1. Introduction
Chorea Acanthocytosis (ChAc) is a rare neurodegenerative disease that affects 500–1000 persons worldwide (OMIM ID: 200150) [
1]. It is part of the group of neuroacanthocytosis syndromes, which include McLeod syndrome, Huntington disease-like syndrome 2 and pantothenate kinase-associated neurodegeneration. The inheritance pattern is autosomal recessive and more than 134 causative mutations of ChAc are located in the locus of
VPS13A (9q21.2) [
2]. Patients affected by the disease present various clinical features including orofacial dyskinesia and choreatiform movements with typical drops of the upper body. Most patients of the disease share signs of peripheral neuropathy. Other features are irregular but may include epilepsy in the early stages and Parkinsonism as the disease progresses. The affected neurons mainly responsible for the choreatiform movement disorder are medium spiny neurons (MSN) of the basal ganglia [
3]. However, many other neuronal subtypes are affected as well, such as dopaminergic neurons or motoneurons, explaining the plethora of maladies including parkinsonism and/or peripheral neuropathy [
4].
The affected gene locus is conserved in higher organisms and various functions have been attributed to its product in lower organisms. Pioneering work on these functional aspects was conducted in yeast, where vacuole sorting, sporulation and inter organelle signaling was described [
5,
6]. More recently, further functional aspects of VPS13A were revealed by description of its function as a lipid transport protein, where it is located at various membrane contact sites—most notably the endoplasmic reticulum and mitochondria [
7,
8,
9,
10].
Structural analysis showed that Vps13 has an APT1 and SHR_BD domain that is responsible for interaction with lipids, among which PI3P is pivotal, due to its ability to guide organelles and facilitate the recruitment of other proteins [
11].
The main focus of interest in the scientific community so far has been the prominent feature presented by the acanthocytes and thus the molecular understanding of the disease mainly arose from work in red blood cells or related hematic models. Current understanding is highlighting two major pathways being affected. Changes in the activity of PI3K and the subsequent kinases RAC1 and PAK1 were shown to have a strong effect on the polymerization of cortical actin, hence causing an acanthocytic shape in erythrocytes [
12,
13,
14]. Also downstream of PI3K is the serum and glucocorticoid inducible kinase SGK1, which regulates the activity of NFκB signaling, ultimately regulating the Ca
2+ channel subunit Orai1 and store operated Ca
2+ entry (SOCE). Hence, it was shown that chorein deficiency resulted in lower Orai1 expression and reduced SOCE, which was at least partially restored upon lithium treatment [
15,
16]. This finding was later confirmed in medium spiny neurons [
17].
The second pillar of the underlying pathophysiology is the SRC-kinase Lyn. Upon depletion of chorein, Lyn becomes hyper active and subsequently phosphorylates its targets, including the anion transport protein SLC4A1 (band 3) [
18]. This was shown to be the major driving force in impairing autophagy [
19]. Furthermore, the hyperactive Lyn was shown to be responsible for the hyperexcitability, which was reported in ChAc patient induced pluripotent stem cell (iPSC) derived MSN [
20]. Treatment of affected cell cultures with the SRC-kinase inhibitor 4-Amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo(3,4-d)pyrimidine (PP2) reverted the phenotypes back to wild type levels.
In our recent study on human patient-derived MSNs, however, we were able to show specific neuronal impairments of mitochondrial and lysosomal morphologies and trafficking features, which were not affected by Lyn kinase inhibition using PP2 pointing towards cell type specific effects [
21]. The current study focuses on the effects of chorein deficiency in patient iPSC derived spinal motoneurons, the cell type causing the hallmark symptom of peripheral motor neuropathy.
3. Discussion
Major advances in understanding the pathomechanism of ChAc and the function of chorein and its orthologues have been made recently [
7,
8,
9,
10,
25]. Still, the main focus of the scientific community lies on the investigation of erythrocyte and medium spiny neuron (MSN) phenotypes as the primarily affected cell types. ChAc patients suffer from a variety of maladies, of which peripheral (motor) neuropathy is a main symptom. In this article, we comparatively investigate for the first time the influence of chorein deficiency on dopaminergic neurons and spinal motoneurons, to gain a better understanding on the late stage ChAc, which can present with Parkinsonism as well as peripheral neuropathy.
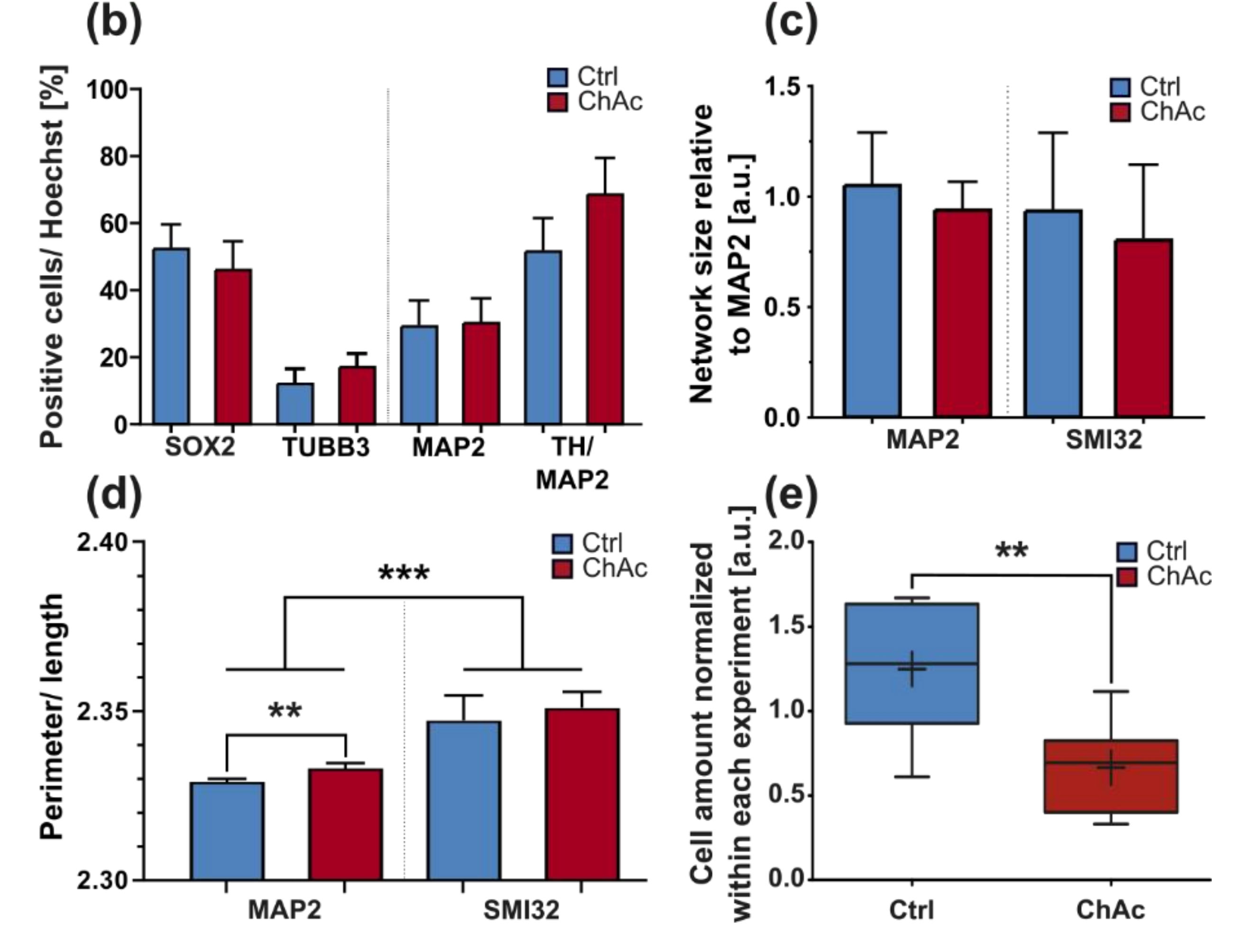
We were not able to identify any abnormalities in the differentiation capacity of ChAc iPSC into midbrain/hindbrain neurons, which is in agreement with previous publications that showed a normal differentiation towards MSN [
20,
21]. In line with this observation, the age of onset and the clinical presentation of ChAc patients strongly suggests a degenerative progression rather than a developmental defect.
Using a MFC system to separate axons from dendrites as well as neurons from non-neuronal cells was documented before and is a well-established technique [
22,
26]. Motoneurons are particularly well suited to penetrate the long distance of the micro channels, because of their sensitivity to the chemotactic guidance of GDNF and BDNF and their nature of generating very long axons and growth cues. We were able to separate them from the other neuronal subtypes and non-neuronal cells using this culture system.
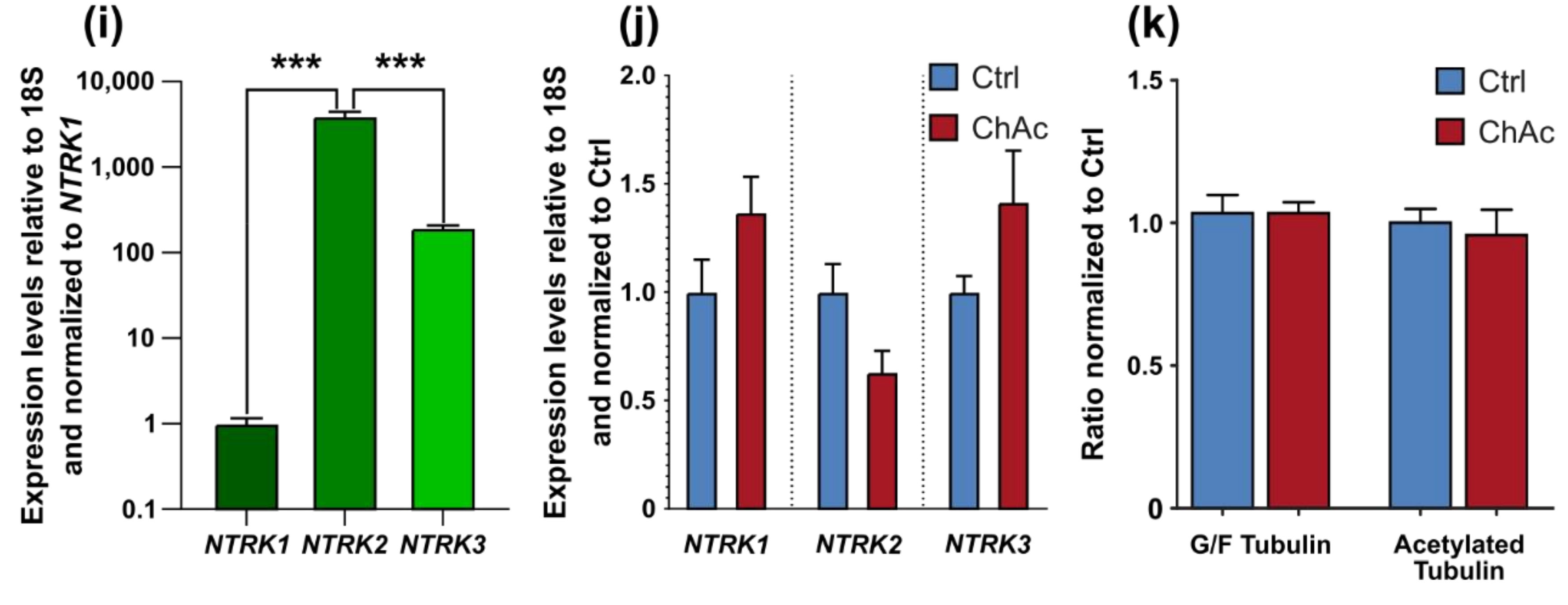
Interestingly, the growth of ChAc axons through the MFCs depends on BDNF substitution. This could either hint to a lack of the endogenous production of BDNF by the cells or a defective signaling which requires elevated BDNF levels to begin with. The concentration of BDNF used in this assay is frequently used in these culture systems [
27]. BDNF levels in the brain are dependent on external stimuli. During context-dependent fear-conditioning tests in mice, the baseline BDNF concentration in the cerebrospinal fluid was estimated at 1–10 pg/mL [
28]. Endogenous levels of BDNF in the human brain are not well documented, however, we believe that the herein used concentration is exceeding the average concentration and therefore should result in a constant activation of BDNF receptors. Since under these BDNF-excessive conditions, there was no ChAc-specific phenotype present and the expression levels of
NTRK1,
NTRK2 and
NTRK3 were not different, we think the resulting effects are due to changes downstream in the BDNF signaling cascade.
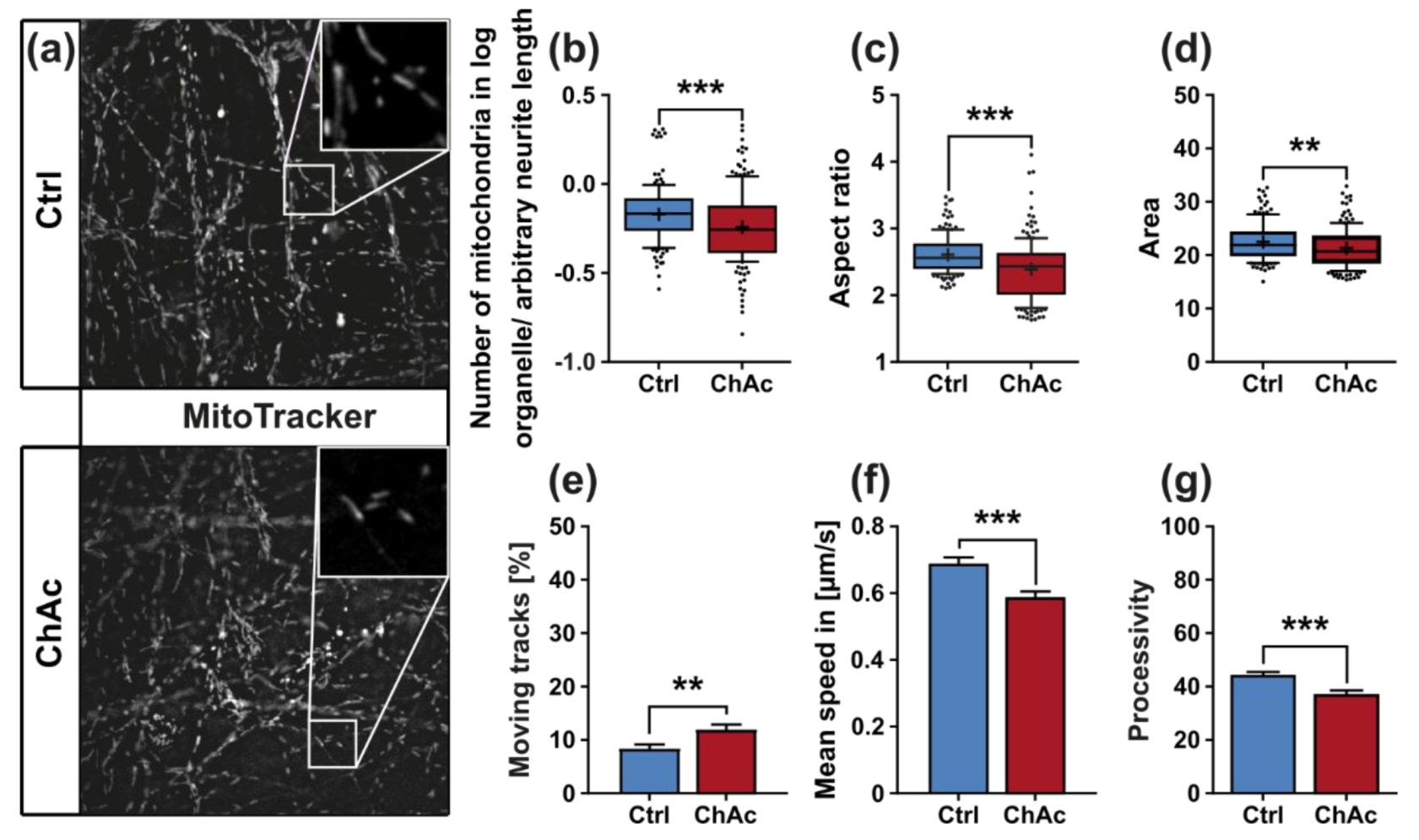
We found that the mitochondria in unguided cultures share similar morphological features in midbrain/hindbrain neurons with mitochondria in MSN [
21]. The trafficking phenotype of midbrain/hindbrain neurons was similar to the one seen in MSN as well. The striking hyperpolarization we observed in the distal axonal sites was reported in MSN as well [
21]. We believe that an increased mitochondrion potential can also lead to neurodegeneration, as was reported in the literature [
29,
30]. Interestingly, it is also described as a measure to counter apoptosis [
31]. Since all mitochondria of the distal axon have to pass proximal site, it remains unclear whether the observed shift in potential is upstream or downstream of the proximally observed trafficking phenotypes. Together with the previously described changes in mitochondria trafficking, especially the increase in retrograde transport, is very likely reflecting increased mitochondrial damage, which urges the cell to recycle it´s mitochondria—a feature, which is usually done in the perinuclear region. In summary, these results indicate that motoneurons suffer from mitochondrial damage in a similar pattern as observed in MSN. We believe that the differences are reflecting the divergent natures of these different neuronal subtypes.
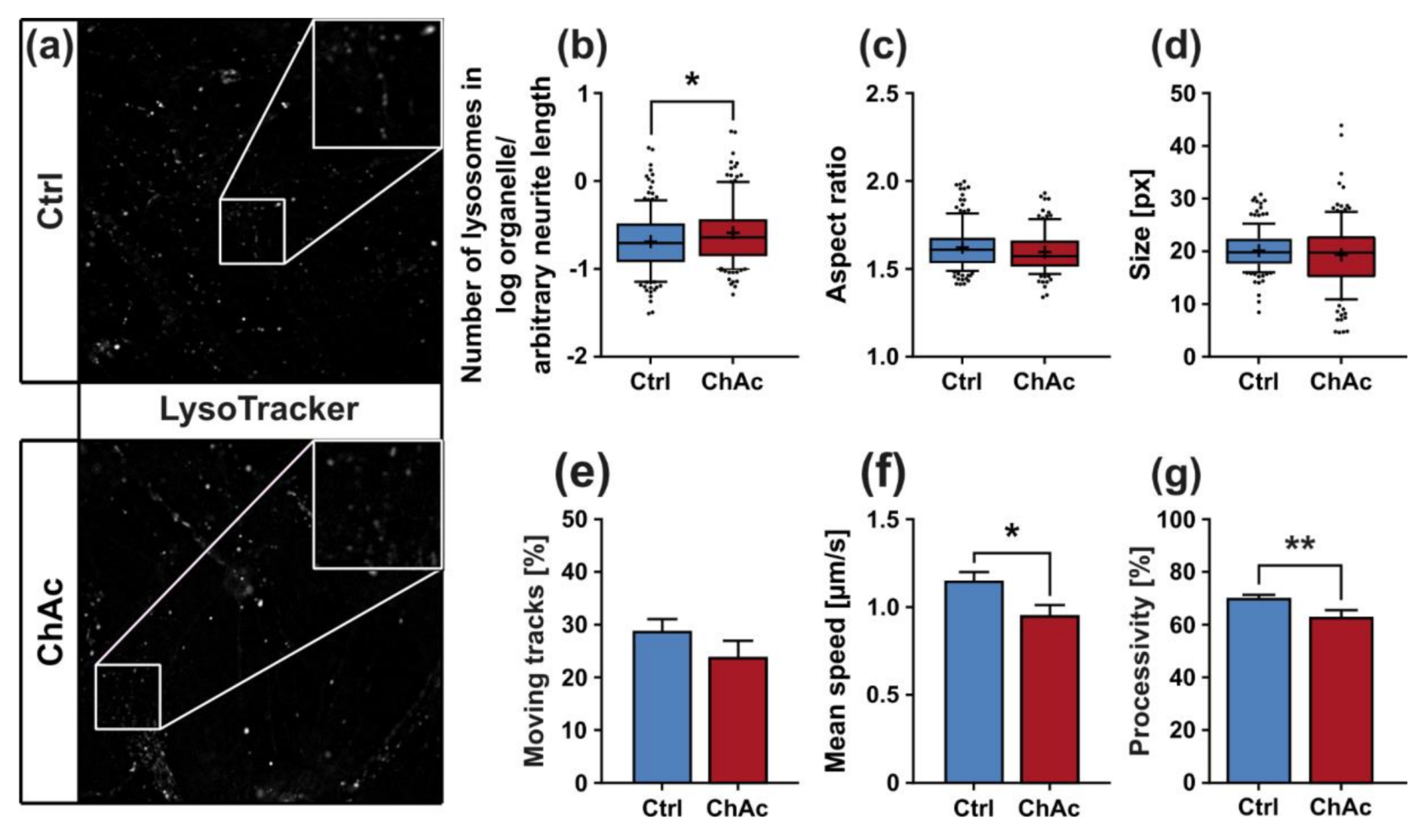
The morphological analysis of endo-/lysosomes revealed an increase in number of organelles. In order to shed light if this is due to an overproduction of lysosomal vesicles or due a deficit in downstream fusion of these compartments, further experiments need to be conducted. Even though we did not comprehensively investigate the underlying mechanism, both possibilities suggest an impairment of this pathway, as was described previously [
25,
32]. The altered transport features of these compartments can be a secondary effect due to mitochondrial failure to provide enough ATP to the cells or it can be indicative that the underlying rails—the microtubules—are defective. However, we did not find a significant difference in microtubule polymerization as well as acetylation (
Figure 2k). Nonetheless, it is still possible that other substantial changes in microtubule modification are present, e.g., phosphorylation, tyrosinylation or alterations of microtubule binding proteins such as Tau.
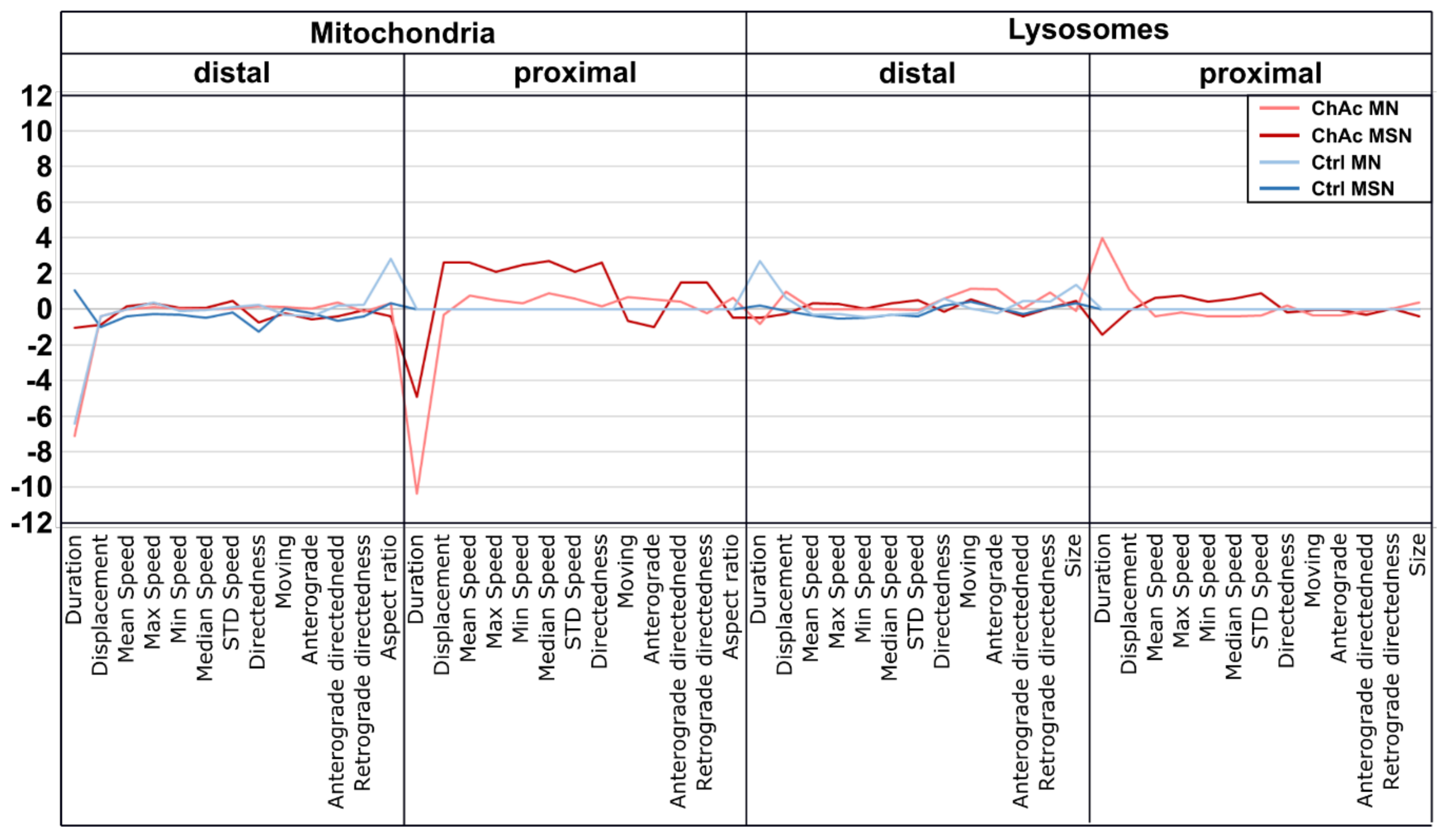
We identified differences for mitochondria as well as lysosomes during morphological and trafficking characterization between unguided cultures and MFCs. These could have different reasons. As mentioned above, the exclusive analysis of motoneurons in MFCs could be one reason for this, as well as unspecific factors due to the culture conditions per se. However, these differences could also point towards differences between axons and dendrites. The latter would fit to the data obtained in unguided cultures (
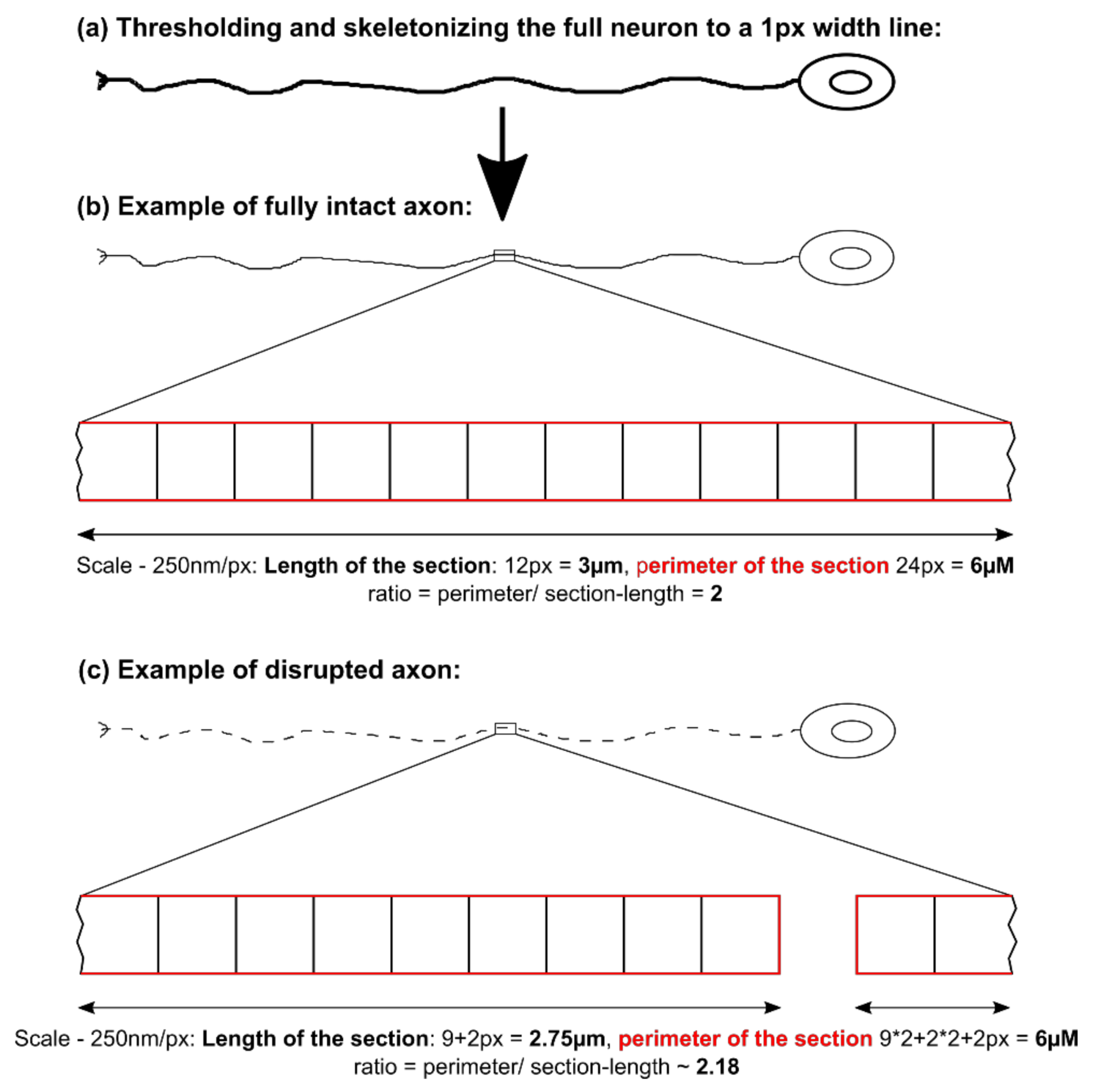
Figure 1) showing a disease phenotype only in dendrites (MAP2 fragmentation) but not in axons (no differences in SMI32 fragmentation between ChAc and Ctrl.) and reduced amount of dendrites (
Figure 2d) while having increased axon lengths (
Figure 2c). However, further studies need to elaborate on this in more details.
The high content disease profile we generated for ChAc patients’ midbrain/hindbrain neurons indicated that different neuronal subtypes share common phenotypes, which are very distinct from other neurodegenerative diseases such as ALS [
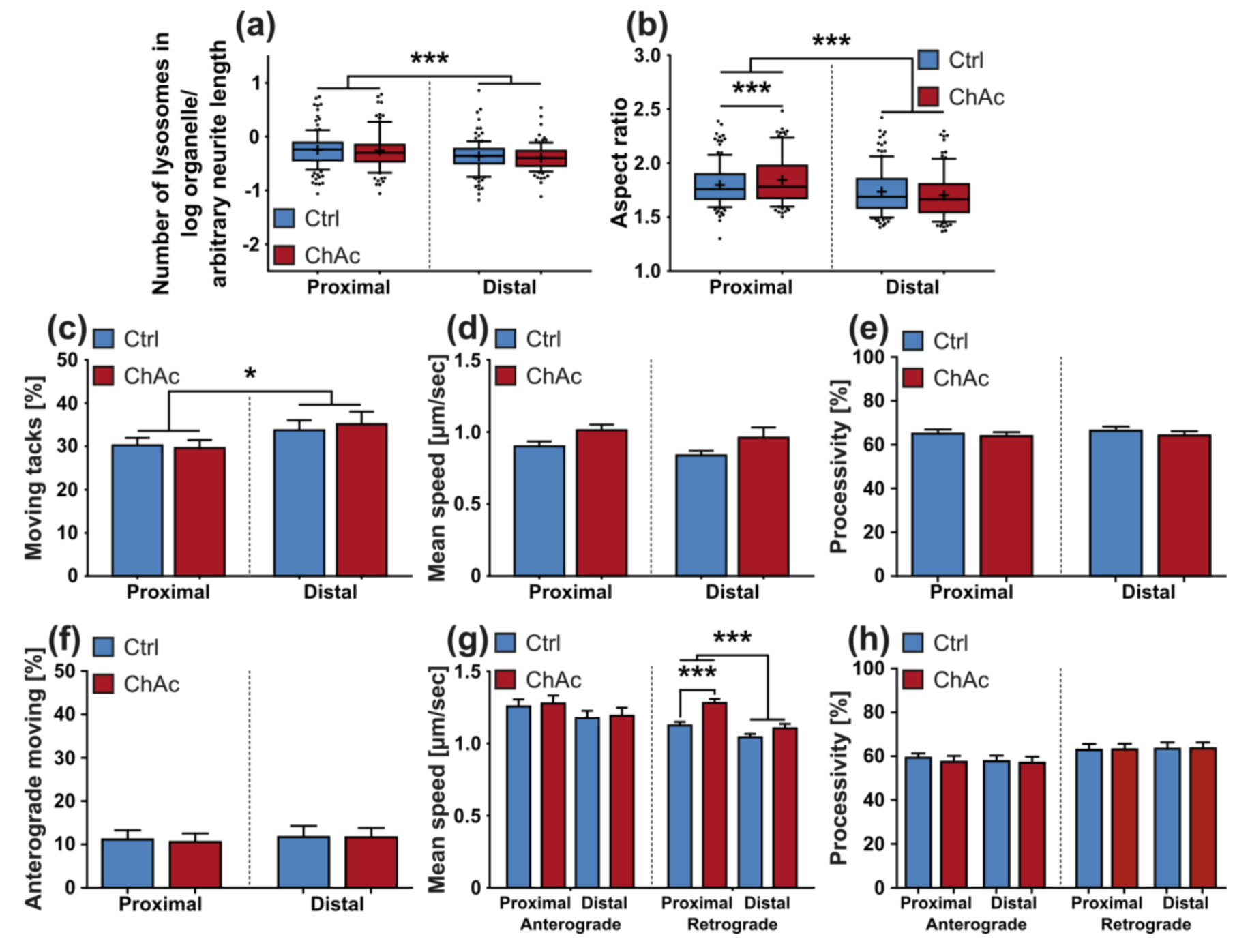
33]. The intensity of the phenotypes corresponds to the clinical severity of the mutations. While the previously published FUS-P525L mutation has a very early disease onset and a fast progression, ChAc has milder course and is usually later onset. In contrast to the ALS profile, which harbors a strong distal phenotype, which is sometimes referred to as “dying-back”, the more pronounced phenotypes in ChAc patient’s neurons were observed in the proximal axon. Even though the singular most striking phenotype of the distal axon was the hyperpolarization of mitochondria, we cannot be certain that this is not a downstream symptom of the mitochondrial impairments in proximal axons (
Figure 7). We therefore believe that—given the current data set—the neurodegenerative progression of neurons affected by ChAc starts at their proximal site.
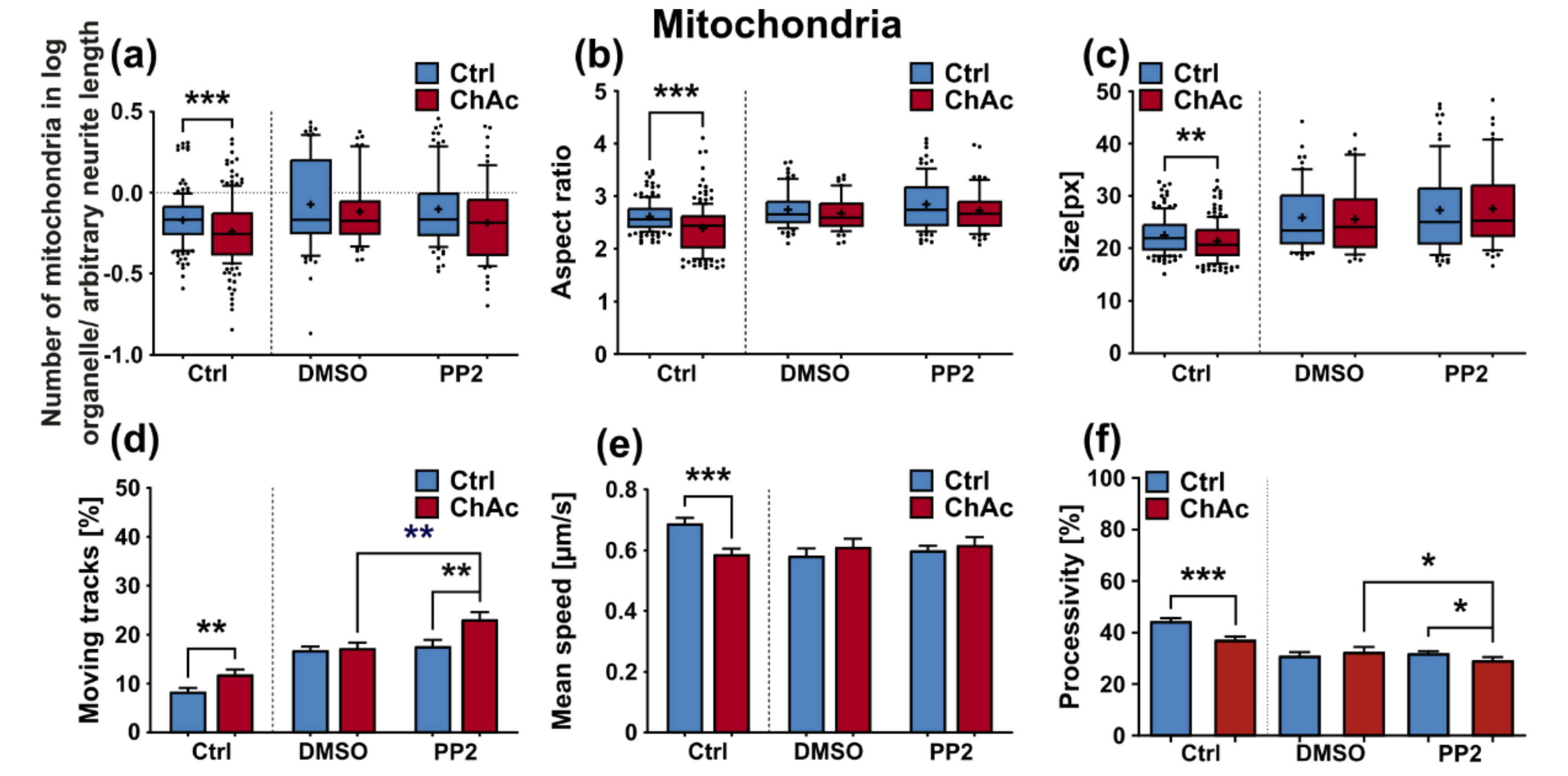
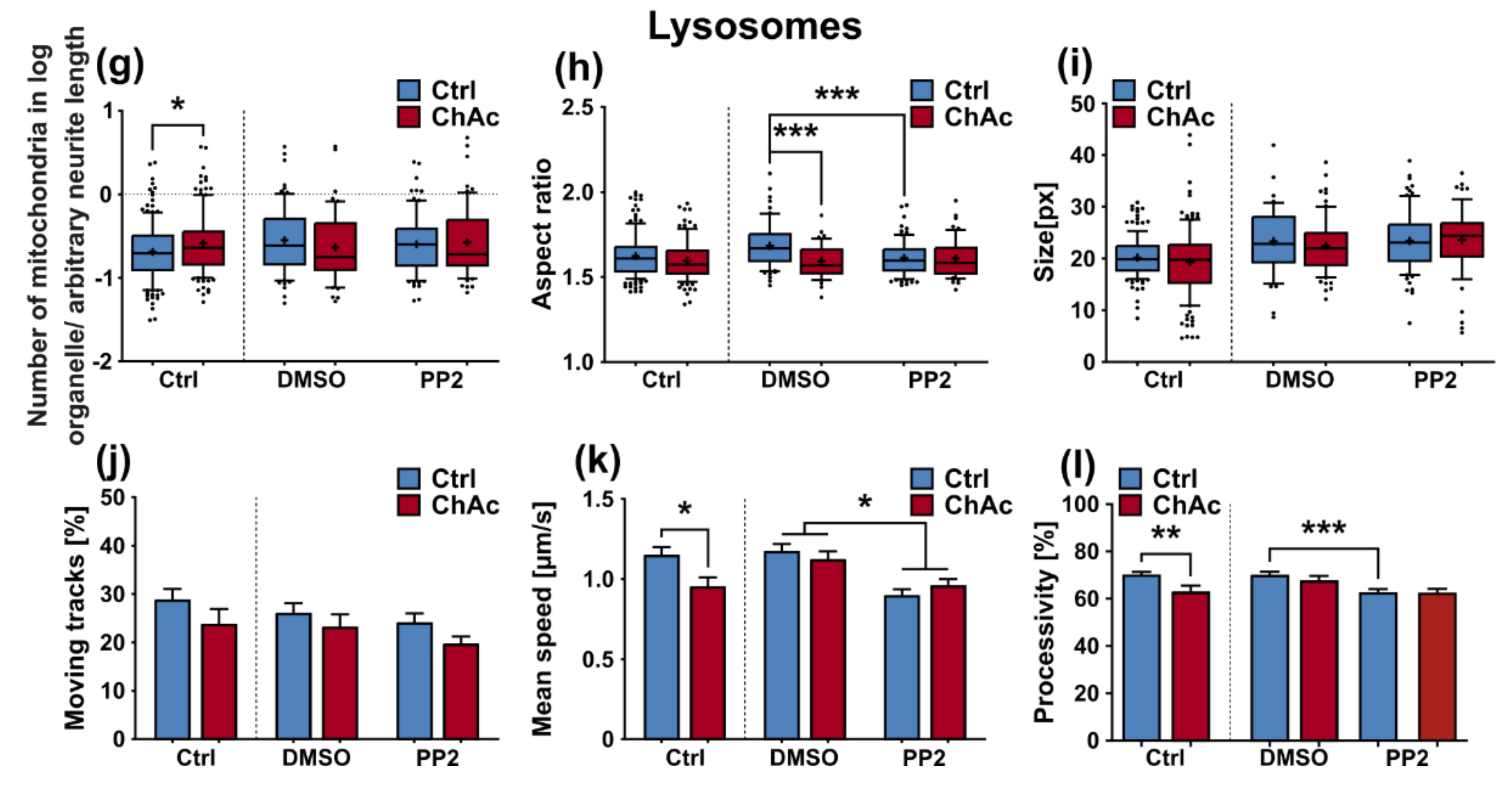
We were not able to revert the disease phenotypes back to control conditions by means of treatment with PP2. Some readouts suggested that the effect attributed to the carrier, DMSO, was larger than that of the Srk-kinase inhibitor (
Figure 8c,d,f). We observed a similar behavior when investigating MSN previously [
21]. Since the observed phenotypes resemble the ones we described in MSN, this further strengthens our believe that the underlying mechanism that leads to these subtle organelle impairments is mediated by a pathway different to Lyn-kinase or is indeed affected by parts which are further upstream. One other candidate for this different pathway is the PI3K signaling pathway, which was already investigated in the context of survival and cytoskeletal organization of erythrocytes [
3]. Our findings suggest that the same pathomechanism affects different neuronal subtypes that are similar to MSN but manifest in different clinical symptoms—e.g., peripheral neuropathy for motoneurons and parkinsonism for dopaminergic neurons but might be different to other tissues. These findings highlight the complex requirements for an effective treatment of this multifaceted disease, which calls for taking clinical pathological phenotypes that are not concerned with the primarily affected neuronal sub populations into account.
4. Materials and Methods
4.1. Materials Patient Characteristics
Patient characteristics are shown in
Table 1. All procedures were in accordance with the Helsinki convention and approved by the Ethical Committee of the Technische Universität Dresden (EK45022009).
4.2. iPSC Derivation
We generated iPSC from patients shown in
Table 1. Derivation and characterization of these lines was described previously [
20]. In short, we transfected fibroblasts using four monocistronic retroviral vectors carrying the Yamanaka factors—OCT4, SOX2, KLF4 and MYCC (addgene #17217, #17218, #17219, #17220, #8449, #8454). We cultivated emerging colonies on MMC (Tocris Bioscience, Bristol, UK)-treated mouse-embryonic-fibroblasts in embryonic stem cell (ESC)-medium (77.8% KO-DMEM (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 20% KO-Serum (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 1% non-essential amino acids (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 1% Penicillin/ Streptomycin (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 0.2% β-mercaptoethanol (Thermo Fisher Scientific, Waltham, Massachusetts, USA)), supplemented with bFGF (Sigma Aldrich, St. Louis, Missouri, USA). We then characterized of these lines by alkaline phosphatase staining, confirmation of exogene silencing/activation of endogenous transcription factors by a quantitative polymerase chain reaction, immunocytochemistry of embryonic stem cell markers, 3-germ-layer-differentiation and confirmation of ChAc-phenotype by chorein western blot. We used two clones from each individual in all of the following experiments. We subsequently differentiated the obtained iPSC lines into small-molecule neuronal-precursor-cells (smNPC).
4.3. Derivation of smNPCs
We conducted our experiments in a smNPC-derived neuronal cell model system, since they are shown to generate a reproducible amount of midbrain dopaminergic neurons as well as hindbrain specific motoneurons—which is ideal for our investigative scope of not-primarily affected neuronal subtypes in ChAc. We derived smNPC from iPSCs, as reported previously [
22,
26,
28]. In short, we cleaned spontaneous differentiated colonies off adherent iPSC cultures and incubated with 2 mg/mL type IV collagenase for one hour. We then collected detached colonies and cultivated them as embryoid bodies in ESC-media supplemented with 1 µM Dorsomorphin (Tocris Bioscience, Bristol, UK), 10 µM SB-431542 (Tocris Bioscience, Bristol, UK), 3 µM CHIR99021 (Cayman chemical company, Ann Arbor, Michigan, USA) and 0.5 μM purmorphamine (PMA) (Cayman chemical company, Ann Arbor, Michigan, USA). We replaced the ESC-media with N2B27 (49% DMEM/F12, 48.5% NeuroBasal, 0.5% N2-supplement, 1% B27-supplement, 1% Penicillin/Streptomycin/L-glutamine) after 2 days. Dorsomorphin and SB-431542 were omitted from day 4 and 150 µM ascorbic acid (AA) (Sigma Aldrich, St. Louis, Missouri, USA) was additionally added. On day 6 we triturated the embryoid bodies and seeded them on matrigel (BD Bioscience, Franklin Lakes, New Jersey, USA) coated plates, which were prepared one day prior to thawing or passaging. We cultivated the lines until passage 10 times to obtain a stable smNPC phenotype, and this was confirmed by quantitative reverse transcription polymerase chain reaction qPCR.
4.4. Culture of smNPCs
We expanded smNPCs continuously on matrigel coated plates in N2B27 media supplemented with 3 µM CHIR99021, 150 µM AA and 0.5 µM PMA, which we changed every other day during proliferation phase. At each split we seeded 100,000 cell/cm². When confluence was reached after roughly one week, we passaged the cultures or initiated differentiation. We washed the cells twice with warm PBS and treated them for 10 min at 37 °C with accutase. The enzymatic reaction was quenched by adding double the volume of warm DMEM/F12 (Thermo Fisher Scientific, Waltham, Massachusetts, USA). We collected the cell suspension and centrifuged at 250 g for 5 min. We then discarded the supernatant and resuspended the cells in 1 mL of warm N2B27 media supplemented with 3 µM CHIR99021, 150 µM AA and 0.5 µM PMA. We assessed concentration by diluting the suspension 1:100 and counting the cells with a Neubauer slide.
4.5. Midbrain/Hindbrain Differentiation
We differentiated smNPC cultures into mature neurons using a multistep protocol according to Reinhardt et al. [
26]. Briefly, we seeded smNPCs in a 6-well and changed media every other day. For the first 2 days, we cultivated cells under normal proliferation conditions using N2B27 supplemented with 3 µM CHIR99021, 150 µM AA and 0.5 µM PMA. On day 2 we changed medium to induction conditions and applied N2B27 supplemented with 200 µM AA, 1 µM PMA and 10 ng/mL FGF8 (R&D Systems, Minneapolis, Minnesota, USA). On day 10, we passaged cells and reseeded them into their final format. For ICC, we seeded cells in 4-well plates with coated cover slips at a density of 100,000 cells/well. For selective motoneuron analysis, we used MFC and injected 300,000 cells in one macro channel of each mounted silicon device. From then on, we fed the cultures every other day with maturation media consisting of N2B27 supplemented with 20 ng/mL BDNF (Promega, Madison, Wisconsin, USA), 10 ng/mL GNDF (Sigma Aldrich, St. Louis, Missouri, USA), 1 ng/mL TGFβ3, (Peprotech, Rocky Hill, USA) 0.5 mM dbcAMP (Sigma Aldrich, St. Louis, Missouri, USA) and 200 µM AA. To achieve growth-through in our MFCs, we applied a growth factor gradient. The “soma”-site was supplemented with 1 ng/mL of TGFβ3, 0.5 mM dbcAMP and 200 µM AA, while the “synapse”-site received a complete media including 20 ng/mL BDNF and 10 ng/mL GNDF. We cultivated the maturating neuronal cultures for three weeks before immunocytochemistry or live cell imaging.
4.6. Immunocytochemistry
For ICC analysis, we washed mature neurons once with PBS and then fixated them by incubation with pre warmed 4% PFA solution for 12 min. After fixation, we washed the cells three times with PBS and subjected them to permeabilization for 10 min at room temperature using following solution (0,2% Triton X-100 (Thermo Fisher Scientifc, Waltham, Massachusetts, USA) in PBS). After permeabilization, we applied blocking buffer for 1 h at room temperature. The blocking buffer contained: 1% bovine serum albumin, 5% donkey serum, 0.3 M glycine (Carl Roth GmbH & Co. KG, Germany) and 0.025% Triton X-100 in PBS. We solved primary antibodies in blocking buffer and incubated the cells with them overnight in a 4 °C fridge: (anti-SOX2 (mouse, R&D Systems, USA) 1: 500; anti-NESTIN (rabbit, Merck, USA) 1: 500; anti-TUBB3 (chicken, Merck, USA) 1: 500; anti-GALC (mouse, Merck, USA) 1: 500; anti-GFAP (rabbit, abcam, USA) 1:600; anti-MAP2 (mouse, BD Bioscience, USA) 1: 500; anti-TH (rabbit, Pel Freez biologicals, Rogers, Arkansas, USA) 1: 500; anti SMI-32 (chicken, Covance, Princeton, New Jersey, USA) 1: 10,000). The next day, we washed the cells three times with PBS before secondary antibodies were incubated for 1 h at room temperature in the dark (all Alexa Fluor, 1: 500; Thermo Fisher Scientific, Waltham, Massachusetts, USA). Following this, we washed the cells three times with PBS and a counter-stained with Hoechst 33342 (0.75 µL/mL PBS; Thermo Fisher Scientific, Waltham, Massachusetts, USA) for 5 min. Cells were again washed two times with PBS before we transferred the coverslips onto object slides with Fluoromount (SouthernBiotech, Birmingham, Alabama, USA) as a mounting media. After one day, we sealed the rims of the coverslips with nail polish to prevent excessive drying.
4.7. Quantitative Reverse Transcription Polymerase Chain Reaction
We performed qPCR in order to investigate the expression of
NTRK1,
2 and
3. We cultivated mature midbrain/hindbrain neuronal cultures of ChAc patients and controls for three weeks in 6-well plates. We washed the cells with pre-warmed PBS and isolated their mRNA using the RNeasy Mini Kit (Qaigen, Hilden, The Netherlands) according to the manufacturer’s instructions. We measured mRNA concentration with a photometer and surveilled the quality by analyzing the 260 nm/230 nm and 260 nm/280 nm absorbance ratios. We then proceeded to generate cDNA from 1000 ng of mRNA using the QuantiTect Reverse Transcription kit (Qiagen, Hilden, The Netherlands) according to the manufacturer’s instructions. qPCR was conducted by using 1 µl of the previously yielded cDNA and the QuantiTect SYBR Green PCR kit (Qiagen, Hilden, The Netherlands). We designed primer-pairs specific for
NTRK1,
NTRK2 and
NTRK3. We used 18S rRNA as a reference gene. For more details regarding the primers used, please refer to
Table 2. We calculated expression values with the ΔΔ
ct method. We analyzed each sample at least with technical duplicates.
4.8. Western Blot
In order to visualize and quantify the G/F tubulin as well as acetylated tubulin, we used Western blots. We obtained fractioned protein samples as previously described, to yield globular tubulin containing extracts and filamentous tubulin containing ones [
34]. In short, we firstly washed two 6-wells of matured neurons with ice-cold phosphatase inhibitor. We then discarded the solution and incubated the cells for 5 min with 200 µL of cold G-actin extraction buffer (0.3% Triton X-100, 5 mM Tris, pH 7.4, 2 mM EGTA, 300 mM sucrose, 2 µM phalloidin, and freshly added Protease Inhibitor Cocktail (1:100)). We collected the soluble fraction stored it at −80 °C, while 200 µL of cold RIPA buffer (1% TritonX-100; 50 mM Tris-HCl; 0.15 M NaCl; 1% Natriumdeoxycholate; 0.1% SDS; 1 mM EDTA; 1 mM DTT; pH 7.4 and freshly added Protease Inhibitor Cocktail (1: 100)) was put in the well. We scraped the cells off the well and collected them in a 1.5 mL reaction tube and incubated them on ice for 15 min. We centrifuged the tube for 2 min at > 10,000
g at 4 °C to separate the insoluble fraction and transferred the supernatant into a new tube, while the pellet was discarded. We stored this so obtained F-actin containing fraction at −80 °C. We gained western blots samples for acetylated tubulin from whole cell lysates from maturated neuronal cultures. In short, we washed cells with PBS containing phosphatase inhibitor (Thermo Fisher Scientific, Waltham, Massachusetts, USA), scraped them into 1.5 mL reaction tubes (Eppendorf, Hamburg, Germany) and centrifuged them for 5 min at 200
g at 4°C. We then removed the supernatant and resuspended the cell pellet with RIPA buffer, thoroughly mixed the tube and incubated it for 15 min on ice. We then centrifuged the lysate for 5 min at >10,000
g at 4 °C and stored the protein containing supernatant at −80 °C, while discarding the DNA containing pellet. We measured protein concentration using Roti Nanoquant (Carl Roth, Karlsruhe, Germany) according to the manufacturers recommendations with a Tecan Sunrise plate reader (Absorbance: 450 nm and 590 nm).
For gel electrophoresis, we loaded same sample volumes per lane—in case of G-tubulin extract and F-tubulin extract—or 15 µg of protein for acetylated tubulin into 12% Bis-Tris precast gels (Life technologies, Carlsbad, California, USA). We conducted electrophoresis at 120 V for one and a half hours. We then blotted the samples onto PVDF membranes (Life Technologies, Carlsbad, California, USA) using an iBlot (Thermo Fisher Scientific, Waltham, Massachusetts, USA; Program 2, 9 min) and a discontinuous buffer condition. We used 25 mM Tris-HCl, 192 mM glycine and 15% methanol as anode buffer and 25 mM Tris-HCl, 192 mM glycine and 0.1% SDS as cathode buffer. We blocked membranes for 2 h with blocking solution (5% Amersham ECL Prime Blocking Reagent, GE Healthcare, Chicago, Illinois, USA). We used mouse anti-alpha tubulin (1: 5000, #7291, Abcam, Cambridge, UK), mouse anti-acetylated tubulin (1: 5000; T7451, Sigma Aldrich, St. Louis, Missouri, USA), hrp-conjugated anti-mouse secondary antibody (1: 5000, Dianova, Hamburg, Germany) and electrochemiluminescence ECL solution (GE healthcare, Chicago, Illinois, USA) for visualization on an LAS 3000 imager (0.8 aperture, 10 s increment, sensitivity high 0.8 aperture, Fuji, Minato, Tokyo, Japan). We performed evaluation of the western blots in the TotalLab Quant software (Totallab, Newcastle-Upon-Tyne, UK). For the quantification of acetylated tubulin, we first developed a membrane with the anti-acetylated tubulin antibody and then proceeded to strip the antibody off by applying two times stripping buffer (1% Tween 20, 0.1% SDS and 1.5% glycine in de-ionized water at pH 2.2) for 1 h. We then washed the membranes twice with PBS for 10 min and twice with PBS-Tween for 5 min. Then we continued by blocking and anti-alpha tubulin incubation procedure as described above. Thereafter, we calculated the ratio of both exposures.
4.9. Live Cell Imaging
After the maturation split, we grew the neuronal cultures for a total of three weeks. We used MitoTracker DeepRed, LysoTracker Red DND-99 or JC-1 (50nM, 50nM or 200nM, respectively; all Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer’s recommendation and imaged the cells with a TIRF 6000. We kept exposure time at 115 ms obtaining video stacks at 3.3 fps for acquisition of two channels, 100× 1.4 Zeiss Plan-Apochromat, 400 pictures (2 min duration) hardware synchronized. We used the same illumination intensity and exposure time when taking images of JC-1 incubated cultures in the red and green fluorescence channels. We used self-made Fiji macros to determine the shape of the organelles and “TrackMate” plugin for tracking (settings: Dog detector, blob diameter 1.6 µm, quality threshold 100, kalman tracker, initial search radius 2 µm, kalman radius 1.25 µm, Max Gap distance 2). We used defined fields-of-view of micro channels of MFCs close to the channel entry at the “soma”-site and close to the channel exit at the “synapse”-site. Data extraction and mining was conducted using KNIME.
4.10. Statistics
We used GraphPad Prism 7 (Graphpad Software Inc., San Diego, California, USA) for statistical analysis. To evaluate the differentiation capacity and phenotype description of the live cell imaging experiments we calculated Student’s t test. When significant differences in the variances between groups were observed, we applied Welch’s correction. For MFC-based live cell imaging experiments as well as PP2 treatment experiments a 2 × 2-way ANOVA was designed with either Šidák’s test to calculate main effects or Tukey’s range test to compare all means.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}