Hepatitis C Virus Entry: An Intriguingly Complex and Highly Regulated Process


Abstract
1. Introduction
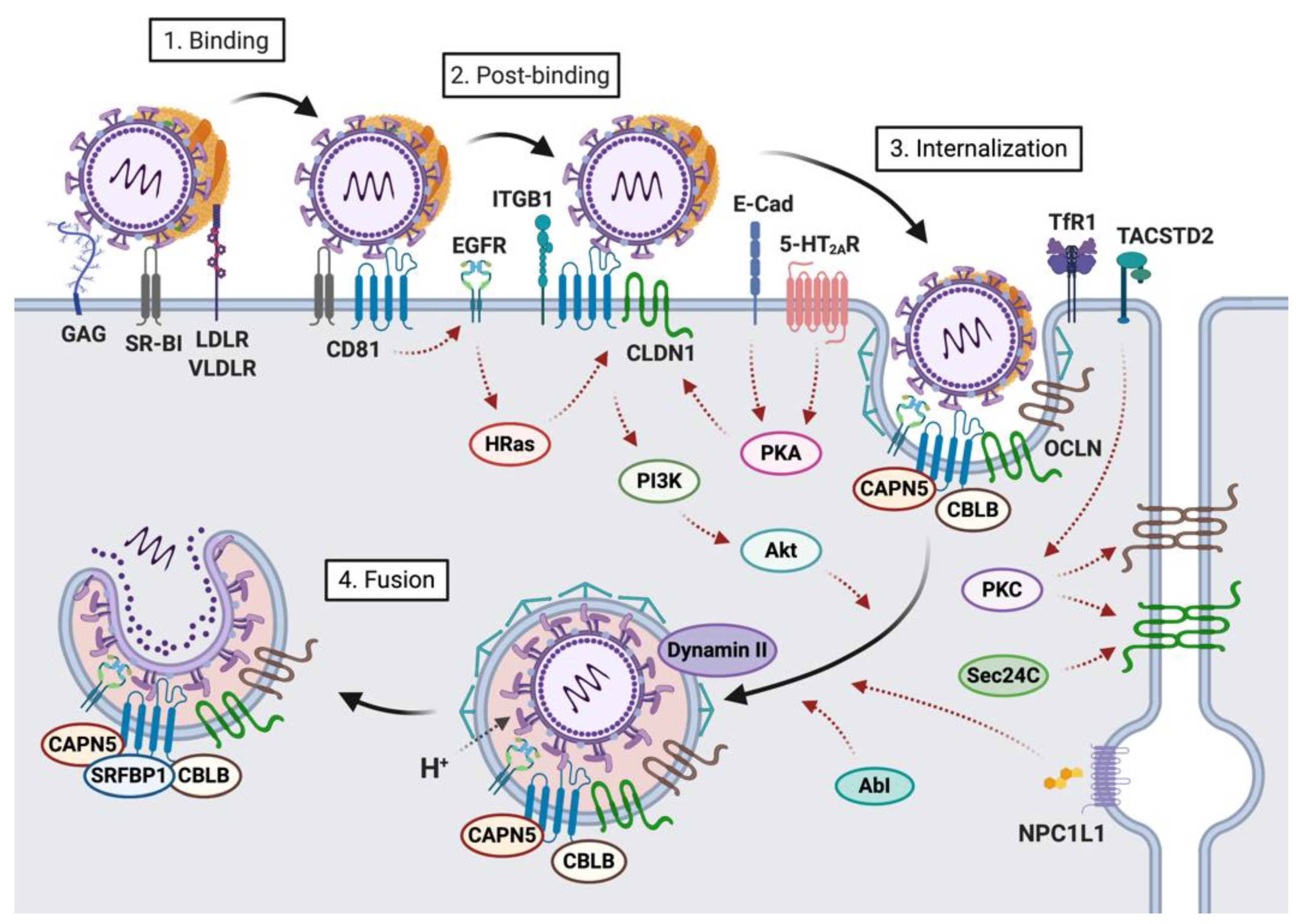
2. Host Factors Involved in the First Steps of HCV-Hepatocyte Interactions
2.1. Host Factors Involved in Viral Attachment to the Hepatocyte Basolateral Membrane
2.2. Host Factors Involved in Viral Internalization
3. Viral Determinants of Fusion: E1 and E2 Glycoproteins
4. HCV Entry into Hepatocytes: A Highly Regulated Process
4.1. The Regulatory Role of CD81-Tetraspanin Platforms
4.2. Regulation of Tight Junction Proteins
5. Viral and Host Factors Involved in Viral Cell-to-Cell Transmission
6. Therapeutic Potential of Host-Targeting Entry Inhibitors
7. Conclusions and Perspectives
Funding
Conflicts of Interest
Abbreviations
| Apo | Apolipoprotein |
| CAPN5 | Calpain-5 |
| CBLB | Casitas B-lineage lymphoma proto-oncogene B |
| CD81 | Cluster of differentiation 81 |
| CLDN | Claudin |
| DAA | Direct-acting antiviral |
| EGFR | Epidermal growth factor receptor |
| EL | Extracellular loop |
| HCV | Hepatitis C virus |
| HCVcc | Cell culture-derived HCV |
| HCVpp | HCV pseudoparticles |
| HDL | High density lipoprotein |
| HSPG | Highly sulfated heparan sulfate proteoglycan |
| LDL | Low density lipoprotein |
| LDLR | Low density lipoprotein receptor |
| LVP | Lipoviral particle |
| mAb | Monoclonal antibody |
| NPC1L1 | Niemann-Pick C1-like 1 |
| OCLN | Occludi |
| PKA | Protein kinase A |
| SR-BI | Scavenger receptor class B type I |
| TfR | Transferrin receptor |
| VLDL | Very low density lipoprotein |
References
- World Health Organization. Global Hepatitis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Hatzakis, A.; Lazarus, J.V.; Cholongitas, E.; Baptista-Leite, R.; Boucher, C.; Busoi, C.S.; Deuffic-Burban, S.; Chhatwal, J.; Esmat, G.; Hutchinson, S.; et al. Securing sustainable funding for viral hepatitis elimination plans. Liver Int. 2020, 40, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L. Challenges and Promise of a Hepatitis C Virus Vaccine. Cold Spring Harb. Perspect. Med. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Roingeard, P.; Beaumont, E. Hepatitis C vaccine: 10 good reasons for continuing. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Murray, C.L.; Thiel, H.J.; Rice, C.M. Flaviviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 712–746. [Google Scholar]
- Thomssen, R.; Bonk, S.; Propfe, C.; Heermann, K.-H.; Koechel, H.G.; Uy, A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. 1992, 181, 293–300. [Google Scholar] [CrossRef]
- Thomssen, R.; Bonk, S.; Thiele, A. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. 1993, 182, 329–334. [Google Scholar] [CrossRef]
- Miyamoto, H.; Okamoto, H.; Sato, K.; Tanaka, T.; Mishiro, S. Extraordinarily low density of hepatitis C virus estimated by sucrose gradient centrifugation and the polymerase chain reaction. J. Gen. Virol. 1992, 73, 715–718. [Google Scholar] [CrossRef]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef]
- Nielsen, S.U.; Bassendine, M.F.; Burt, A.D.; Martin, C.; Pumeechockchai, W.; Toms, G.L. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 2006, 80, 2418–2428. [Google Scholar] [CrossRef]
- Felmlee, D.J.; Sheridan, D.A.; Bridge, S.H.; Nielsen, S.U.; Milne, R.W.; Packard, C.J.; Caslake, M.J.; McLauchlan, J.; Toms, G.L.; Neely, R.D.; et al. Intravascular transfer contributes to postprandial increase in numbers of very-low-density hepatitis C virus particles. Gastroenterology 2010, 139, 1774–1783.e6. [Google Scholar] [CrossRef]
- Piver, E.; Boyer, A.; Gaillard, J.; Bull, A.; Beaumont, E.; Roingeard, P.; Meunier, J.C. Ultrastructural organisation of HCV from the bloodstream of infected patients revealed by electron microscopy after specific immunocapture. Gut 2017, 66, 1487–1495. [Google Scholar] [CrossRef]
- Merz, A.; Long, G.; Hiet, M.S.; Brugger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Lussignol, M.; Kopp, M.; Molloy, K.; Vizcay-Barrena, G.; Fleck, R.A.; Dorner, M.; Bell, K.L.; Chait, B.T.; Rice, C.M.; Catanese, M.T. Proteomics of HCV virions reveals an essential role for the nucleoporin Nup98 in virus morphogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, 2484–2489. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Felmlee, D.J.; Baumert, T.F. Hepatitis C virus entry. Curr Top. Microbiol. Immunol. 2013, 369, 87–112. [Google Scholar] [CrossRef]
- Gerold, G.; Moeller, R.; Pietschmann, T. Hepatitis C Virus Entry: Protein Interactions and Fusion Determinants Governing Productive Hepatocyte Invasion. Cold Spring Harb. Perspect. Med. 2020, 10. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Crouchet, E.; Baumert, T.F.; Schuster, C. Host-Targeting Agents to Prevent and Cure Hepatitis C Virus Infection. Viruses 2015, 7, 5659–5685. [Google Scholar] [CrossRef]
- Wrensch, F.; Crouchet, E.; Ligat, G.; Zeisel, M.B.; Keck, Z.Y.; Foung, S.K.H.; Schuster, C.; Baumert, T.F. Hepatitis C Virus (HCV)-Apolipoprotein Interactions and Immune Evasion and Their Impact on HCV Vaccine Design. Front. Immunol. 2018, 9, 1436. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Fofana, I.; Fafi-Kremer, S.; Baumert, T.F. Hepatitis C virus entry into hepatocytes: Molecular mechanisms and targets for antiviral therapies. J. Hepatol. 2011, 54, 566–576. [Google Scholar] [CrossRef]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef]
- Da Costa, D.; Turek, M.; Felmlee, D.J.; Girardi, E.; Pfeffer, S.; Long, G.; Bartenschlager, R.; Zeisel, M.B.; Baumert, T.F. Reconstitution of the entire hepatitis C virus life cycle in non-hepatic cells. J. Virol. 2012, 86, 11919–11925. [Google Scholar] [CrossRef] [PubMed]
- Dorner, M.; Horwitz, J.A.; Donovan, B.M.; Labitt, R.N.; Budell, W.C.; Friling, T.; Vogt, A.; Catanese, M.T.; Satoh, T.; Kawai, T.; et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature 2013, 501, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; von Schaewen, M.; Hrebikova, G.; Heller, B.; Sandmann, L.; Plaas, M.; Ploss, A. Mice Expressing Minimally Humanized CD81 and Occludin Genes Support Hepatitis C Virus Uptake In Vivo. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.P.; Durso, R.J.; Arrigale, R.R.; Donovan, G.P.; Maddon, P.J.; Dragic, T.; Olson, W.C. L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proc. Natl. Acad. Sci. USA 2003, 100, 4498–4503. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, S.; Zhang, J.; Baribaud, F.; Chen, Z.; Leslie, G.J.; Lin, G.; Granelli-Piperno, A.; Doms, R.W.; Rice, C.M.; McKeating, J.A. Hepatitis C Virus Glycoproteins Interact with DC-SIGN and DC-SIGNR. J. Virol. 2003, 77, 4070–4080. [Google Scholar] [CrossRef]
- Lozach, P.Y.; Lortat-Jacob, H.; de Lacroix de Lavalette, A.; Staropoli, I.; Foung, S.; Amara, A.; Houles, C.; Fieschi, F.; Schwartz, O.; Virelizier, J.L.; et al. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J. Biol. Chem. 2003, 278, 20358–20366. [Google Scholar] [CrossRef]
- Lozach, P.Y.; Amara, A.; Bartosch, B.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Cosset, F.L.; Altmeyer, R. C-type lectins L-SIGN and DC-SIGN capture and transmit infectious hepatitis C virus pseudotype particles. J. Biol. Chem. 2004, 279, 32035–32045. [Google Scholar] [CrossRef]
- Saito, H.; Dhanasekaran, P.; Nguyen, D.; Baldwin, F.; Weisgraber, K.H.; Wehrli, S.; Phillips, M.C.; Lund-Katz, S. Characterization of the heparin binding sites in human apolipoprotein E. J. Biol. Chem. 2003, 278, 14782–14787. [Google Scholar] [CrossRef]
- Maillard, P.; Huby, T.; Andreo, U.; Moreau, M.; Chapman, J.; Budkowska, A. The interaction of natural hepatitis C virus with human scavenger receptor SR-BI/Cla1 is mediated by ApoB-containing lipoproteins. FASEB J. 2006, 20, 735–737. [Google Scholar] [CrossRef]
- Hishiki, T.; Shimizu, Y.; Tobita, R.; Sugiyama, K.; Ogawa, K.; Funami, K.; Ohsaki, Y.; Fujimoto, T.; Takaku, H.; Wakita, T.; et al. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J. Virol. 2010, 84, 12048–12057. [Google Scholar] [CrossRef]
- Liu, S.; McCormick, K.D.; Zhao, W.; Zhao, T.; Fan, D.; Wang, T. Human apolipoprotein E peptides inhibit hepatitis C virus entry by blocking virus binding. Hepatology 2012, 56, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Cun, W.; Wu, X.; Shi, Q.; Tang, H.; Luo, G. Hepatitis C Virus Attachment Mediated by Apolipoprotein E Binding to Cell Surface Heparan Sulfate. J. Virol. 2012, 86, 7256–7267. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wu, X.; Tang, H.; Luo, G. Apolipoprotein E mediates attachment of clinical hepatitis C virus to hepatocytes by binding to cell surface heparan sulfate proteoglycan receptors. PLoS ONE 2013, 8, e67982. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Schnober, E.K.; Zhang, F.; Linhardt, R.J.; Depla, E.; Boson, B.; Cosset, F.L.; Patel, A.H.; Blum, H.E.; Baumert, T.F. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J. Virol. 2006, 80, 10579–10590. [Google Scholar] [CrossRef]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef]
- Morikawa, K.; Zhao, Z.; Date, T.; Miyamoto, M.; Murayama, A.; Akazawa, D.; Tanabe, J.; Sone, S.; Wakita, T. The roles of CD81 and glycosaminoglycans in the adsorption and uptake of infectious HCV particles. J. Med. Virol. 2007, 79, 714–723. [Google Scholar] [CrossRef]
- Shi, Q.; Jiang, J.; Luo, G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J. Virol. 2013, 87, 6866–6875. [Google Scholar] [CrossRef]
- Lefevre, M.; Felmlee, D.J.; Parnot, M.; Baumert, T.F.; Schuster, C. Syndecan 4 is involved in mediating HCV entry through interaction with lipoviral particle-associated apolipoprotein E. PLoS ONE 2014, 9, e95550. [Google Scholar] [CrossRef]
- Fan, H.; Qiao, L.; Kang, K.D.; Fan, J.; Wei, W.; Luo, G. Attachment and Postattachment Receptors Important for Hepatitis C Virus Infection and Cell-to-Cell Transmission. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.X. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar] [CrossRef]
- Monazahian, M.; Bohme, I.; Bonk, S.; Koch, A.; Scholz, C.; Grethe, S.; Thomssen, R. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J. Med. Virol. 1999, 57, 223–229. [Google Scholar] [CrossRef]
- Wunschmann, S.; Medh, J.D.; Klinzmann, D.; Schmidt, W.N.; Stapleton, J.T. Characterization of hepatitis C virus (HCV) and HCV E2 interactions with CD81 and the low-density lipoprotein receptor. J. Virol. 2000, 74, 10055–10062. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.; Castet, V.; Fournier-Wirth, C.; Pichard-Garcia, L.; Avner, R.; Harats, D.; Roitelman, J.; Barbaras, R.; Graber, P.; Ghersa, P.; et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 2007, 46, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Cerino, R.; Arcuri, M.; Hoffmann, M.; Schurmann, P.; Adah, M.I.; Gissler, B.; Zhao, X.; Ghisetti, V.; Lavezzo, B.; et al. Scavenger receptor class B type I and hepatitis C virus infection of primary tupaia hepatocytes. J. Virol. 2005, 79, 5774–5785. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Graziani, R.; von Hahn, T.; Moreau, M.; Huby, T.; Paonessa, G.; Santini, C.; Luzzago, A.; Rice, C.M.; Cortese, R.; et al. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J. Virol. 2007, 81, 8063–8071. [Google Scholar] [CrossRef]
- Dreux, M.; Dao Thi, V.L.; Fresquet, J.; Guerin, M.; Julia, Z.; Verney, G.; Durantel, D.; Zoulim, F.; Lavillette, D.; Cosset, F.L.; et al. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog. 2009, 5, e1000310. [Google Scholar] [CrossRef]
- Dao Thi, V.L.; Granier, C.; Zeisel, M.B.; Guerin, M.; Mancip, J.; Granio, O.; Penin, F.; Lavillette, D.; Bartenschlager, R.; Baumert, T.F.; et al. Characterization of Hepatitis C Virus Particle Subpopulations Reveals Multiple Usage of the Scavenger Receptor BI for Entry Steps. J. Biol. Chem. 2012, 287, 31242–31257. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F.-L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C.; et al. Scavenger receptor BI is a key host factor for Hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef]
- Zahid, M.N.; Turek, M.; Xiao, F.; Thi, V.L.; Guerin, M.; Fofana, I.; Bachellier, P.; Thompson, J.; Delang, L.; Neyts, J.; et al. The postbinding activity of scavenger receptor class B type I mediates initiation of hepatitis C virus infection and viral dissemination. Hepatology 2013, 57, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Grigorov, B.; Reungoat, E.; Gentil Dit Maurin, A.; Varbanov, M.; Blaising, J.; Michelet, M.; Manuel, R.; Parent, R.; Bartosch, B.; Zoulim, F.; et al. Hepatitis C virus infection propagates through interactions between Syndecan-1 and CD81 and impacts the hepatocyte glycocalyx. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Albecka, A.; Belouzard, S.; de Beeck, A.O.; Descamps, V.; Goueslain, L.; Bertrand-Michel, J.; Terce, F.; Duverlie, G.; Rouille, Y.; Dubuisson, J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 2012, 55, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qiao, L.; Hou, Z.; Luo, G. TIM-1 Promotes Hepatitis C Virus Cell Attachment and Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Kachko, A.; Costafreda, M.I.; Zubkova, I.; Jacques, J.; Takeda, K.; Wells, F.; Kaplan, G.; Major, M.E. Determinants in the Ig Variable Domain of Human HAVCR1 (TIM-1) Are Required To Enhance Hepatitis C Virus Entry. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Verney, G.; Dreux, M.; Donot, P.; Morice, Y.; Penin, F.; Pawlotsky, J.M.; Lavillette, D.; Cosset, F.L. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol 2005, 79, 8217–8229. [Google Scholar] [CrossRef] [PubMed]
- Voisset, C.; Callens, N.; Blanchard, E.; De Beeck, A.O.; Dubuisson, J.; Vu-Dac, N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 2005, 280, 7793–7799. [Google Scholar] [CrossRef]
- Von Hahn, T.; Lindenbach, B.D.; Boullier, A.; Quehenberger, O.; Paulson, M.; Rice, C.M.; McKeating, J.A. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 2006, 43, 932–942. [Google Scholar] [CrossRef]
- Andreo, U.; Maillard, P.; Kalinina, O.; Walic, M.; Meurs, E.; Martinot, M.; Marcellin, P.; Budkowska, A. Lipoprotein lipase mediates hepatitis C virus (HCV) cell entry and inhibits HCV infection. Cell. Microbiol. 2007, 9, 2445–2456. [Google Scholar] [CrossRef]
- Maillard, P.; Walic, M.; Meuleman, P.; Roohvand, F.; Huby, T.; Le Goff, W.; Leroux-Roels, G.; Pecheur, E.I.; Budkowska, A. Lipoprotein lipase inhibits hepatitis C virus (HCV) infection by blocking virus cell entry. PLoS ONE 2011, 6, e26637. [Google Scholar] [CrossRef]
- Shimizu, Y.; Hishiki, T.; Sugiyama, K.; Ogawa, K.; Funami, K.; Kato, A.; Ohsaki, Y.; Fujimoto, T.; Takaku, H.; Shimotohno, K. Lipoprotein lipase and hepatic triglyceride lipase reduce the infectivity of hepatitis C virus (HCV) through their catalytic activities on HCV-associated lipoproteins. Virology 2010, 407, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, J.; Feng, Y.; Cai, H.; Li, Y.P.; Peng, T. Long-chain fatty acyl-coenzyme A suppresses hepatitis C virus infection by targeting virion-bound lipoproteins. Antiviral. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bertaux, C.; Dragic, T. Different domains of CD81 mediate distinct stages of hepatitis C virus pseudoparticle entry. J. Virol. 2006, 80, 4940–4948. [Google Scholar] [CrossRef][Green Version]
- Krieger, S.E.; Zeisel, M.B.; Davis, C.; Thumann, C.; Harris, H.J.; Schnober, E.K.; Mee, C.; Soulier, E.; Royer, C.; Lambotin, M.; et al. Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology 2010, 51, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef]
- Douam, F.; Dao Thi, V.L.; Maurin, G.; Fresquet, J.; Mompelat, D.; Zeisel, M.B.; Baumert, T.F.; Cosset, F.L.; Lavillette, D. Critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 2014, 59, 776–788. [Google Scholar] [CrossRef]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; et al. CD81 and claudin 1 coreceptor association: Role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar] [CrossRef]
- Harris, H.J.; Davis, C.; Mullins, J.G.; Hu, K.; Goodall, M.; Farquhar, M.J.; Mee, C.J.; McCaffrey, K.; Young, S.; Drummer, H.; et al. Claudin association with CD81 defines hepatitis C virus entry. J. Biol. Chem 2010, 285, 21092–21102. [Google Scholar] [CrossRef]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef]
- Baktash, Y.; Madhav, A.; Coller, K.E.; Randall, G. Single Particle Imaging of Polarized Hepatoma Organoids upon Hepatitis C Virus Infection Reveals an Ordered and Sequential Entry Process. Cell Host Microbe 2018, 23, 382–394.e385. [Google Scholar] [CrossRef]
- Bruening, J.; Lasswitz, L.; Banse, P.; Kahl, S.; Marinach, C.; Vondran, F.W.; Kaderali, L.; Silvie, O.; Pietschmann, T.; Meissner, F.; et al. Hepatitis C virus enters liver cells using the CD81 receptor complex proteins calpain-5 and CBLB. PLoS Pathog. 2018, 14, e1007111. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Huby, T.; Stamataki, Z.; Vanwolleghem, T.; Meuleman, P.; Farquhar, M.; Schwarz, A.; Moreau, M.; Owen, J.S.; Leroux-Roels, G.; et al. Scavenger receptor BI and BII expression levels modulate Hepatitis C virus infectivity. J. Virol. 2007, 81, 3162–3169. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.K.; Grove, J.; Hu, K.; Mee, C.J.; Balfe, P.; McKeating, J.A. Hepatoma cell density promotes claudin-1 and scavenger receptor BI expression and hepatitis C virus internalization. J. Virol. 2009, 83, 12407–12414. [Google Scholar] [CrossRef] [PubMed]
- Kalemera, M.; Mincheva, D.; Grove, J.; Illingworth, C.J.R. Building a mechanistic mathematical model of hepatitis C virus entry. PLoS Comput. Biol. 2019, 15, e1006905. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Brazzoli, M.; Bianchi, A.; Filippini, S.; Weiner, A.; Zhu, Q.; Pizza, M.; Crotta, S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol. 2008, 82, 8316–8329. [Google Scholar] [CrossRef]
- Diao, J.; Pantua, H.; Ngu, H.; Komuves, L.; Diehl, L.; Schaefer, G.; Kapadia, S.B. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J. Virol. 2012, 86, 10935–10949. [Google Scholar] [CrossRef]
- Kim, S.; Ishida, H.; Yamane, D.; Yi, M.; Swinney, D.C.; Foung, S.; Lemon, S.M. Contrasting roles of mitogen-activated protein kinases in cellular entry and replication of hepatitis C virus: MKNK1 facilitates cell entry. J. Virol. 2013, 87, 4214–4224. [Google Scholar] [CrossRef]
- Cukierman, L.; Meertens, L.; Bertaux, C.; Kajumo, F.; Dragic, T. Residues in a highly conserved claudin-1 motif are required for hepatitis C virus entry and mediate the formation of cell-cell contacts. J. Virol. 2009, 83, 5477–5484. [Google Scholar] [CrossRef]
- Hopcraft, S.E.; Evans, M.J. Selection of a hepatitis C virus with altered entry factor requirements reveals a genetic interaction between the E1 glycoprotein and claudins. Hepatology 2015. [Google Scholar] [CrossRef]
- Haid, S.; Grethe, C.; Dill, M.T.; Heim, M.; Kaderali, L.; Pietschmann, T. Isolate-dependent use of claudins for cell entry by hepatitis C virus. Hepatology 2014, 59, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Yuan, F.; Li, Y.; Zhu, F.; Hou, P.; Li, J.; Song, X.; Ding, M.; Deng, H. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J. Virol. 2007, 81, 12465–12471. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Bertaux, C.; Cukierman, L.; Cormier, E.; Lavillette, D.; Cosset, F.L.; Dragic, T. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 2008, 82, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Brass, A.L.; Ng, A.; Hu, Z.; Xavier, R.J.; Liang, T.J.; Elledge, S.J. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. USA 2009, 106, 16410–16415. [Google Scholar] [CrossRef]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef]
- Huang, J.; Yin, P.; Zhang, L. COPII cargo claudin-12 promotes hepatitis C virus entry. J. Viral Hepat. 2019, 26, 308–312. [Google Scholar] [CrossRef]
- Liu, S.; Kuo, W.; Yang, W.; Liu, W.; Gibson, G.A.; Dorko, K.; Watkins, S.C.; Strom, S.C.; Wang, T. The second extracellular loop dictates Occludin-mediated HCV entry. Virology 2010, 407, 160–170. [Google Scholar] [CrossRef]
- Sourisseau, M.; Michta, M.L.; Zony, C.; Israelow, B.; Hopcraft, S.E.; Narbus, C.M.; Parra Martin, A.; Evans, M.J. Temporal analysis of hepatitis C virus cell entry with occludin directed blocking antibodies. PLoS Pathog. 2013, 9, e1003244. [Google Scholar] [CrossRef]
- Benedicto, I.; Molina-Jimenez, F.; Bartosch, B.; Cosset, F.L.; Lavillette, D.; Prieto, J.; Moreno-Otero, R.; Valenzuela-Fernandez, A.; Aldabe, R.; Lopez-Cabrera, M.; et al. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J. Virol. 2009, 83, 8012–8020. [Google Scholar] [CrossRef]
- Ujino, S.; Nishitsuji, H.; Hishiki, T.; Sugiyama, K.; Takaku, H.; Shimotohno, K. Hepatitis C virus utilizes VLDLR as a novel entry pathway. Proc. Natl. Acad. Sci. USA 2016, 113, 188–193. [Google Scholar] [CrossRef]
- Min, S.; Lim, Y.S.; Shin, D.; Park, C.; Park, J.B.; Kim, S.; Windisch, M.P.; Hwang, S.B. Abl Tyrosine Kinase Regulates Hepatitis C Virus Entry. Front. Microbiol. 2017, 8, 1129. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA 2013, 110, 10777–10782. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B., Jr.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.J.; Hu, K.; Harris, H.J.; Davis, C.; Brimacombe, C.L.; Fletcher, S.J.; Baumert, T.F.; Rappoport, J.Z.; Balfe, P.; McKeating, J.A. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol. 2012, 86, 4305–4316. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, E.; Belouzard, S.; Goueslain, L.; Wakita, T.; Dubuisson, J.; Wychowski, C.; Rouille, Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006, 80, 6964–6972. [Google Scholar] [CrossRef]
- Matsuda, M.; Suzuki, R.; Kataoka, C.; Watashi, K.; Aizaki, H.; Kato, N.; Matsuura, Y.; Suzuki, T.; Wakita, T. Alternative endocytosis pathway for productive entry of hepatitis C virus. J. Gen. Virol. 2014, 95, 2658–2667. [Google Scholar] [CrossRef]
- Sharma, N.R.; Mateu, G.; Dreux, M.; Grakoui, A.; Cosset, F.L.; Melikyan, G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011, 286, 30361–30376. [Google Scholar] [CrossRef]
- Lavillette, D.; Pecheur, E.I.; Donot, P.; Fresquet, J.; Molle, J.; Corbau, R.; Dreux, M.; Penin, F.; Cosset, F.L. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 2007, 81, 8752–8765. [Google Scholar] [CrossRef]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar] [CrossRef]
- Falson, P.; Bartosch, B.; Alsaleh, K.; Tews, B.A.; Loquet, A.; Ciczora, Y.; Riva, L.; Montigny, C.; Montpellier, C.; Duverlie, G.; et al. Hepatitis C Virus Envelope Glycoprotein E1 Forms Trimers at the Surface of the Virion. J. Virol. 2015, 89, 10333–10346. [Google Scholar] [CrossRef] [PubMed]
- Garry, R.F.; Dash, S. Proteomics computational analyses suggest that hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class II fusion proteins. Virology 2003, 307, 255–265. [Google Scholar] [CrossRef]
- Perin, P.M.; Haid, S.; Brown, R.J.; Doerrbecker, J.; Schulze, K.; Zeilinger, C.; von Schaewen, M.; Heller, B.; Vercauteren, K.; Luxenburger, E.; et al. Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 2016, 63, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Chi, X.; Yang, W.; Zhong, J. Functional Analysis of Hepatitis C Virus (HCV) Envelope Protein E1 Using a trans-Complementation System Reveals a Dual Role of a Putative Fusion Peptide of E1 in both HCV Entry and Morphogenesis. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Lombana, L.; Ortega-Atienza, S.; Gomez-Gutierrez, J.; Yelamos, B.; Peterson, D.L.; Gavilanes, F. The deletion of residues 268-292 of E1 impairs the ability of HCV envelope proteins to induce pore formation. Virus Res. 2016, 217, 63–70. [Google Scholar] [CrossRef][Green Version]
- Banda, D.H.; Perin, P.M.; Brown, R.J.P.; Todt, D.; Solodenko, W.; Hoffmeyer, P.; Kumar Sahu, K.; Houghton, M.; Meuleman, P.; Muller, R.; et al. A central hydrophobic E1 region controls the pH range of hepatitis C virus membrane fusion and susceptibility to fusion inhibitors. J. Hepatol. 2019, 70, 1082–1092. [Google Scholar] [CrossRef]
- El Omari, K.; Iourin, O.; Kadlec, J.; Sutton, G.; Harlos, K.; Grimes, J.M.; Stuart, D.I. Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat. Commun. 2014, 5, 4874. [Google Scholar] [CrossRef]
- Li, Y.; Modis, Y. A novel membrane fusion protein family in Flaviviridae? Trends Microbiol. 2014, 22, 176–182. [Google Scholar] [CrossRef]
- Maurin, G.; Fresquet, J.; Granio, O.; Wychowski, C.; Cosset, F.L.; Lavillette, D. Identification of interactions in the E1E2 heterodimer of hepatitis C virus important for cell entry. J. Biol. Chem. 2011, 286, 23865–23876. [Google Scholar] [CrossRef]
- Douam, F.; Fusil, F.; Enguehard, M.; Dib, L.; Nadalin, F.; Schwaller, L.; Hrebikova, G.; Mancip, J.; Mailly, L.; Montserret, R.; et al. A protein coevolution method uncovers critical features of the Hepatitis C Virus fusion mechanism. PLoS Pathog. 2018, 14, e1006908. [Google Scholar] [CrossRef]
- Gerold, G.; Meissner, F.; Bruening, J.; Welsch, K.; Perin, P.M.; Baumert, T.F.; Vondran, F.W.; Kaderali, L.; Marcotrigiano, J.; Khan, A.G.; et al. Quantitative Proteomics Identifies Serum Response Factor Binding Protein 1 as a Host Factor for Hepatitis C Virus Entry. Cell Rep. 2015, 12, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Perugini, V.; Montpellier, C.; Delgrange, D.; Wychowski, C.; Helle, F.; Pillez, A.; Drobecq, H.; Le Naour, F.; Charrin, S.; Levy, S.; et al. The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS ONE 2008, 3, e1866. [Google Scholar] [CrossRef] [PubMed]
- Montpellier, C.; Tews, B.A.; Poitrimole, J.; Rocha-Perugini, V.; D’Arienzo, V.; Potel, J.; Zhang, X.A.; Rubinstein, E.; Dubuisson, J.; Cocquerel, L. Interacting regions of CD81 and two of its partners, EWI-2 and EWI-2wint, and their effect on hepatitis C virus infection. J. Biol. Chem. 2011, 286, 13954–13965. [Google Scholar] [CrossRef] [PubMed]
- Potel, J.; Rassam, P.; Montpellier, C.; Kaestner, L.; Werkmeister, E.; Tews, B.A.; Couturier, C.; Popescu, C.I.; Baumert, T.F.; Rubinstein, E.; et al. EWI-2wint promotes CD81 clustering that abrogates Hepatitis C Virus entry. Cell. Microbiol. 2013, 15, 1234–1252. [Google Scholar] [CrossRef] [PubMed]
- Earnest, J.T.; Hantak, M.P.; Li, K.; McCray, P.B., Jr.; Perlman, S.; Gallagher, T. The tetraspanin CD9 facilitates MERS-coronavirus entry by scaffolding host cell receptors and proteases. PLoS Pathog. 2017, 13, e1006546. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Lang, T. Tetraspanin Assemblies in Virus Infection. Front. Immunol. 2018, 9, 1140. [Google Scholar] [CrossRef] [PubMed]
- Hantak, M.P.; Qing, E.; Earnest, J.T.; Gallagher, T. Tetraspanins: Architects of Viral Entry and Exit Platforms. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Li, Y.; Zhang, L. Sec24C-Dependent Transport of Claudin-1 Regulates Hepatitis C Virus Entry. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Sekhar, V.; Pollicino, T.; Diaz, G.; Engle, R.E.; Alayli, F.; Melis, M.; Kabat, J.; Tice, A.; Pomerenke, A.; Altan-Bonnet, N.; et al. Infection with hepatitis C virus depends on TACSTD2, a regulator of claudin-1 and occludin highly downregulated in hepatocellular carcinoma. PLoS Pathog. 2018, 14, e1006916. [Google Scholar] [CrossRef]
- Cao, L.; Chen, J.; Wang, Y.; Yang, Y.; Qing, J.; Rao, Z.; Chen, X.; Lou, Z. Identification of serotonin 2A receptor as a novel HCV entry factor by a chemical biology strategy. Protein Cell 2019, 10, 178–195. [Google Scholar] [CrossRef]
- Riva, L.; Song, O.R.; Prentoe, J.; Helle, F.; L’Homme, L.; Gattolliat, C.H.; Vandeputte, A.; Feneant, L.; Belouzard, S.; Baumert, T.F.; et al. Identification of Piperazinylbenzenesulfonamides as New Inhibitors of Claudin-1 Trafficking and Hepatitis C Virus Entry. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.J.; Harris, H.J.; Diskar, M.; Jones, S.; Mee, C.J.; Nielsen, S.U.; Brimacombe, C.L.; Molina, S.; Toms, G.L.; Maurel, P.; et al. Protein kinase A-dependent step(s) in hepatitis C virus entry and infectivity. J. Virol. 2008, 82, 8797–8811. [Google Scholar] [CrossRef]
- Li, Q.; Sodroski, C.; Lowey, B.; Schweitzer, C.J.; Cha, H.; Zhang, F.; Liang, T.J. Hepatitis C virus depends on E-cadherin as an entry factor and regulates its expression in epithelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2016, 113, 7620–7625. [Google Scholar] [CrossRef]
- Timpe, J.M.; Stamataki, Z.; Jennings, A.; Hu, K.; Farquhar, M.J.; Harris, H.J.; Schwarz, A.; Desombere, I.; Roels, G.L.; Balfe, P.; et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 2008, 47, 17–24. [Google Scholar] [CrossRef]
- Brimacombe, C.L.; Grove, J.; Meredith, L.W.; Hu, K.; Syder, A.J.; Flores, M.V.; Timpe, J.M.; Krieger, S.E.; Baumert, T.F.; Tellinghuisen, T.L.; et al. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J. Virol. 2011, 85, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Fofana, I.; Thumann, C.; Mailly, L.; Alles, R.; Robinet, E.; Meyer, N.; Schaeffer, M.; Habersetzer, F.; Doffoel, M.; et al. Synergy of entry inhibitors with direct-acting antivirals uncovers novel combinations for prevention and treatment of hepatitis C. Gut 2015, 64, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Loureiro, J.; Jones, C.T.; Dorner, M.; von Hahn, T.; Rice, C.M. Different requirements for scavenger receptor class B type I in hepatitis C virus cell-free versus cell-to-cell transmission. J. Virol. 2013, 87, 8282–8293. [Google Scholar] [CrossRef] [PubMed]
- Hueging, K.; Doepke, M.; Vieyres, G.; Bankwitz, D.; Frentzen, A.; Doerrbecker, J.; Gumz, F.; Haid, S.; Wolk, B.; Kaderali, L.; et al. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J. Virol. 2014, 88, 1433–1446. [Google Scholar] [CrossRef] [PubMed]
- Gondar, V.; Molina-Jimenez, F.; Hishiki, T.; Garcia-Buey, L.; Koutsoudakis, G.; Shimotohno, K.; Benedicto, I.; Majano, P.L. Apolipoprotein E, but Not Apolipoprotein B, Is Essential for Efficient Cell-to-Cell Transmission of Hepatitis C Virus. J. Virol. 2015, 89, 9962–9973. [Google Scholar] [CrossRef]
- Zhao, F.; Zhao, T.; Deng, L.; Lv, D.; Zhang, X.; Pan, X.; Xu, J.; Long, G. Visualizing the Essential Role of Complete Virion Assembly Machinery in Efficient Hepatitis C Virus Cell-to-Cell Transmission by a Viral Infection-Activated Split-Intein-Mediated Reporter System. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Lee, J.Y.; Acosta, E.G.; Stoeck, I.K.; Long, G.; Hiet, M.S.; Mueller, B.; Fackler, O.T.; Kallis, S.; Bartenschlager, R. Apolipoprotein E likely contributes to a maturation step of infectious hepatitis C virus particles and interacts with viral envelope glycoproteins. J. Virol. 2014, 88, 12422–12437. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.; Dumans, A.; Beaumont, E.; Etienne, L.; Roingeard, P.; Meunier, J.C. The association of hepatitis C virus glycoproteins with apolipoproteins E and B early in assembly is conserved in lipoviral particles. J. Biol. Chem. 2014, 289, 18904–18913. [Google Scholar] [CrossRef] [PubMed]
- Witteveldt, J.; Evans, M.J.; Bitzegeio, J.; Koutsoudakis, G.; Owsianka, A.M.; Angus, A.G.; Keck, Z.Y.; Foung, S.K.; Pietschmann, T.; Rice, C.M.; et al. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J. Gen. Virol. 2009, 90, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Catanese, M.T.; Law, L.M.; Khetani, S.R.; Syder, A.J.; Ploss, A.; Oh, T.S.; Schoggins, J.W.; MacDonald, M.R.; Bhatia, S.N.; et al. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat. Biotechnol. 2010, 28, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Decembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef]
- Longatti, A.; Boyd, B.; Chisari, F.V. Virion-independent transfer of replication-competent hepatitis C virus RNA between permissive cells. J. Virol. 2015, 89, 2956–2961. [Google Scholar] [CrossRef]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.C.; Ray, R.; Ray, R.B. Knockdown of Autophagy Inhibits Infectious Hepatitis C Virus Release by the Exosomal Pathway. J. Virol. 2016, 90, 1387–1396. [Google Scholar] [CrossRef]
- Medvedev, R.; Hildt, E.; Ploen, D. Look who’s talking-the crosstalk between oxidative stress and autophagy supports exosomal-dependent release of HCV particles. Cell Biol. Toxicol. 2017, 33, 211–231. [Google Scholar] [CrossRef]
- Xiao, F.; Fofana, I.; Heydmann, L.; Barth, H.; Soulier, E.; Habersetzer, F.; Doffoel, M.; Bukh, J.; Patel, A.H.; Zeisel, M.B.; et al. Hepatitis C virus cell-cell transmission and resistance to direct-acting antiviral agents. PLoS Pathog. 2014, 10, e1004128. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, C.C.; Tawar, R.G.; Mailly, L.; Thumann, C.; Heydmann, L.; Durand, S.C.; Xiao, F.; Robinet, E.; Pessaux, P.; Zeisel, M.B.; et al. Humanisation of a claudin-1-specific monoclonal antibody for clinical prevention and cure of HCV infection without escape. Gut 2018, 67, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Lupberger, J.; Fofana, I.; Baumert, T.F. Host-targeting agents for prevention and treatment of chronic hepatitis C-perspectives and challenges. J. Hepatol. 2013, 58, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Ansuini, H.; Graziani, R.; Huby, T.; Moreau, M.; Ball, J.K.; Paonessa, G.; Rice, C.M.; Cortese, R.; Vitelli, A.; et al. Role of scavenger receptor class B type I in hepatitis C virus entry: Kinetics and molecular determinants. J. Virol. 2010, 84, 34–43. [Google Scholar] [CrossRef]
- Syder, A.J.; Lee, H.; Zeisel, M.B.; Grove, J.; Soulier, E.; Macdonald, J.; Chow, S.; Chang, J.; Baumert, T.F.; McKeating, J.A.; et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J. Hepatol. 2011, 54, 48–55. [Google Scholar] [CrossRef]
- Lacek, K.; Vercauteren, K.; Grzyb, K.; Naddeo, M.; Verhoye, L.; Slowikowski, M.P.; Fafi-Kremer, S.; Patel, A.H.; Baumert, T.F.; Folgori, A.; et al. Novel human SR-BI antibodies prevent infection and dissemination of HCV in vitro and in humanized mice. J. Hepatol. 2012, 57, 17–23. [Google Scholar] [CrossRef]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef]
- Flint, M.; von Hahn, T.; Zhang, J.; Farquhar, M.; Jones, C.T.; Balfe, P.; Rice, C.M.; McKeating, J.A. Diverse CD81 proteins support hepatitis C virus infection. J. Virol. 2006, 80, 11331–11342. [Google Scholar] [CrossRef]
- Molina, S.; Castet, V.; Pichard-Garcia, L.; Wychowski, C.; Meurs, E.; Pascussi, J.M.; Sureau, C.; Fabre, J.M.; Sacunha, A.; Larrey, D.; et al. Serum-derived hepatitis C virus infection of primary human hepatocytes is tetraspanin CD81 dependent. J. Virol. 2008, 82, 569–574. [Google Scholar] [CrossRef]
- Fofana, I.; Xiao, F.; Thumann, C.; Turek, M.; Zona, L.; Tawar, R.G.; Grunert, F.; Thompson, J.; Zeisel, M.B.; Baumert, T.F. A novel monoclonal anti-CD81 antibody produced by genetic immunization efficiently inhibits Hepatitis C virus cell-cell transmission. PLoS ONE 2013, 8, e64221. [Google Scholar] [CrossRef]
- Fofana, I.; Krieger, S.E.; Grunert, F.; Glauben, S.; Xiao, F.; Fafi-Kremer, S.; Soulier, E.; Royer, C.; Thumann, C.; Mee, C.J.; et al. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology 2010, 39, 953–964. [Google Scholar] [CrossRef]
- Mailly, L.; Xiao, F.; Lupberger, J.; Wilson, G.K.; Aubert, P.; Duong, F.H.; Calabrese, D.; Leboeuf, C.; Fofana, I.; Thumann, C.; et al. Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat. Biotechnol. 2015, 33, 549–554. [Google Scholar] [CrossRef]
- Michta, M.L.; Hopcraft, S.E.; Narbus, C.M.; Kratovac, Z.; Israelow, B.; Sourisseau, M.; Evans, M.J. Species-specific regions of occludin required by hepatitis C virus for cell entry. J. Virol. 2010, 84, 11696–11708. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Shirasago, Y.; Kondoh, M.; Suzuki, T.; Wakita, T.; Hanada, K.; Yagi, K.; Fukasawa, M. Monoclonal antibodies against occludin completely prevented hepatitis C virus infection in a mouse model. J. Virol. 2018. [Google Scholar] [CrossRef]
- Okai, K.; Ichikawa-Tomikawa, N.; Saito, A.C.; Watabe, T.; Sugimoto, K.; Fujita, D.; Ono, C.; Fukuhara, T.; Matsuura, Y.; Ohira, H.; et al. A novel occludin-targeting monoclonal antibody prevents hepatitis C virus infection in vitro. Oncotarget 2018, 9, 16588–16598. [Google Scholar] [CrossRef][Green Version]
- Shimizu, Y.; Shirasago, Y.; Suzuki, T.; Hata, T.; Kondoh, M.; Hanada, K.; Yagi, K.; Fukasawa, M. Characterization of monoclonal antibodies recognizing each extracellular loop domain of occludin. J. Biochem. 2019. [Google Scholar] [CrossRef]
- Shimizu, Y.; Yoneda, K.; Shirasago, Y.; Suzuki, T.; Tada, M.; Ishii-Watabe, A.; Sugiyama, K.; Suzuki, T.; Wakita, T.; Yagi, K.; et al. Human-rat chimeric anti-occludin monoclonal antibodies inhibit hepatitis C virus infection. Biochem. Biophys. Res. Commun. 2019, 514, 785–790. [Google Scholar] [CrossRef]
- Fuerst, T.R.; Pierce, B.G.; Keck, Z.Y.; Foung, S.K.H. Designing a B Cell-Based Vaccine against a Highly Variable Hepatitis C Virus. Front. Microbiol. 2017, 8, 2692. [Google Scholar] [CrossRef]
- Duncan, J.D.; Urbanowicz, R.A.; Tarr, A.W.; Ball, J.K. Hepatitis C Virus Vaccine: Challenges and Prospects. Vaccines (Basel) 2020, 8, 90. [Google Scholar] [CrossRef]


{kind=link}
| Host Factor | HCV Entry Steps | Host-Targeting Agents | References |
|---|---|---|---|
| Scavenger receptor BI (SR-BI) | Attachment, postbinding, cell-to-cell transmission | Anti-SR-BI mAbs ITX5061 | [46,47,48,49,50,51,52,53,128,146,147,148] |
| CD81 | Postbinding, endocytosis, signaling, cell-cell transmission | Anti-CD81 mAbs | [37,47,67,78,96,149,150,151,152] |
| Claudin-1 (CLDN1) | Postbinding, endocytosis, cell-cell transmission | Anti-CLDN1 mAbs | [21,66,69,70,96,143,144,153,154] |
| Occludin (OCLN) | Postbinding, endocytosis, cell-cell transmission | Anti-OCLN mAbs | [22,89,90,155,156,157,158,159] |
| Epidermal growth factor receptor (EGFR) | Postbinding, endocytosis, signaling, cell-cell transmission | Anti-EGFR mAbs Erlotinib | [77,79] |
| Niemann-Pick C1-like 1 (NPC1L1) | Postbinding, fusion, cell-cell transmission | Ezetimide | [95] |
| 5-HT2AR | Endocytosis, fusion | Phenoxybenzamine | [122] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colpitts, C.C.; Tsai, P.-L.; Zeisel, M.B. Hepatitis C Virus Entry: An Intriguingly Complex and Highly Regulated Process. Int. J. Mol. Sci. 2020, 21, 2091. https://doi.org/10.3390/ijms21062091
Colpitts CC, Tsai P-L, Zeisel MB. Hepatitis C Virus Entry: An Intriguingly Complex and Highly Regulated Process. International Journal of Molecular Sciences. 2020; 21(6):2091. https://doi.org/10.3390/ijms21062091
Chicago/Turabian StyleColpitts, Che C., Pei-Ling Tsai, and Mirjam B. Zeisel. 2020. "Hepatitis C Virus Entry: An Intriguingly Complex and Highly Regulated Process" International Journal of Molecular Sciences 21, no. 6: 2091. https://doi.org/10.3390/ijms21062091
APA StyleColpitts, C. C., Tsai, P.-L., & Zeisel, M. B. (2020). Hepatitis C Virus Entry: An Intriguingly Complex and Highly Regulated Process. International Journal of Molecular Sciences, 21(6), 2091. https://doi.org/10.3390/ijms21062091