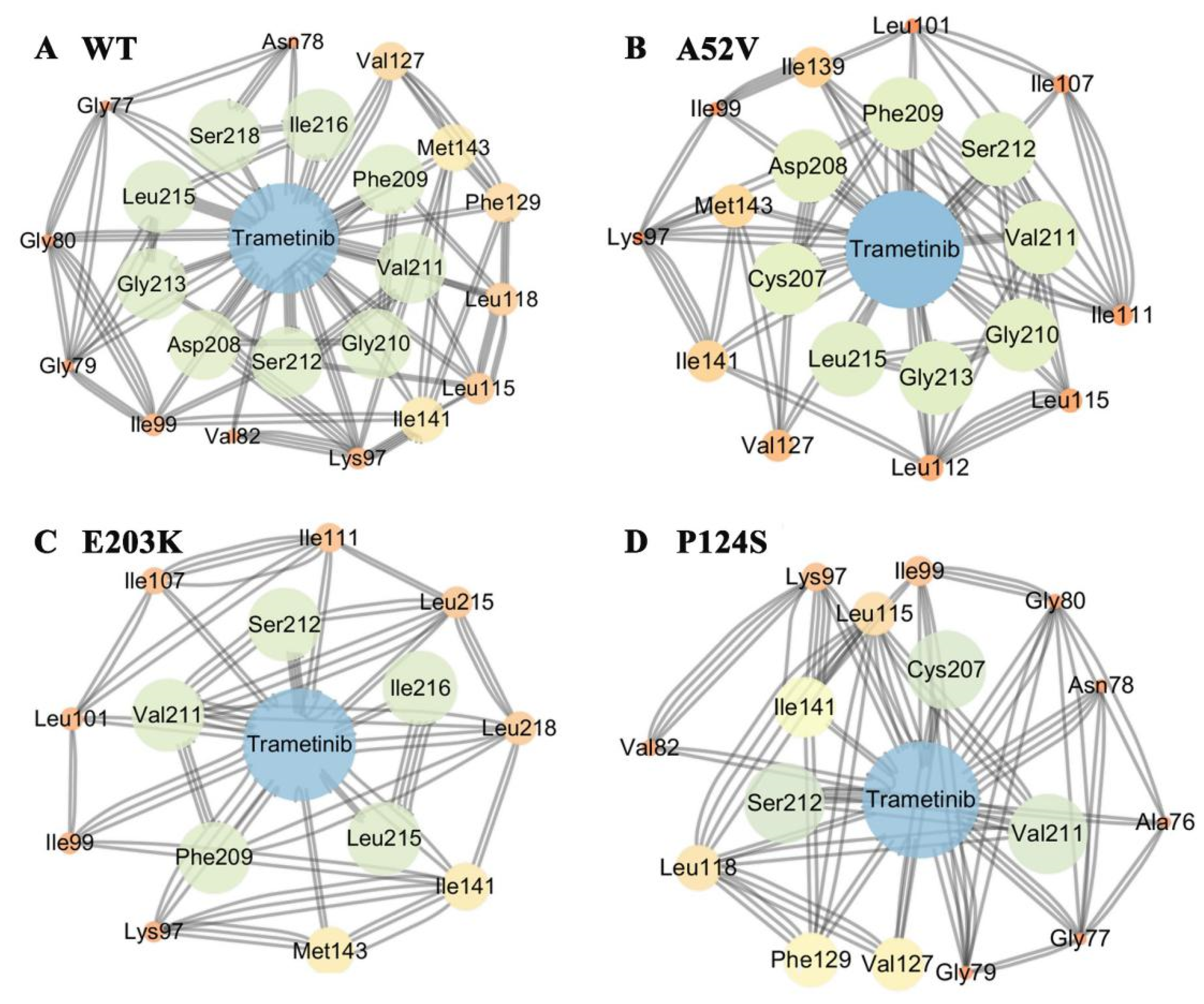
2.1. Binding Mode of Trametinib to MEK1
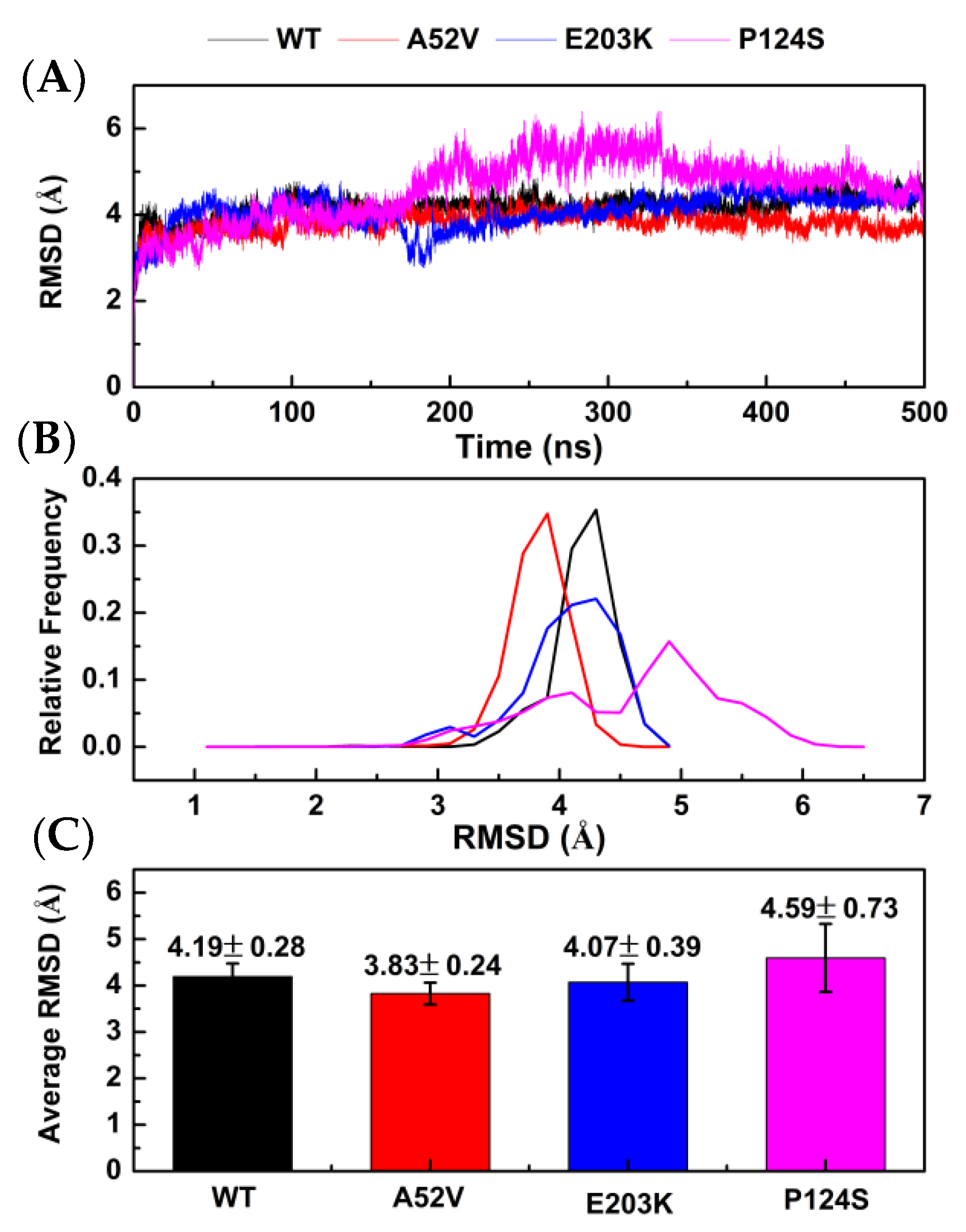
2.2. Conformational Changes for Trametinib Bound to the WT MEK1 and Three Mutants
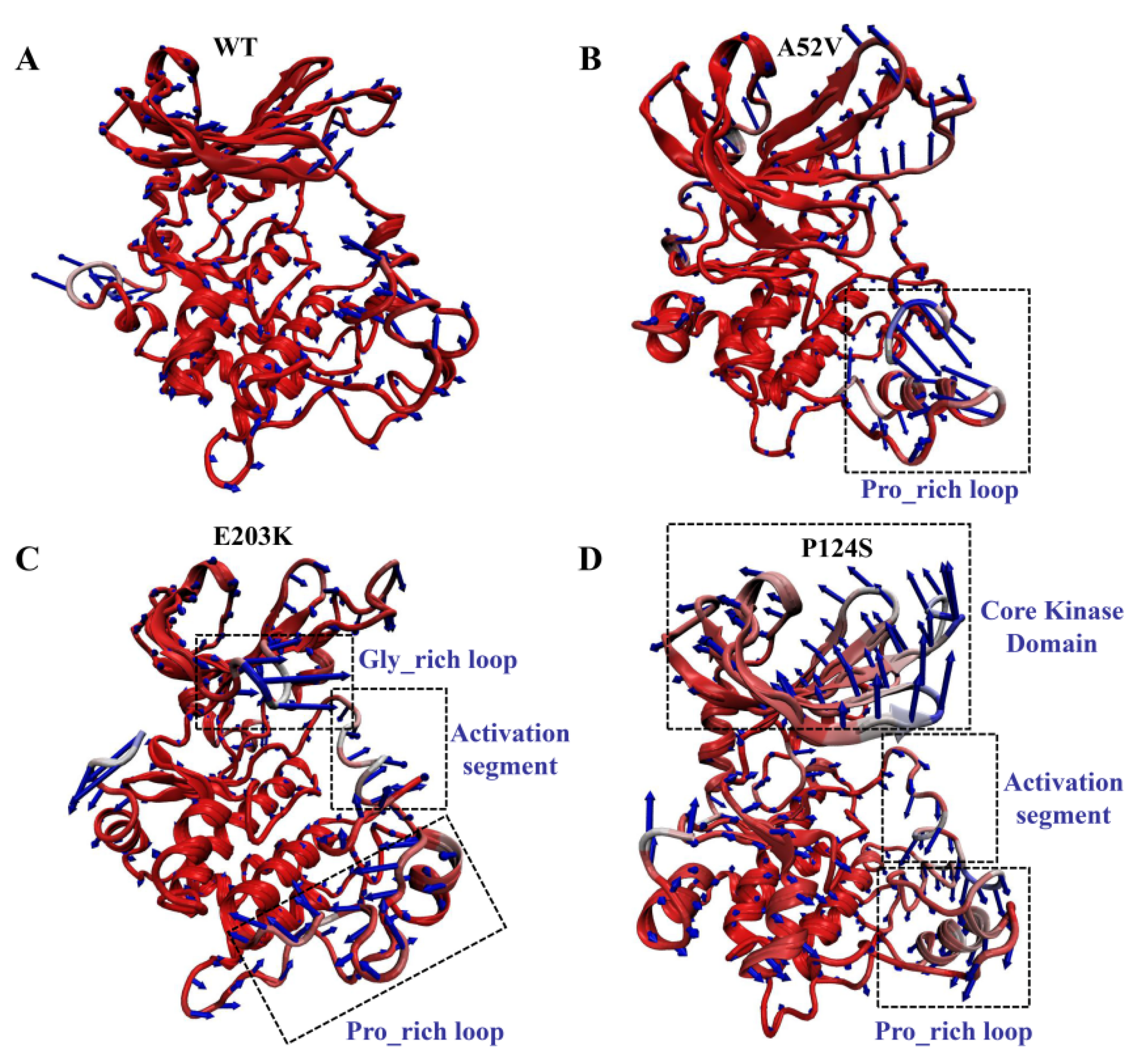
2.3. Structural Motion of the WT and Mutants MEK1 after Binding with Trametinib
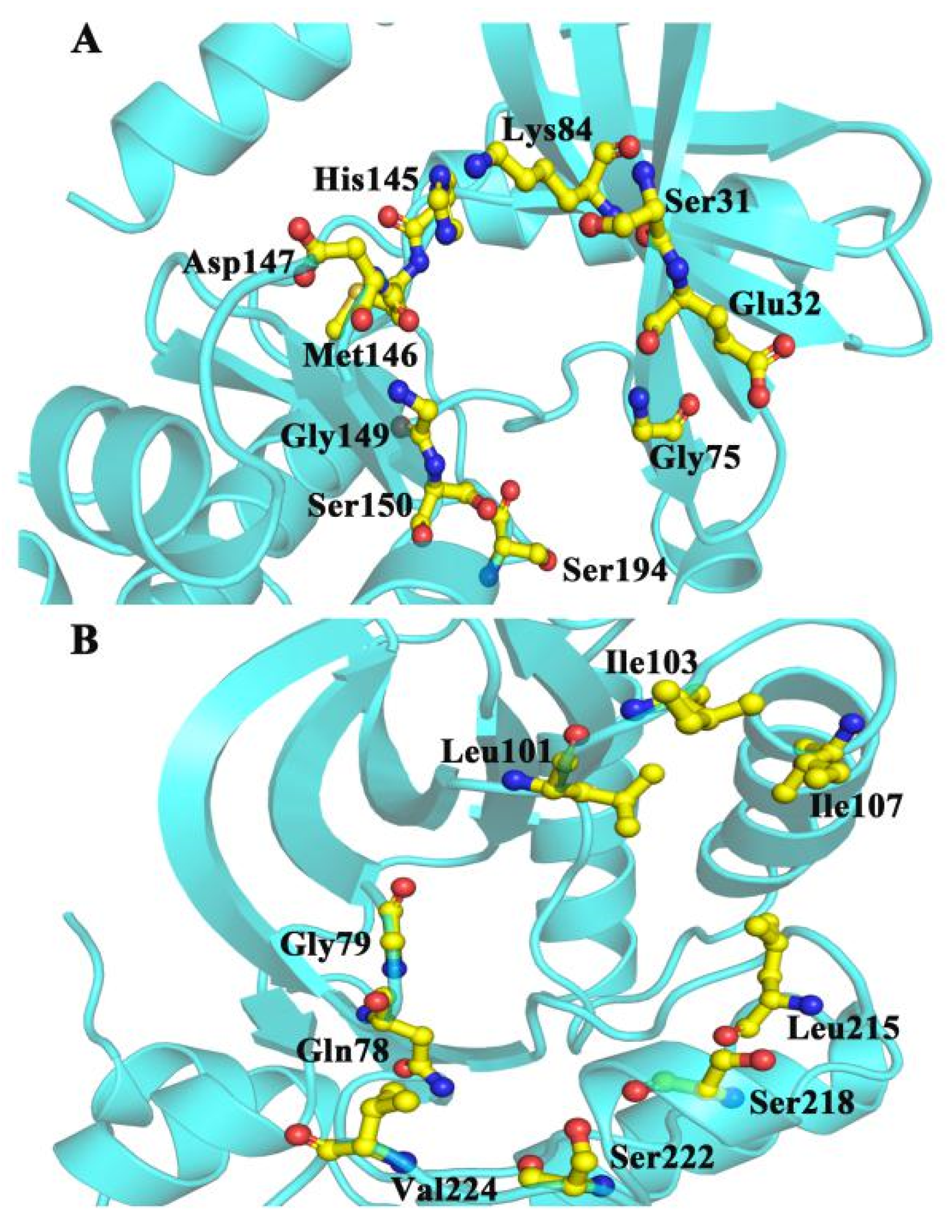
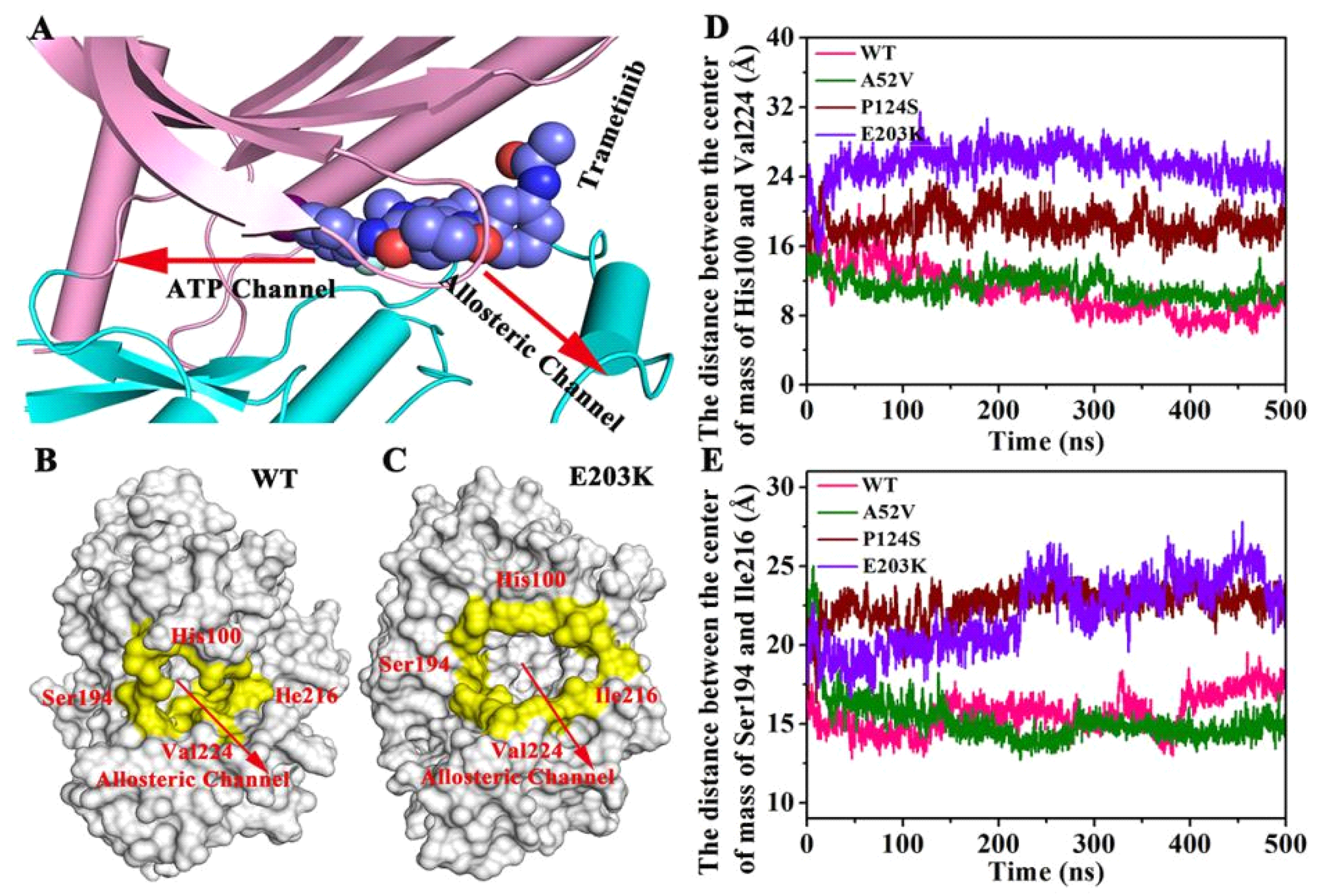
2.4. CAVER 3.0 Identifies the Allosteric Channel of MEK1
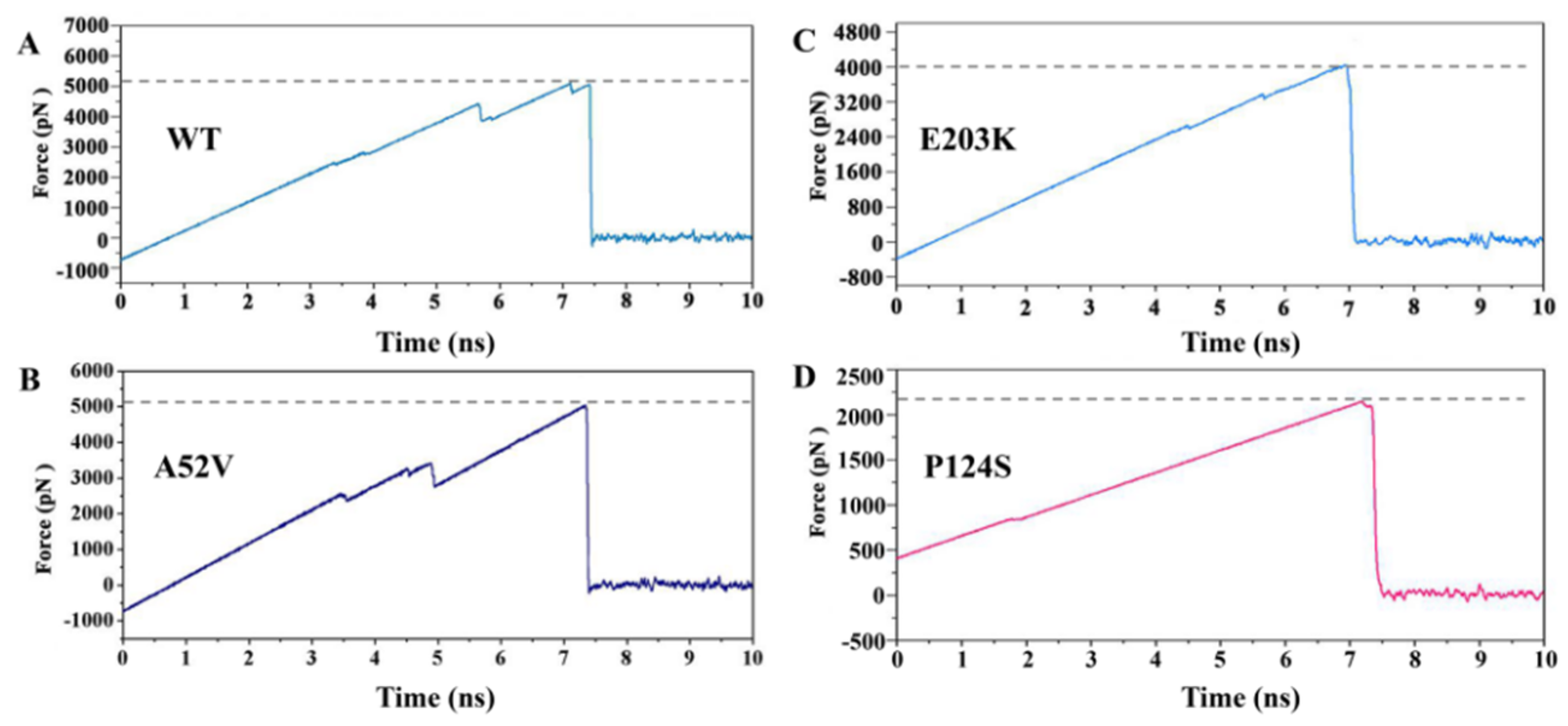
2.5. Dissociation of Trametinib from the WT MEK1 and Its Three Mutants
3. Methods
3.1. Preparation of the Protein Structures
3.2. Docking Study
3.3. MD Simulations
3.4. Protein Structure Network Analysis
3.5. Cross Correlation Analysis and Principal Component Analysis (PCA)
3.6. MM-PBSA Calculations
3.7. Steered Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zheng, C.F.; Guan, K.L. Cloning and characterization of two distinct human extracellular signal-regulated kinase activator kinases, MEK1 and MEK2. J. Biol. Chem. 1993, 268, 11435–11439. [Google Scholar] [PubMed]
- Dhanasekaran, N.; Premkumar Reddy, E. Signaling by dual specificity kinases. Oncogene 1998, 17, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Robarge, K.D.; Lee, W.; Eigenbrot, C.; Ultsch, M.; Wiesmann, C.; Heald, R.; Price, S.; Hewitt, J.; Jackson, P.; Savy, P.; et al. Structure based design of novel 6,5 heterobicyclic mitogen-activated protein kinase kinase (MEK) inhibitors leading to the discovery of imidazo[1,5-a] pyrazine G-479. Bioorg. Med. Chem. Lett. 2014, 24, 4714–4723. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.Y.; Quirion, R. The ERK/MAPK pathway, as a target for the treatment of neuropathic pain. Expert Opin. Ther. Targets 2005, 9, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Ohren, J.F.; Chen, H.F.; Pavlovsky, A.; Whitehead, C.; Zhang, E.L.; Kuffa, P.; Yan, C.H.; McConnell, P.; Spessard, C.; Banotai, C.; et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol. 2004, 11, 1192–1197. [Google Scholar] [CrossRef]
- Lugowska, I.; Kosela-Paterczyk, H.; Kozak, K.; Rutkowski, P. Trametinib: A MEK inhibitor for management of metastatic melanoma. Oncotargets Ther. 2015, 8, 2251–2259. [Google Scholar] [CrossRef]
- Umapathy, G.; Guan, J.; Gustafsson, D.E.; Javanmardi, N.; Cervantes-Madrid, D.; Djos, A.; Martinsson, T.; Palmer, R.H.; Hallberg, B. MEK inhibitor trametinib does not prevent the growth of anaplastic lymphoma kinase (ALK)–addicted neuroblastomas. Sci. Signal. 2017, 10, eaam7550. [Google Scholar] [CrossRef]
- Kang, S.H.; Keam, B.; Ahn, Y.O.; Park, H.R.; Kim, M.; Kim, T.M.; Kim, D.W.; Heo, D.S. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating anti-tumor immunity in head and neck squamous cell carcinoma. Oncoimmunology 2019, 8, e1515057. [Google Scholar] [CrossRef]
- Daud, A.; Gill, J.; Kamra, S.; Chen, L.; Ahuja, A. Indirect treatment comparison of dabrafenib plus trametinib versus vemurafenib plus cobimetinib in previously untreated metastatic melanoma patients. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef]
- Galvan-Banqueri, M.; Ubago-Perez, R.; Molina-Lopez, T. The relative clinical efficacy of trametinib-dabrafenib and cobimetinib-vemurafenib in advanced melanoma: An indirect comparison. J. Clin. Pharm. Ther. 2016, 41, 285–289. [Google Scholar] [CrossRef]
- Sun, J.; Zager, J.S.; Eroglu, Z. Encorafenib/binimetinib for the treatment of BRAF-mutant advanced, unresectable, or metastatic melanoma: Design, development, and potential place in therapy. Oncotargets Ther. 2018, 11, 9081–9089. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.; Tsai, K. Management of Treatment-Related Adverse Events with Agents Targeting the MAPK Pathway in Patients with Metastatic Melanoma. Oncologist 2017, 22, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Napolitano, S.; Vitagliano, D.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; Ciardiello, D.; Ciardiello, F.; Martinelli, E. Primary and Acquired Resistance of Colorectal Cancer Cells to Anti-EGFR Antibodies Converge on MEK/ERK Pathway Activation and Can Be Overcome by Combined MEK/EGFR Inhibition. Clin. Cancer Res. 2014, 20, 3775–3786. [Google Scholar] [CrossRef]
- Knispel, S.; Zimmer, L.; Kanaki, T.; Ugurel, S.; Schadendorf, D.; Livingstone, E. The safety and efficacy of dabrafenib and trametinib for the treatment of melanoma. Expert Opin. Drug Saf. 2018, 17, 73–87. [Google Scholar] [CrossRef]
- Verduzco, D.; Kuenzi, B.M.; Kinose, F.; Sondak, V.K.; Eroglu, Z.; Rix, U.; Smalley, K.S.M. Ceritinib Enhances the Efficacy of Trametinib in BRAF/NRAS-Wild-Type Melanoma Cell Lines. Mol. Cancer Ther. 2018, 17, 73–83. [Google Scholar] [CrossRef]
- Fu, H.; Cheng, L.X.; Jin, Y.C.; Cheng, L.; Liu, M.; Chen, L.B. MAPK Inhibitors Enhance HDAC Inhibitor-Induced Redifferentiation in Papillary Thyroid Cancer Cells Harboring BRAF(V600E): An In Vitro. Mol. Ther. Oncol. 2019, 12, 235–245. [Google Scholar] [CrossRef]
- Janne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crino, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer The SELECT-1 Randomized Clinical Trial. JAMA J. Am. Med. Assoc. 2017, 317, 1844–1853. [Google Scholar] [CrossRef]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Roskoski, R. Allosteric MEK1/2 inhibitors including cobimetanib and trametinib in the treatment of cutaneous melanomas. Pharmacol. Res. 2017, 117, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Grisham, R.N.; Sylvester, B.E.; Won, H.; McDermott, G.; DeLair, D.; Ramirez, R.; Yao, Z.; Shen, R.L.; Dao, F.; Bogomolniy, F.; et al. Extreme Outlier Analysis Identifies Occult Mitogen-Activated Protein Kinase Pathway Mutations in Patients With Low-Grade Serous Ovarian Cancer. J. Clin. Oncol. 2015, 33, 4099. [Google Scholar] [CrossRef]
- Aguilar-Arevalo, A.A.; Brown, B.C.; Bugel, L.; Cheng, G.; Church, E.D.; Conrad, J.M.; Dharmapalan, R.; Djurcic, Z.; Finley, D.A.; Ford, R.; et al. First measurement of the muon antineutrino double-differential charged-current quasielastic cross section. Phys. Rev. D 2013, 88. [Google Scholar] [CrossRef]
- Nikolaev, S.I.; Rimoldi, D.; Iseli, C.; Valsesia, A.; Robyr, D.; Gehrig, C.; Harshman, K.; Guipponi, M.; Bukach, O.; Zoete, V.; et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat. Genet. 2012, 44, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.J.; Chang, M.T.; Johnsen, H.C.; Gao, S.P.; Sylvester, B.E.; Sumer, S.O.; Zhang, H.X.; Solit, D.B.; Taylor, B.S.; Schultz, N.; et al. 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Han, W.W.; Zhu, J.X.; Wang, S.; Xu, D. Understanding the Phosphorylation Mechanism by Using Quantum Chemical Calculations and Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 3565–3573. [Google Scholar] [CrossRef]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.P.A.; Katzmann, D.J.; Sosa, C.P. Multilevel Parallelization of AutoDock 4.2. J. Cheminform. 2011, 3. [Google Scholar] [CrossRef]
- Jin, H.Y.; Zhou, Z.H.; Wang, D.M.; Guan, S.S.; Han, W.W. Molecular Dynamics Simulations of Acylpeptide Hydrolase Bound to Chlorpyrifosmethyl Oxon and Dichlorvos. Int. J. Mol. Sci. 2015, 16, 6217–6234. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Zhang, H.X.; Zheng, Q.C. A unique activation-promotion mechanism of the influenza B M2 proton channel uncovered by multiscale simulations. Phys. Chem. Chem. Phys. 2019, 21, 2984–2991. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.S.; Zhao, L.; Jin, H.Y.; Shan, N.; Han, W.W.; Wang, S.; Shan, Y.M. Binding modes of phosphotriesterase-like lactonase complexed with -nonanoic lactone and paraoxon using molecular dynamics simulations. J. Biomol. Struct. Dyn. 2017, 35, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wan, Y.Z.; Zhu, J.X.; Yu, Z.F.; Tian, X.P.; Han, J.R.; Zhang, Z.M.; Han, W.W. Theoretical Study on Zearalenol Compounds Binding with Wild Type Zearalenone Hydrolase and V153H Mutant. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Schuttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sec. D Struct. Biol. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.V.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 5741. [Google Scholar] [CrossRef]
- O’Driscoll, P.; Merenyi, E.; Karmonik, C.; Grossman, R. SOM and MCODE Methods of Defining Functional Clusters in MRI of the Brain. In Proceedings of the 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Chicago, IL, USA, 26–30 August 2014; pp. 734–737. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An Nlog (N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Kang, Y.B.; Haghighi, P.D.; Burstein, F. CFinder: An intelligent key concept finder from text for ontology development. Expert Syst. Appl. 2014, 41, 4494–4504. [Google Scholar] [CrossRef]
- Qian, M.D.; Shan, Y.M.; Guan, S.S.; Zhang, H.; Wang, S.; Han, W.W. Structural Basis of Fullerene Derivatives as Novel Potent Inhibitors of Protein Tyrosine Phosphatase 1B: Insight into the Inhibitory Mechanism through Molecular Modeling Studies. J. Chem. Inf. Model. 2016, 56, 2024–2034. [Google Scholar] [CrossRef]
- Hou, T.J.; Wang, J.M.; Li, Y.Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Probability for α_Helix (%) | Probability for 310_Helix (%) |
|---|---|---|
| N214-I216 | P193-N195 | |
| WT | 54.6 | 62.0 |
| A52V | 94.7 | 79.6 |
| E203K | 2.8 | 17.1 |
| P124S | 8.8 | 44.2 |
| Hydrogen Bonds | WT (%) | A52V (%) | P124S (%) | E203K (%) | |
|---|---|---|---|---|---|
| Donor | Accepter | ||||
| Gly210:N | MOL:F34 | 81.8 ± 0.4 | 81.0 ± 0.4 | 23.9 ± 0.4 | 67.4 ± 0.5 |
| Gly213:N | MOL:C10 | 40.8 ± 0.5 | 44.5 ± 0.5 | -- | -- |
| Val211:N | MOL:O8 | 84.0 ± 0.4 | 80.9 ± 0.4 | 21.1 ± 0.4 | 79.6 ± 0.4 |
| Ser212:N | MOL:O8 | 91.4 ± 0.3 | 81.4 ± 0.4 | 23.7 ± 0.4 | -- |
| Ser212:OG | MOL:O8 | 58.4 ± 0.5 | 80.5 ± 0.4 | 45.9 ± 0.5 | 22.2 ± 0.4 |
| Ser212:CB | MOL:O8 | 77.0 ± 0.4 | 62.5 ± 0.5 | 37.1 ± 0.5 | -- |
| Ser212:OG | MOL:C7 | 81.9 ± 0.4 | 73.3 ± 0.4 | 33.4 ± 0.5 | 58.6 ± 0.5 |
| Lys97:NZ | MOL:O12 | 71.6 ± 0.5 | 86.8 ± 0.3 | 65.3 ± 0.5 | 46.3 ± 0.5 |
| Phe209:N | MOL:F34 | 37.9 ± 0.5 | 47.4 ± 0.5 | 26.0 ± 0.4 | -- |
| Lys97:CD | MOL:O12 | 72.6 ± 0.4 | 40.8 ± 0.5 | 20.4 ± 0.4 | 39.3 ± 0.5 |
| Lys97:CE | MOL:O12 | 66.6 ± 0.5 | 32.7 ± 0.5 | 23.4 ± 0.4 | 37.6 ± 0.5 |
| Gly210:CA | MOL:F34 | 73.1 ± 0.4 | 66.9 ± 0.5 | -- | 41.9 ± 0.5 |
| MOL:C35 | Asp208:N | 31.6 ± 0.5 | 20.8 ± 0.4 | -- | -- |
| Val211:N | MOL:F34 | -- | 36.1 ± 0.5 | -- | -- |
| Ser212:CA | MOL:O8 | 81.9 ± 0.4 | 73.5 ± 0.4 | -- | 28.9 ± 0.5 |
| MOL:N22 | His100:O | 35.7 ± 0.5 | 24.5 ± 0.4 | -- | -- |
| Asp208:N | MOL:C36 | 36.3 ± 0.5 | 36.6 ± 0.5 | -- | -- |
| System | WT | A52V | P124S | E203K |
|---|---|---|---|---|
| ΔEele | −85.0 ± 10.4 | −87.9 ± 8.3 | −70.0 ± 7.5 | −69.9 ± 6.2 |
| ΔEvdw | −54.6 ± 4.9 | −51.7 ± 4.7 | −41.0 ± 3.0 | −43.7 ± 3.9 |
| ΔGnp | −5.6 ± 2.0 | −5.4 ± 1.6 | −4.8 ± 2.2 | −5.0 ± 1.7 |
| ΔGpb | 90.1 ± 13.6 | 93.2 ± 12.1 | 75.1 ± 10.8 | 76.5 ± 15.3 |
| TΔS | −60.2 ± 5.5 | −59.1 ± 4.9 | −45.9 ± 3.4 | −48.7 ± 3.2 |
| ΔGbind | −55.0 ± 3.9 | −53.9 ± 2.5 | −40.7 ± 2.1 | −42.2 ± 3.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.; Li, C.; Yang, H.; Guo, X.; Huang, T.; Han, W. Computational Study on the Effect of Inactivating/Activating Mutations on the Inhibition of MEK1 by Trametinib. Int. J. Mol. Sci. 2020, 21, 2167. https://doi.org/10.3390/ijms21062167
Zhu J, Li C, Yang H, Guo X, Huang T, Han W. Computational Study on the Effect of Inactivating/Activating Mutations on the Inhibition of MEK1 by Trametinib. International Journal of Molecular Sciences. 2020; 21(6):2167. https://doi.org/10.3390/ijms21062167
Chicago/Turabian StyleZhu, Jingxuan, Congcong Li, Hengzheng Yang, Xiaoqing Guo, Tianci Huang, and Weiwei Han. 2020. "Computational Study on the Effect of Inactivating/Activating Mutations on the Inhibition of MEK1 by Trametinib" International Journal of Molecular Sciences 21, no. 6: 2167. https://doi.org/10.3390/ijms21062167
APA StyleZhu, J., Li, C., Yang, H., Guo, X., Huang, T., & Han, W. (2020). Computational Study on the Effect of Inactivating/Activating Mutations on the Inhibition of MEK1 by Trametinib. International Journal of Molecular Sciences, 21(6), 2167. https://doi.org/10.3390/ijms21062167