Insights into the Therapeutic Potential of Glucocorticoid Receptor Modulators for Neurodegenerative Diseases


 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Hypercorticosteronemia in Experimental Neurodegenerative Disorders
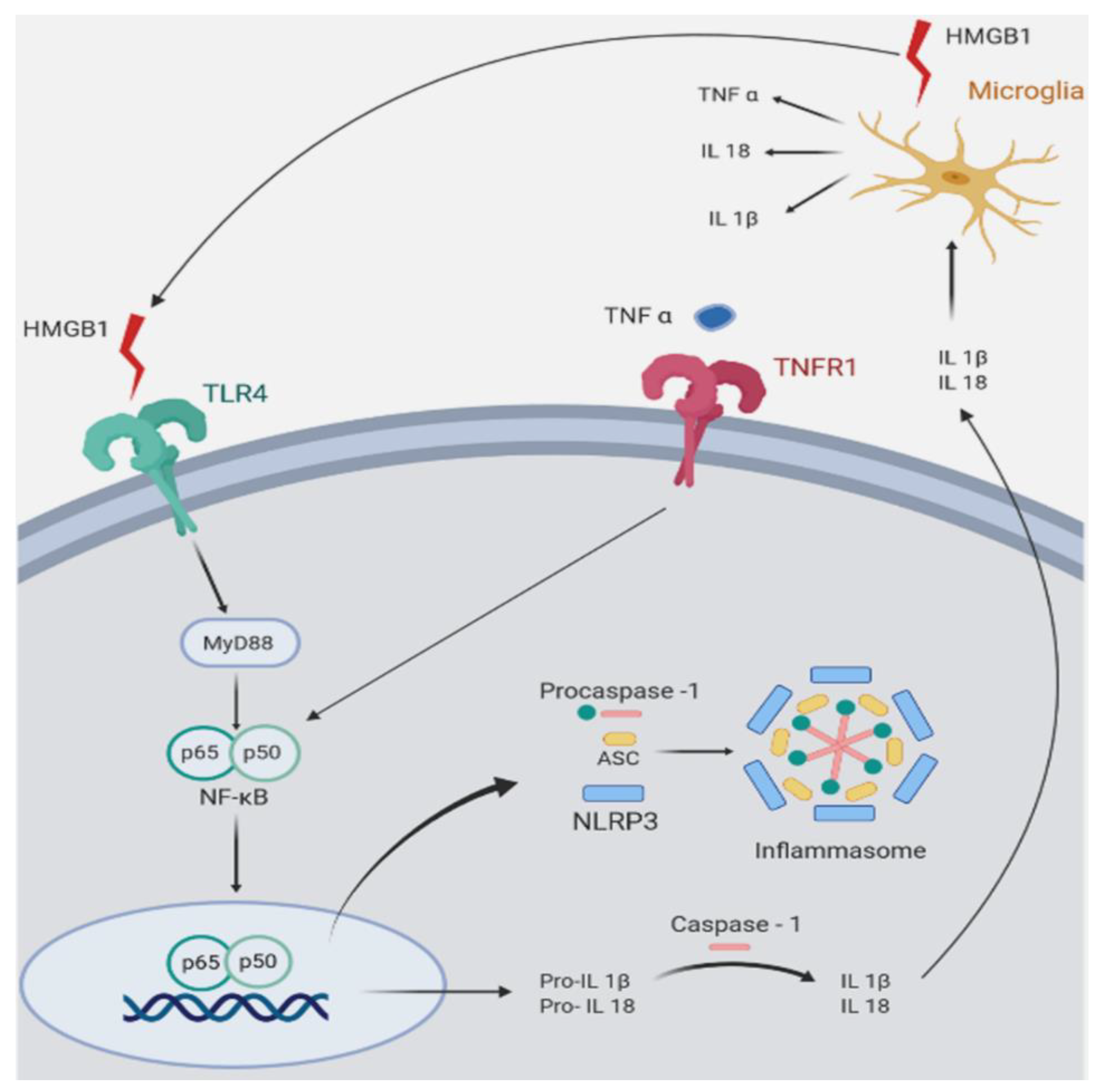
3. Glucocorticoids, Inflammation, and CNS Disorders
4. Effect of Glucocorticoid Receptor Antagonists in CNS Disorders

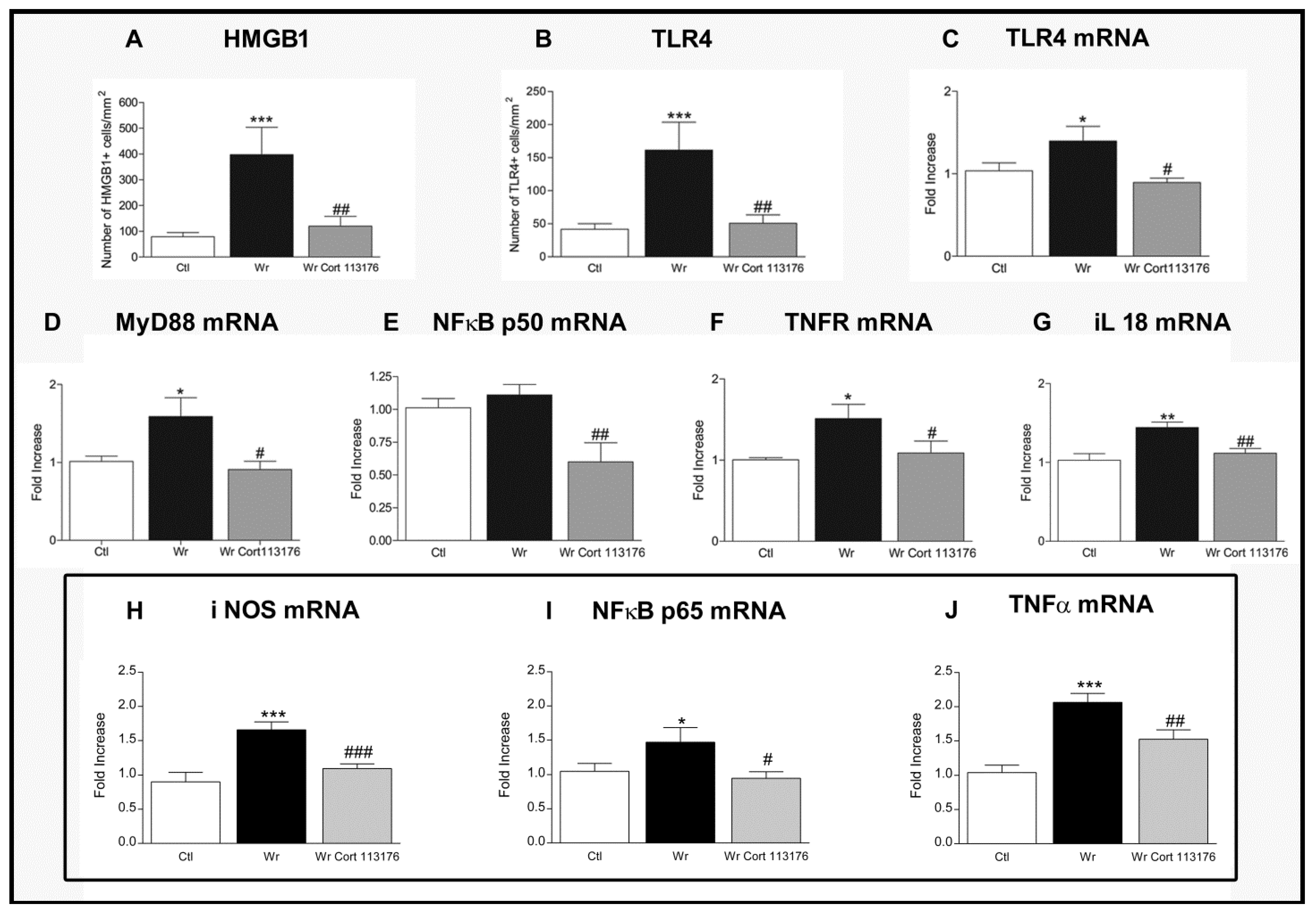
5. Efficacy of a Glucocorticoid Receptor Antagonist in Wobbler Neurodegeneration
6. GRA and Neurodegenerative Diseases: Lessons from Animal Models
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACTH | adrenocorticotrophic hormone |
| AKT | a serine/thronine kinase |
| ALS | amyotrophic lateral sclerosis |
| APP | amyloid precursor protein |
| CD11b | cluster of differentiation molecule 11 B |
| CNS | central nervous system |
| EAE | experimental autoimmune encephalomyelitis |
| GC/MS | gas chromatography /mass spectrometry |
| GFAP | glial fibrillary acidic protein |
| GLAST | glutamate/aspartate transporter |
| GLT1 | glutamate transporter 1 |
| GR | glucocorticoid receptor |
| GRA | glucocorticoid receptor antagonist |
| GRE | glucocorticoid response element |
| HMGB1 | high mobility group box 1 protein |
| HPA | hypothalamic pituitary adrenal axis |
| HSD | 11β-hydroxusteroid dehydrogenase |
| IL18 | interleukin 18 |
| IL1β | interleukin 1 beta |
| IL6 | interleukin 6 |
| JNK | c-Jun-N-terminal kinase |
| MR | mineralocorticoid receptor |
| MS | multiple sclerosis |
| MyD88 | myeloid differentiation primary response 88 |
| NFκB | nuclear factor kappa B |
| NLPR3 | NLR family pyrin domain containing 3 inflammasome |
| NMDA | n-metyl-D-aspartate |
| iNOS | nitric oxide synthase |
| PVN | paraventricular nucleus |
| SOD1 | superoxide dismutase 1 |
| TDP43 | transactive response DNA-binding protein 42 |
| TLR4 | toll-like receptor 4 |
| TNFR | tumor necrosis factor receptor |
| TNFα | tumor necrosis factor alpha |
| Vps54 | vesicular vacuolar protein sorting 54 |
References
- Sierra, A.; Gottfried-Blackmore, A.; Milner, T.A.; McEwen, B.S.; Bulloch, K. Steroid hormone receptor expression and function in microglia. Glia 2008, 56, 659–674. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.D.; Kogan, J.F.; Marrocco, J.; McEwen, B.S. Genomic and epigenomic mechanisms of glucocorticoids in the brain. Nat. Rev. Endocrinol. 2017, 13, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Joels, M.; de Kloet, E.R. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: The brain mineralocorticoid receptor: A saga in three episodes. J. Endocrinol. 2017, 234, T49–T66. [Google Scholar] [CrossRef]
- Ahima, R.; Krozowski, Z.; Harlan, R. Type I corticosteroid receptor-like immunoreactivity in the rat CNS: Distribution and regulation by corticosteroids. J. Comp. Neurol. 1991, 313, 522–538. [Google Scholar] [CrossRef]
- Reul, J.M.; de Kloet, E.R. Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology 1985, 117, 2505–2511. [Google Scholar] [CrossRef]
- de Kloet, E.R.; Meijer, O.C.; de Nicola, A.F.; de Rijk, R.H.; Joels, M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocrinol. 2018, 49, 124–145. [Google Scholar] [CrossRef]
- Ahima, R.S.; Harlan, R.E. Charting of type II glucocorticoid receptor-like immunoreactivity in the rat central nervous system. Neuroscience 1990, 39, 579–604. [Google Scholar] [CrossRef]
- de Kloet, E.R. From receptor balance to rational glucocorticoid therapy. Endocrinology 2014, 155, 2754–2769. [Google Scholar] [CrossRef]
- Picard, M.; McEwen, B.S.; Epel, E.S.; Sandi, C. An energetic view of stress: Focus on mitochondria. Front. Neuroendocrinol. 2018, 49, 72–85. [Google Scholar] [CrossRef]
- Selye, H. Stress: The physiology and pathology of exposure to stress. Acta Inc. Montreal 1950. [Google Scholar] [CrossRef]
- Edwards, C.R.; Stewart, P.M.; Burt, D.; Brett, L.; McIntyre, M.A.; Sutanto, W.S.; de Kloet, E.R.; Monder, C. Localisation of 11 beta-hydroxysteroid dehydrogenase--tissue specific protector of the mineralocorticoid receptor. Lancet 1988, 2, 986–989. [Google Scholar] [CrossRef]
- Gonzalez, S.L.; Saravia, F.; Gonzalez Deniselle, M.C.; Lima, A.E.; De Nicola, A.F. Glucocorticoid regulation of motoneuronal parameters in rats with spinal cord injury. Cell Mol. Neurobiol. 1999, 19, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Yan, P.; Xiao, Q.; Chen, S.; Lee, K.Y.; Hsu, C.Y.; Xu, J. Methylprednisolone protects oligodendrocytes but not neurons after spinal cord injury. J. Neurosci. 2008, 28, 3141–3149. [Google Scholar] [CrossRef]
- Batista, P.; Pereira, A. Biomarkers in neurodegenerative diseases: Cortisol. J. Mol. Biomark Diagnosis. 2016, 7, 2. [Google Scholar] [CrossRef]
- Popp, J.; Wolfsgruber, S.; Heuser, I.; Peters, O.; Hull, M.; Schroder, J.; Moller, H.J.; Lewczuk, P.; Schneider, A.; Jahn, H.; et al. Cerebrospinal fluid cortisol and clinical disease progression in MCI and dementia of Alzheimer’s type. Neurobiol. Aging 2015, 36, 601–607. [Google Scholar] [CrossRef]
- Sotiropoulos, I.; Catania, C.; Pinto, L.G.; Silva, R.; Pollerberg, G.E.; Takashima, A.; Sousa, N.; Almeida, O.F. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 2011, 31, 7840–7847. [Google Scholar] [CrossRef]
- Soares, N.M.; Pereira, G.M.; Altmann, V.; de Almeida, R.M.M.; Rieder, C.R.M. Cortisol levels, motor, cognitive and behavioral symptoms in Parkinson’s disease: A systematic review. J. Neural. Transm. (Vienna) 2019, 126, 219–232. [Google Scholar] [CrossRef]
- Bartlett, D.M.; Cruickshank, T.M.; Hannan, A.J.; Eastwood, P.R.; Lazar, A.S.; Ziman, M.R. Neuroendocrine and neurotrophic signaling in Huntington’s disease: Implications for pathogenic mechanisms and treatment strategies. Neurosci. Biobehav. Rev. 2016, 71, 444–454. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Yadav, V.; Marks, D.L. Hypothalamic Dysfunction and Multiple Sclerosis: Implications for Fatigue and Weight Dysregulation. Curr. Neurol. Neurosci. Rep. 2016, 16, 98. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo-Monachelli, G.M.; Sivori, M.; Meyer, M.; Sica, R.E.; De Nicola, A.F.; Gonzalez-Deniselle, M.C. Circulating gonadal and adrenal steroids in amyotrophic lateral sclerosis: Possible markers of susceptibility and outcome. Horm. Metab. Res. 2014, 46, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Patacchioli, F.R.; Monnazzi, P.; Scontrini, A.; Tremante, E.; Caridi, I.; Brunetti, E.; Buttarelli, F.R.; Pontieri, F.E. Adrenal dysregulation in amyotrophic lateral sclerosis. J. Endocrinol. Invest. 2003, 26, RC23–RC25. [Google Scholar] [CrossRef] [PubMed]
- Roozendaal, B.; Kim, S.; Wolf, O.T.; Kim, M.S.; Sung, K.K.; Lee, S. The cortisol awakening response in amyotrophic lateral sclerosis is blunted and correlates with clinical status and depressive mood. Psychoneuroendocrinology 2012, 37, 20–26. [Google Scholar] [CrossRef]
- Spataro, R.; Volanti, P.; Vitale, F.; Meli, F.; Colletti, T.; Di Natale, A.; La Bella, V. Plasma cortisol level in amyotrophic lateral sclerosis. J. Neurol Sci 2015, 358, 282–286. [Google Scholar] [CrossRef]
- Yates, K.F.; Sweat, V.; Yau, P.L.; Turchiano, M.M.; Convit, A. Impact of metabolic syndrome on cognition and brain: A selected review of the literature. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2060–2067. [Google Scholar] [CrossRef]
- Moraitis, A.G.; Block, T.; Nguyen, D.; Belanoff, J.K. The role of glucocorticoid receptors in metabolic syndrome and psychiatric illness. J. Steroid Biochem. Mol. Biol. 2017, 165, 114–120. [Google Scholar] [CrossRef]
- Schatzberg, A.F.; Rothschild, A.J.; Langlais, P.J.; Bird, E.D.; Cole, J.O. A corticosteroid/dopamine hypothesis for psychotic depression and related states. J. Psychiatr. Res. 1985, 19, 57–64. [Google Scholar] [CrossRef]
- Casas, S.; Perez, A.F.; Mattiazzi, M.; Lopez, J.; Folgueira, A.; Gargiulo-Monachelli, G.M.; Gonzalez Deniselle, M.C.; De Nicola, A.F. Potential Biomarkers with Plasma Cortisol, Brain-derived Neurotrophic Factor and Nitrites in Patients with Acute Ischemic Stroke. Curr. Neurovasc. Res. 2017, 14, 338–346. [Google Scholar] [CrossRef]
- Ong, L.K. Walker, Frederick, Nilsson, Michael. Is Stroke a Neurodegenerative Condition? A Critical Review of Secondary Neurodegeneration and Amyloid-beta Accumulation after Stroke. AIMS Medical. Science 2017, 4, 1–16. [Google Scholar] [CrossRef]
- Stuart, K.E.; King, A.E.; Fernandez-Martos, C.M.; Summers, M.J.; Vickers, J.C. Environmental novelty exacerbates stress hormones and Abeta pathology in an Alzheimer’s model. Sci. Rep. 2017, 7, 2764. [Google Scholar] [CrossRef] [PubMed]
- Lante, F.; Chafai, M.; Raymond, E.F.; Pereira, A.R.; Mouska, X.; Kootar, S.; Barik, J.; Bethus, I.; Marie, H. Subchronic glucocorticoid receptor inhibition rescues early episodic memory and synaptic plasticity deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2015, 40, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Kootar, S.; Frandemiche, M.L.; Dhib, G.; Mouska, X.; Lorivel, T.; Poupon-Silvestre, G.; Hunt, H.; Tronche, F.; Bethus, I.; Barik, J.; et al. Identification of an acute functional cross-talk between amyloid-beta and glucocorticoid receptors at hippocampal excitatory synapses. Neurobiol. Dis. 2018, 118, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Rodrigues, A.J.; Silva, J.M.; Tronche, F.; Almeida, O.F.; Sousa, N.; Sotiropoulos, I. Chronic Stress and Glucocorticoids: From Neuronal Plasticity to Neurodegeneration. Neural. Plast 2016, 2016, 6391686. [Google Scholar] [CrossRef]
- Canet, G.; Chevallier, N.; Zussy, C.; Desrumaux, C.; Givalois, L. Central Role of Glucocorticoid Receptors in Alzheimer’s Disease and Depression. Front. Neurosci. 2018, 12, 739. [Google Scholar] [CrossRef] [PubMed]
- Bjorkqvist, M.; Petersen, A.; Bacos, K.; Isaacs, J.; Norlen, P.; Gil, J.; Popovic, N.; Sundler, F.; Bates, G.P.; Tabrizi, S.J.; et al. Progressive alterations in the hypothalamic-pituitary-adrenal axis in the R6/2 transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 2006, 15, 1713–1721. [Google Scholar] [CrossRef]
- Acharjee, S.; Nayani, N.; Tsutsui, M.; Hill, M.N.; Ousman, S.S.; Pittman, Q.J. Altered cognitive-emotional behavior in early experimental autoimmune encephalitis--cytokine and hormonal correlates. Brain Behav. Immun. 2013, 33, 164–172. [Google Scholar] [CrossRef]
- Caudal, D.; Alvarsson, A.; Bjorklund, A.; Svenningsson, P. Depressive-like phenotype induced by AAV-mediated overexpression of human alpha-synuclein in midbrain dopaminergic neurons. Exp. Neurol. 2015, 273, 243–252. [Google Scholar] [CrossRef]
- Schmitt-John, T. VPS54 and the wobbler mouse. Front. Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef]
- Gonzalez Deniselle, M.C.; Gonzalez, S.; Piroli, G.; Ferrini, M.; Lima, A.E.; De Nicola, A.F. Glucocorticoid receptors and actions in the spinal cord of the Wobbler mouse, a model for neurodegenerative diseases. J. Steroid. Biochem. Mol. Biol. 1997, 60, 205–213. [Google Scholar] [CrossRef]
- Gonzalez Deniselle, M.C.; Liere, P.; Pianos, A.; Meyer, M.; Aprahamian, F.; Cambourg, A.; Di Giorgio, N.P.; Schumacher, M.; De Nicola, A.F.; Guennoun, R. Steroid Profiling in Male Wobbler Mouse, a Model of Amyotrophic Lateral Sclerosis. Endocrinology 2016, 157, 4446–4460. [Google Scholar] [CrossRef] [PubMed]
- Diana, V.; Ottolina, A.; Botti, F.; Fumagalli, E.; Calcagno, E.; De Paola, M.; Cagnotto, A.; Invernici, G.; Parati, E.; Curti, D.; et al. Neural precursor-derived astrocytes of wobbler mice induce apoptotic death of motor neurons through reduced glutamate uptake. Exp. Neurol. 2010, 225, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Gonzalez Deniselle, M.C.; Hunt, H.; de Kloet, E.R.; De Nicola, A.F. The selective glucocorticoid receptor modulator CORT108297 restores faulty hippocampal parameters in Wobbler and corticosterone-treated mice. J. Steroid. Biochem. Mol. Biol. 2014, 143, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Lara, A.; Hunt, H.; Belanoff, J.; de Kloet, E.R.; Gonzalez Deniselle, M.C.; De Nicola, A.F. The Selective Glucocorticoid Receptor Modulator Cort 113176 Reduces Neurodegeneration and Neuroinflammation in Wobbler Mice Spinal Cord. Neuroscience 2018, 384, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Thielsen, K.D.; Moser, J.M.; Schmitt-John, T.; Jensen, M.S.; Jensen, K.; Holm, M.M. The Wobbler mouse model of amyotrophic lateral sclerosis (ALS) displays hippocampal hyperexcitability, and reduced number of interneurons, but no presynaptic vesicle release impairments. PLoS ONE 2013, 8, e82767. [Google Scholar] [CrossRef]
- Fidler, J.A.; Treleaven, C.M.; Frakes, A.; Tamsett, T.J.; McCrate, M.; Cheng, S.H.; Shihabuddin, L.S.; Kaspar, B.K.; Dodge, J.C. Disease progression in a mouse model of amyotrophic lateral sclerosis: The influence of chronic stress and corticosterone. FASEB J. 2011, 25, 4369–4377. [Google Scholar] [CrossRef]
- Caccamo, A.; Medina, D.X.; Oddo, S. Glucocorticoids exacerbate cognitive deficits in TDP-25 transgenic mice via a glutathione-mediated mechanism: Implications for aging, stress and TDP-43 proteinopathies. J. Neurosci. 2013, 33, 906–913. [Google Scholar] [CrossRef]
- Dammann, C.; Stapelfeld, C.; Maser, E. Expression and activity of the cortisol-activating enzyme 11beta-hydroxysteroid dehydrogenase type 1 is tissue and species-specific. Chem. Biol. Interact. 2019, 303, 57–61. [Google Scholar] [CrossRef]
- Martins, C.S.; Elias, D.; Colli, L.M.; Couri, C.E.; Souza, M.C.; Moreira, A.C.; Foss, M.C.; Elias, L.L.; de Castro, M. HPA axis dysregulation, NR3C1 polymorphisms and glucocorticoid receptor isoforms imbalance in metabolic syndrome. Diabetes Metab. Res. Rev. 2017, 33. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef]
- Cruz-Topete, D.; Myers, P.H.; Foley, J.F.; Willis, M.S.; Cidlowski, J.A. Corticosteroids Are Essential for Maintaining Cardiovascular Function in Male Mice. Endocrinology 2016, 157, 2759–2771. [Google Scholar] [CrossRef] [PubMed]
- Lapp, H.E.; Bartlett, A.A.; Hunter, R.G. Stress and glucocorticoid receptor regulation of mitochondrial gene expression. J. Mol. Endocrinol. 2019, 62, R121–R128. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, S.F.; Munhoz, C.D.; Manley, N.C.; Yen, S.; Sapolsky, R.M. Glucocorticoids increase excitotoxic injury and inflammation in the hippocampus of adult male rats. Neuroendocrinology 2014, 100, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Annis, J.L.; Watkins, L.R.; Maier, S.F. Glucocorticoids mediate stress induction of the alarmin HMGB1 and reduction of the microglia checkpoint receptor CD200R1 in limbic brain structures. Brain Behav. Immun. 2019, 80, 678–687. [Google Scholar] [CrossRef]
- Noorzehi, G.; Pasbakhsh, P.; Borhani-Haghighi, M.; Kashani, I.R.; Madadi, S.; Tahmasebi, F.; Nekoonam, S.; Azizi, M. Microglia polarization by methylprednizolone acetate accelerates cuprizone induced demyelination. J. Mol. Histol. 2018, 49, 471–479. [Google Scholar] [CrossRef]
- Pedrazzoli, M.; Losurdo, M.; Paolone, G.; Medelin, M.; Jaupaj, L.; Cisterna, B.; Slanzi, A.; Malatesta, M.; Coco, S.; Buffelli, M. Glucocorticoid receptors modulate dendritic spine plasticity and microglia activity in an animal model of Alzheimer’s disease. Neurobiol. Dis. 2019, 132, 104568. [Google Scholar] [CrossRef]
- Alexander, J.K.; DeVries, A.C.; Kigerl, K.A.; Dahlman, J.M.; Popovich, P.G. Stress exacerbates neuropathic pain via glucocorticoid and NMDA receptor activation. Brain Behav. Immun. 2009, 23, 851–860. [Google Scholar] [CrossRef]
- Duque Ede, A.; Munhoz, C.D. The Pro-inflammatory Effects of Glucocorticoids in the Brain. Front. Endocrinol. (Lausanne) 2016, 7, 78. [Google Scholar] [CrossRef]
- Kennedy, L. Mifepristone and Cushing Syndrome: A Commentary. Endocr. Pract. 2015, 21, 1175–1177. [Google Scholar] [CrossRef]
- Block, T.S.; Kushner, H.; Kalin, N.; Nelson, C.; Belanoff, J.; Schatzberg, A. Combined Analysis of Mifepristone for Psychotic Depression: Plasma Levels Associated With Clinical Response. Biol. Psychiatry 2018, 84, 46–54. [Google Scholar] [CrossRef]
- Dalm, S.; Karssen, A.M.; Meijer, O.C.; Belanoff, J.K.; de Kloet, E.R. Resetting the Stress System with a Mifepristone Challenge. Cell Mol. Neurobiol. 2019, 39, 503–522. [Google Scholar] [CrossRef] [PubMed]
- Ratka, A.; Sutanto, W.; De Kloet, E.R. Long-lasting glucocorticoid suppression of opioid-induced antinociception. Neuroendocrinology 1988, 48, 439–444. [Google Scholar] [CrossRef] [PubMed]
- DeBattista, C.; Belanoff, J. C-1073 (mifepristone) in the adjunctive treatment of Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Behl, C.; Lezoualc’h, F.; Trapp, T.; Widmann, M.; Skutella, T.; Holsboer, F. Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurons in vitro. Endocrinology 1997, 138, 101–106. [Google Scholar] [CrossRef]
- Baglietto-Vargas, D.; Medeiros, R.; Martinez-Coria, H.; LaFerla, F.M.; Green, K.N. Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol. Psychiatry 2013, 74, 357–366. [Google Scholar] [CrossRef]
- Hu, P.; Oomen, C.; van Dam, A.M.; Wester, J.; Zhou, J.N.; Joels, M.; Lucassen, P.J. A single-day treatment with mifepristone is sufficient to normalize chronic glucocorticoid induced suppression of hippocampal cell proliferation. PLoS ONE 2012, 7, e46224. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Wang, H.Y.; Zhang, C.; Liu, B.P.; Peng, Z.L.; Li, Y.Y.; Liu, F.M.; Song, C. Mifepristone attenuates depression-like changes induced by chronic central administration of interleukin-1beta in rats. Behav. Brain Res. 2018, 347, 436–445. [Google Scholar] [CrossRef]
- Wulsin, A.C.; Herman, J.P.; Solomon, M.B. Mifepristone decreases depression-like behavior and modulates neuroendocrine and central hypothalamic-pituitary-adrenocortical axis responsiveness to stress. Psychoneuroendocrinology 2010, 35, 1100–1112. [Google Scholar] [CrossRef]
- Revsin, Y.; Rekers, N.V.; Louwe, M.C.; Saravia, F.E.; De Nicola, A.F.; de Kloet, E.R.; Oitzl, M.S. Glucocorticoid receptor blockade normalizes hippocampal alterations and cognitive impairment in streptozotocin-induced type 1 diabetes mice. Neuropsychopharmacology 2009, 34, 747–758. [Google Scholar] [CrossRef]
- Zalachoras, I.; Houtman, R.; Atucha, E.; Devos, R.; Tijssen, A.M.; Hu, P.; Lockey, P.M.; Datson, N.A.; Belanoff, J.K.; Lucassen, P.J.; et al. Differential targeting of brain stress circuits with a selective glucocorticoid receptor modulator. Proc. Natl. Acad. Sci. USA 2013, 110, 7910–7915. [Google Scholar] [CrossRef]
- van den Heuvel, J.K.; Boon, M.R.; van Hengel, I.; Peschier-van der Put, E.; van Beek, L.; van Harmelen, V.; van Dijk, K.W.; Pereira, A.M.; Hunt, H.; Belanoff, J.K.; et al. Identification of a selective glucocorticoid receptor modulator that prevents both diet-induced obesity and inflammation. Br. J. Pharmacol. 2016, 173, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Solomon, M.B.; Wulsin, A.C.; Rice, T.; Wick, D.; Myers, B.; McKlveen, J.; Flak, J.N.; Ulrich-Lai, Y.; Herman, J.P. The selective glucocorticoid receptor antagonist CORT 108297 decreases neuroendocrine stress responses and immobility in the forced swim test. Horm. Behav. 2014, 65, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Pineau, F.; Canet, G.; Desrumaux, C.; Hunt, H.; Chevallier, N.; Ollivier, M.; Belanoff, J.K.; Givalois, L. New selective glucocorticoid receptor modulators reverse amyloid-beta peptide-induced hippocampus toxicity. Neurobiol. Aging 2016, 45, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, E.T.; Caldwell, J.L.; Streicher, J.; Ghisays, V.; Balmer, N.J.; Estrada, C.M.; Solomon, M.B. Differential effects of imipramine and CORT118335 (Glucocorticoid receptor modulator/mineralocorticoid receptor antagonist) on brain-endocrine stress responses and depression-like behavior in female rats. Behav. Brain Res. 2018, 336, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Atucha, E.; Zalachoras, I.; van den Heuvel, J.K.; van Weert, L.T.; Melchers, D.; Mol, I.M.; Belanoff, J.K.; Houtman, R.; Hunt, H.; Roozendaal, B.; et al. A Mixed Glucocorticoid/Mineralocorticoid Selective Modulator With Dominant Antagonism in the Male Rat Brain. Endocrinology 2015, 156, 4105–4114. [Google Scholar] [CrossRef] [PubMed]
- Mammi, C.; Marzolla, V.; Armani, A.; Feraco, A.; Antelmi, A.; Maslak, E.; Chlopicki, S.; Cinti, F.; Hunt, H.; Fabbri, A.; et al. A novel combined glucocorticoid-mineralocorticoid receptor selective modulator markedly prevents weight gain and fat mass expansion in mice fed a high-fat diet. Int. J. Obes. (Lond) 2016, 40, 964–972. [Google Scholar] [CrossRef]
- Meyer, M.; Kruse, M.S.; Garay, L.; Lima, A.; Roig, P.; Hunt, H.; Belanoff, J.; de Kloet, E.R.; Deniselle, M.C.G.; De Nicola, A.F. Long-term effects of the glucocorticoid receptor modulator CORT113176 in murine motoneuron degeneration. Brain Res. 2020, 1727, 146551. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef]
- Lo Coco, D.; Veglianese, P.; Allievi, E.; Bendotti, C. Distribution and cellular localization of high mobility group box protein 1 (HMGB1) in the spinal cord of a transgenic mouse model of ALS. Neurosci. Lett. 2007, 412, 73–77. [Google Scholar] [CrossRef]
- Malaspina, A.; Puentes, F.; Amor, S. Disease origin and progression in amyotrophic lateral sclerosis: An immunology perspective. Int. Immunol. 2015, 27, 117–129. [Google Scholar] [CrossRef]
- De Paola, M.; Mariani, A.; Bigini, P.; Peviani, M.; Ferrara, G.; Molteni, M.; Gemma, S.; Veglianese, P.; Castellaneta, V.; Boldrin, V.; et al. Neuroprotective effects of toll-like receptor 4 antagonism in spinal cord cultures and in a mouse model of motor neuron degeneration. Mol. Med. 2012, 18, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci 2018, 19, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, M.A.; Rivest, S. The HPA-Immune Axis and the Immunomodulatory Actions of Glucocorticoids in the Brain. Front. Immunol. 2014, 5, 136. [Google Scholar] [CrossRef] [PubMed]
- Zorn, J.V.; Schür, R.R.; Boks, M.P.; Kahn, R.S.; Joëls, M.; Vinkers., C.H. Cortisol stress reactivity across psychiatric disorders: A systematic review and meta-analysis. Psychoneuroendocrinology 2017, 77, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Duma, D.; Collins, J.B.; Chou, J.W.; Cidlowski, J.A. Sexually dimorphic actions of glucocorticoids provide a link to inflammatory diseases with gender differences in prevalence. Sci. Signal. 2010, 3, ra74. [Google Scholar] [CrossRef] [PubMed]
- Manjaly, Z.R.; Scott, K.M.; Abhinav, K. The sex ratio in amyotrophic lateral sclerosis: A population based study. Amyotroph. Lateral. Scler. 2010, 11, 439–442. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Nicola, A.F.; Meyer, M.; Guennoun, R.; Schumacher, M.; Hunt, H.; Belanoff, J.; de Kloet, E.R.; Gonzalez Deniselle, M.C. Insights into the Therapeutic Potential of Glucocorticoid Receptor Modulators for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 2137. https://doi.org/10.3390/ijms21062137
De Nicola AF, Meyer M, Guennoun R, Schumacher M, Hunt H, Belanoff J, de Kloet ER, Gonzalez Deniselle MC. Insights into the Therapeutic Potential of Glucocorticoid Receptor Modulators for Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(6):2137. https://doi.org/10.3390/ijms21062137
Chicago/Turabian StyleDe Nicola, Alejandro F., Maria Meyer, Rachida Guennoun, Michael Schumacher, Hazel Hunt, Joseph Belanoff, E. Ronald de Kloet, and Maria Claudia Gonzalez Deniselle. 2020. "Insights into the Therapeutic Potential of Glucocorticoid Receptor Modulators for Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 6: 2137. https://doi.org/10.3390/ijms21062137
APA StyleDe Nicola, A. F., Meyer, M., Guennoun, R., Schumacher, M., Hunt, H., Belanoff, J., de Kloet, E. R., & Gonzalez Deniselle, M. C. (2020). Insights into the Therapeutic Potential of Glucocorticoid Receptor Modulators for Neurodegenerative Diseases. International Journal of Molecular Sciences, 21(6), 2137. https://doi.org/10.3390/ijms21062137