Dysregulated Gut Homeostasis Observed Prior to the Accumulation of the Brain Amyloid-β in Tg2576 Mice

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
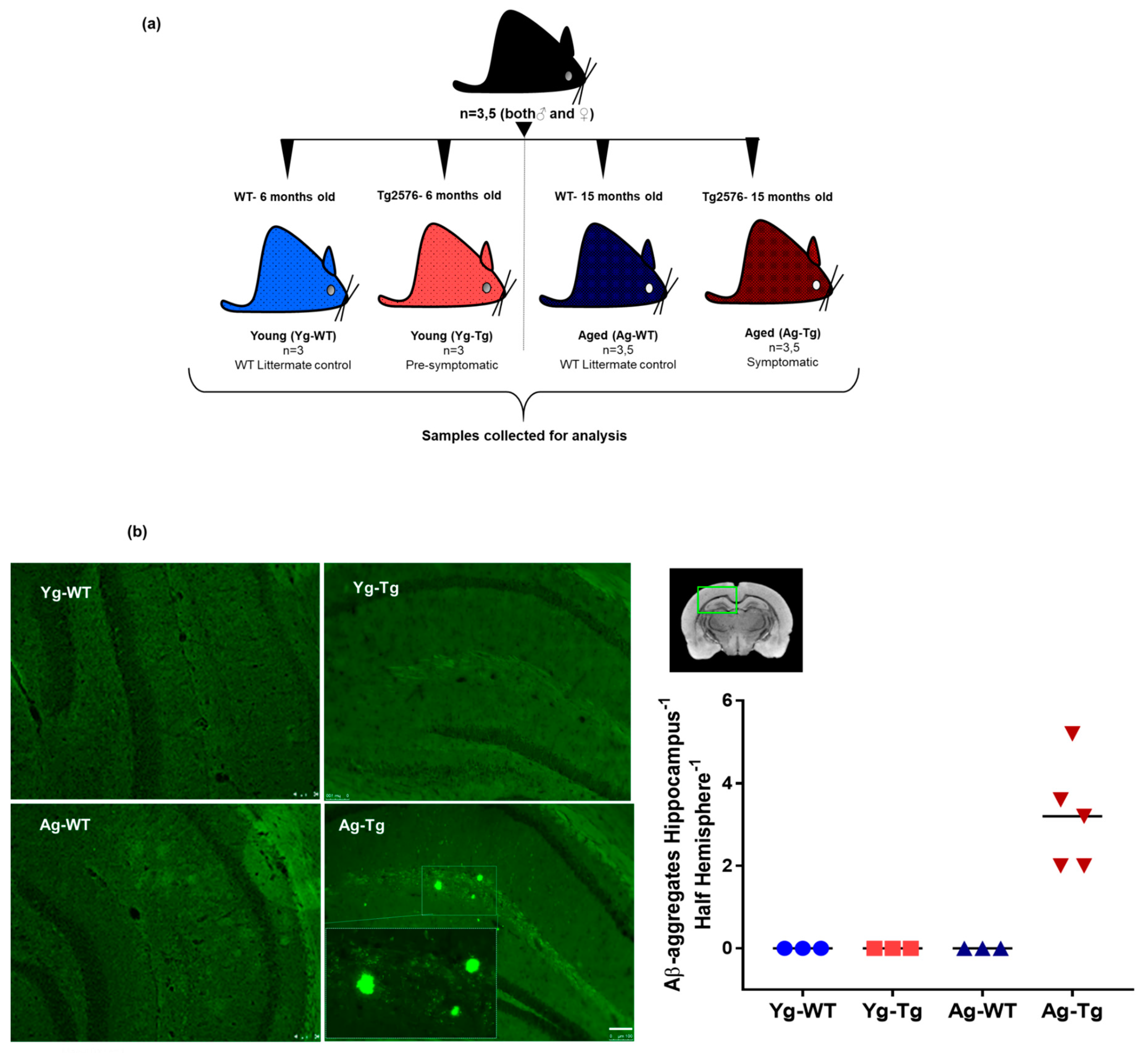
2.1. Cerebral Aβ Plaques are Undetectable in Pre-Symptomatic Mice but Present in Symptomatic Tg2576 Mice
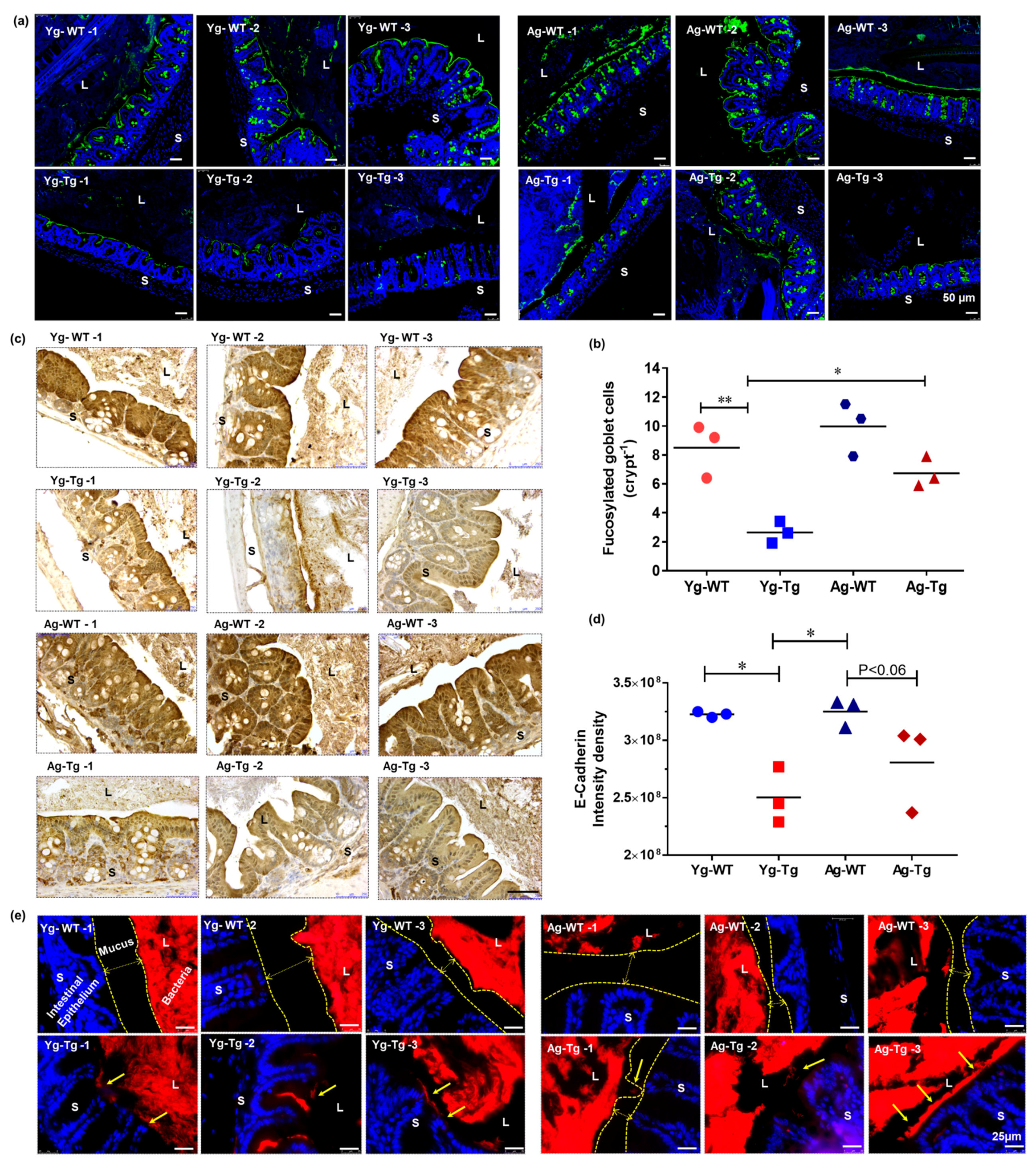
2.2. IEB Dysfunction Occurs before Development of Cerebral Aβ Pathology in Tg2576 Mice
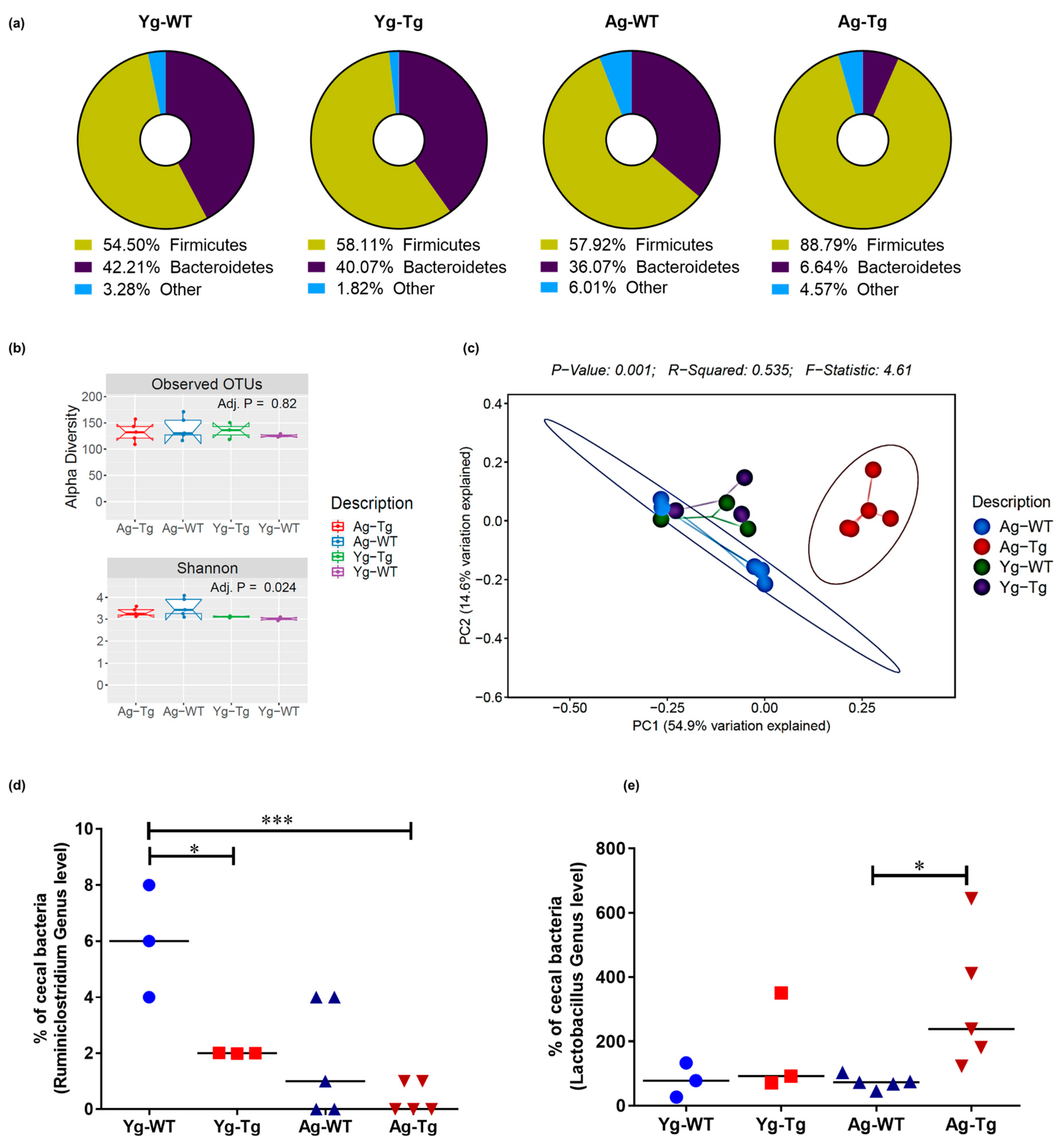
2.3. Gut Microbiota Composition Is Significantly Different in Symptomatic Tg2576 Mice
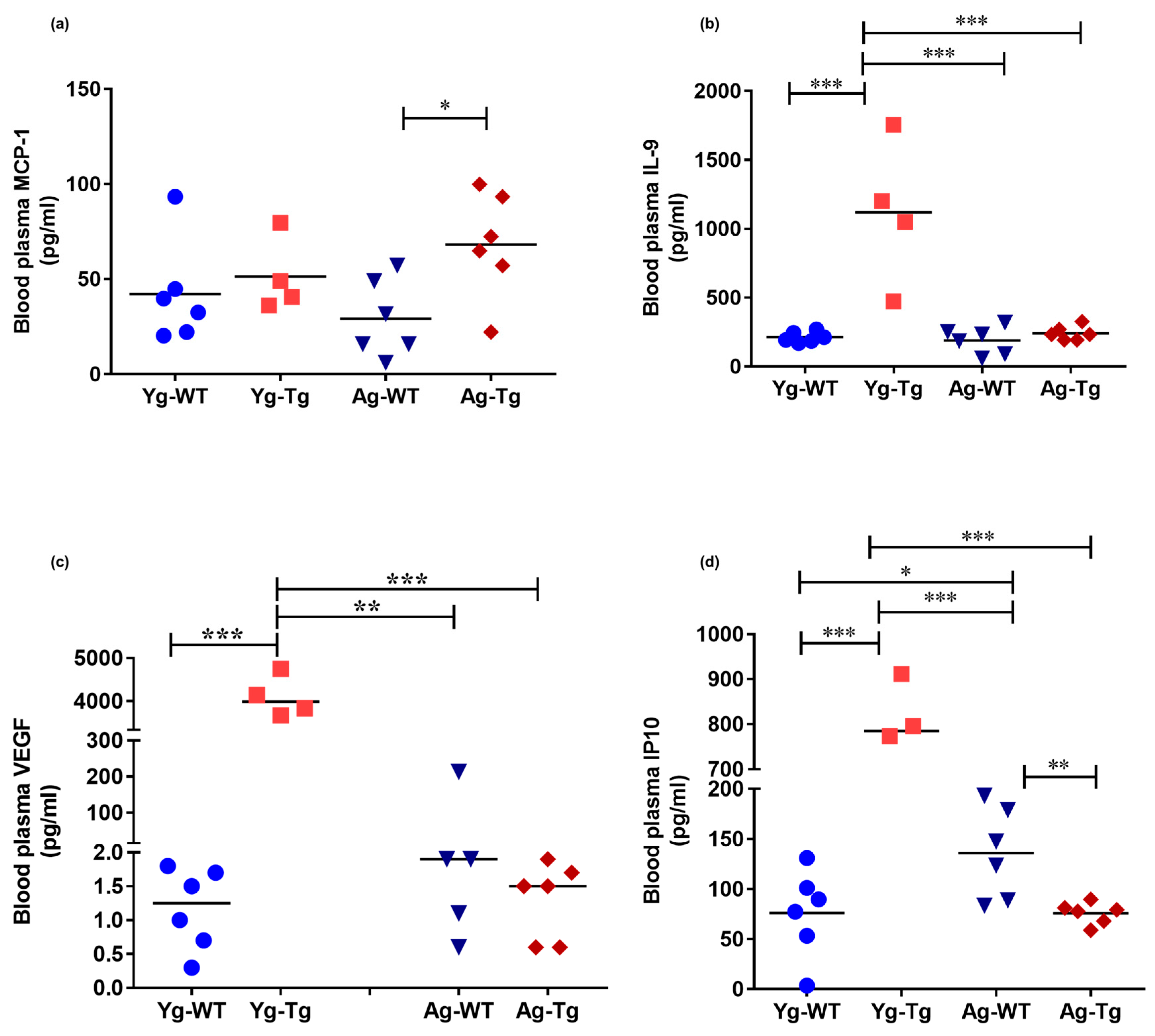
2.4. Plasma Levels of Inflammatory and Angiogenic Cytokines Are Elevated in Pre-Symptomatic Tg2576 Mice
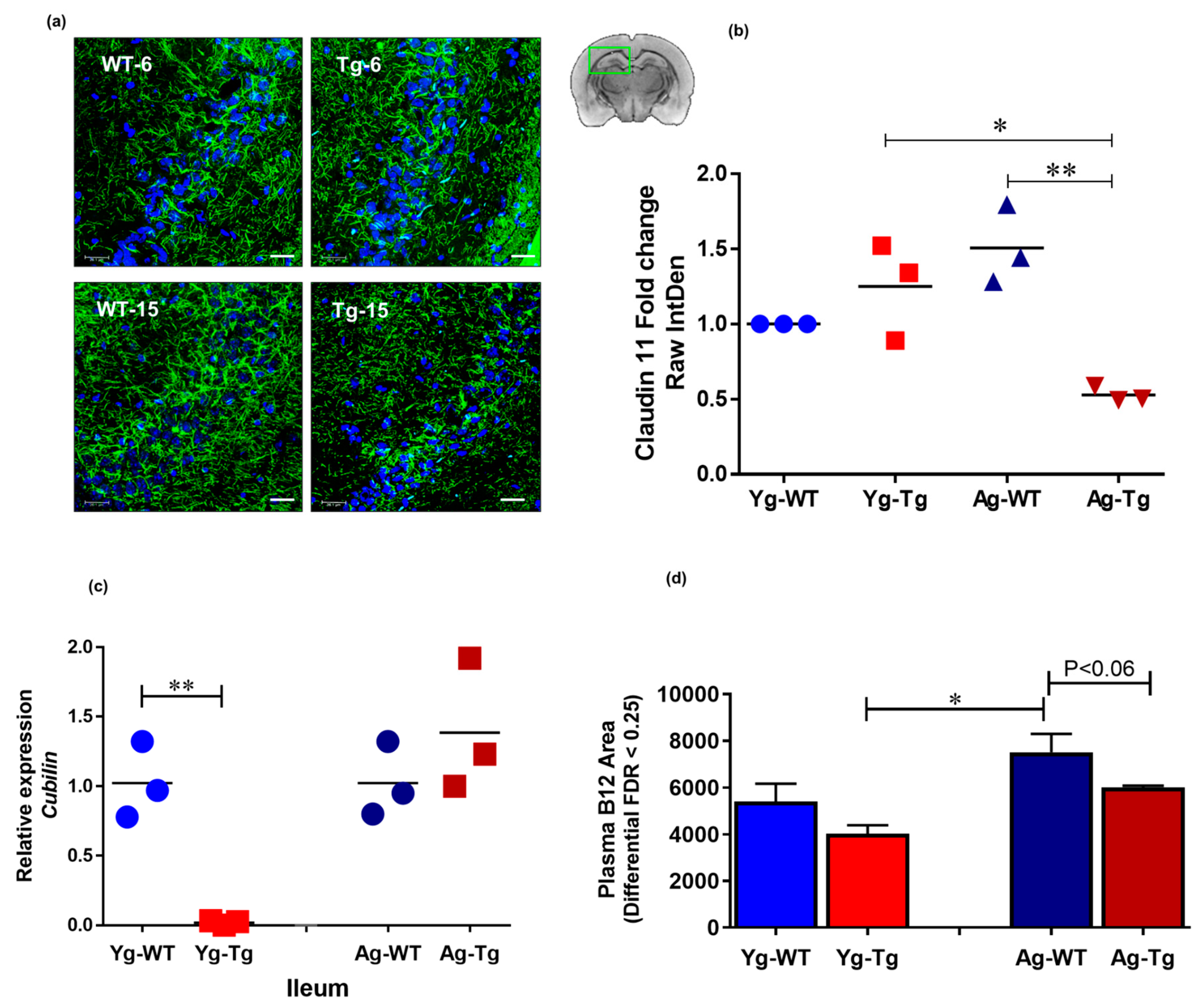
2.5. Reduced Systemic Levels of Vitamin B12 Associated with Reduced Myelin Tight Junction Proteins in the CNS Is Present in Symptomatic Tg2576 Mice
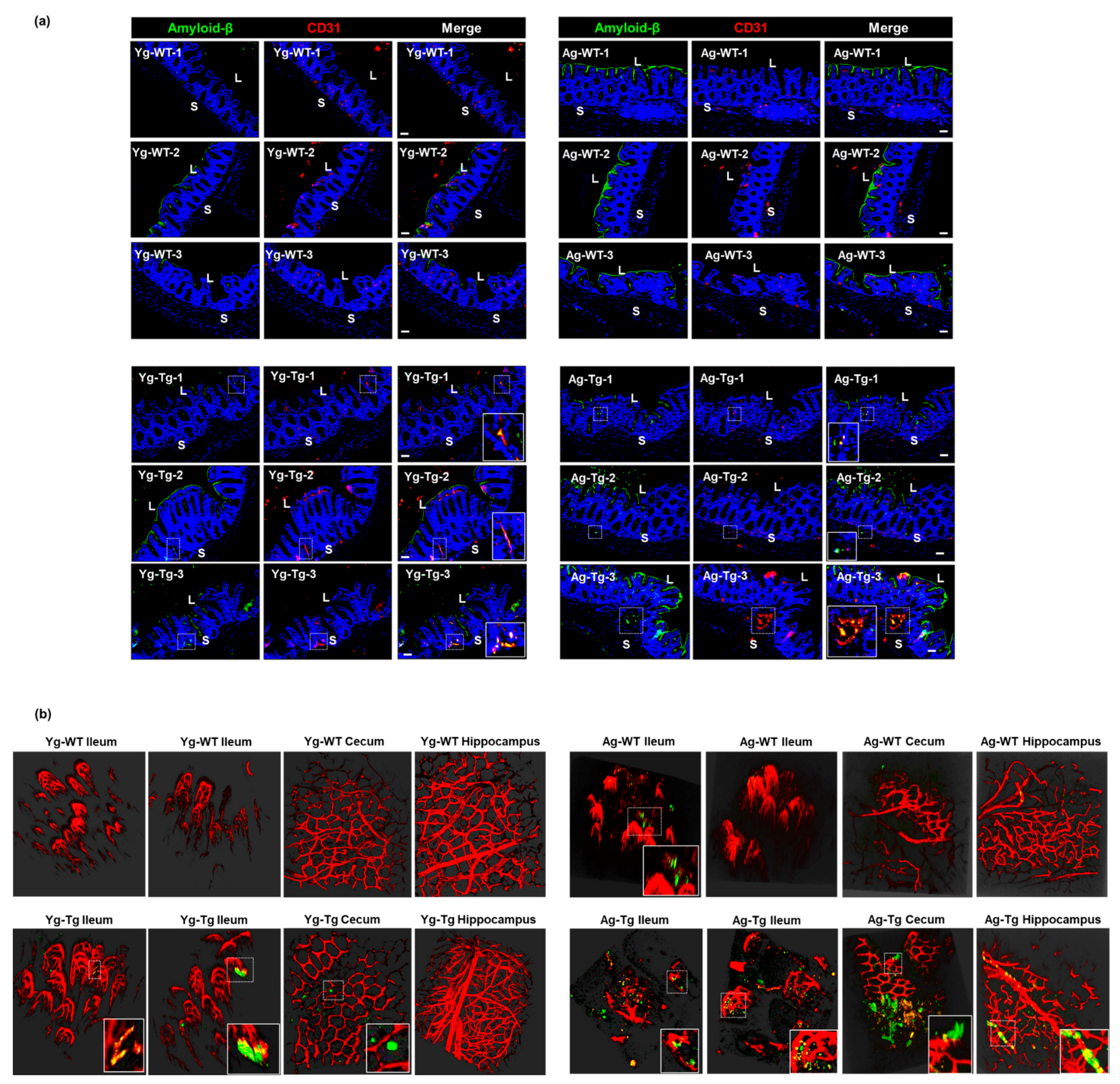
2.6. Aβ Co-Localizes with the Intestinal Vasculature in the Pre-Symptomatic Tg2576 Mice
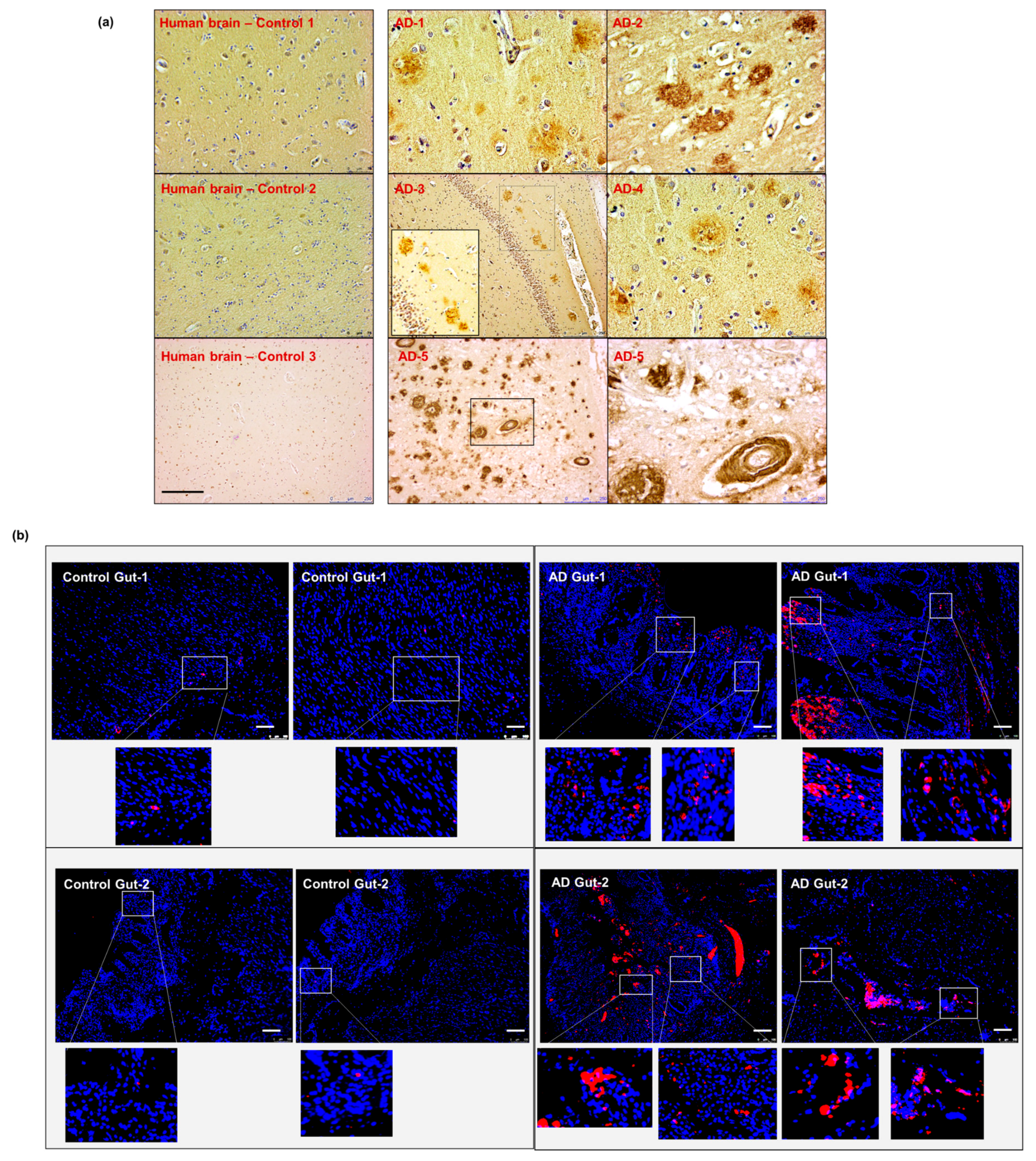
2.7. Intestinal Aβ Deposition Is Present in Human Samples with AD Pathology
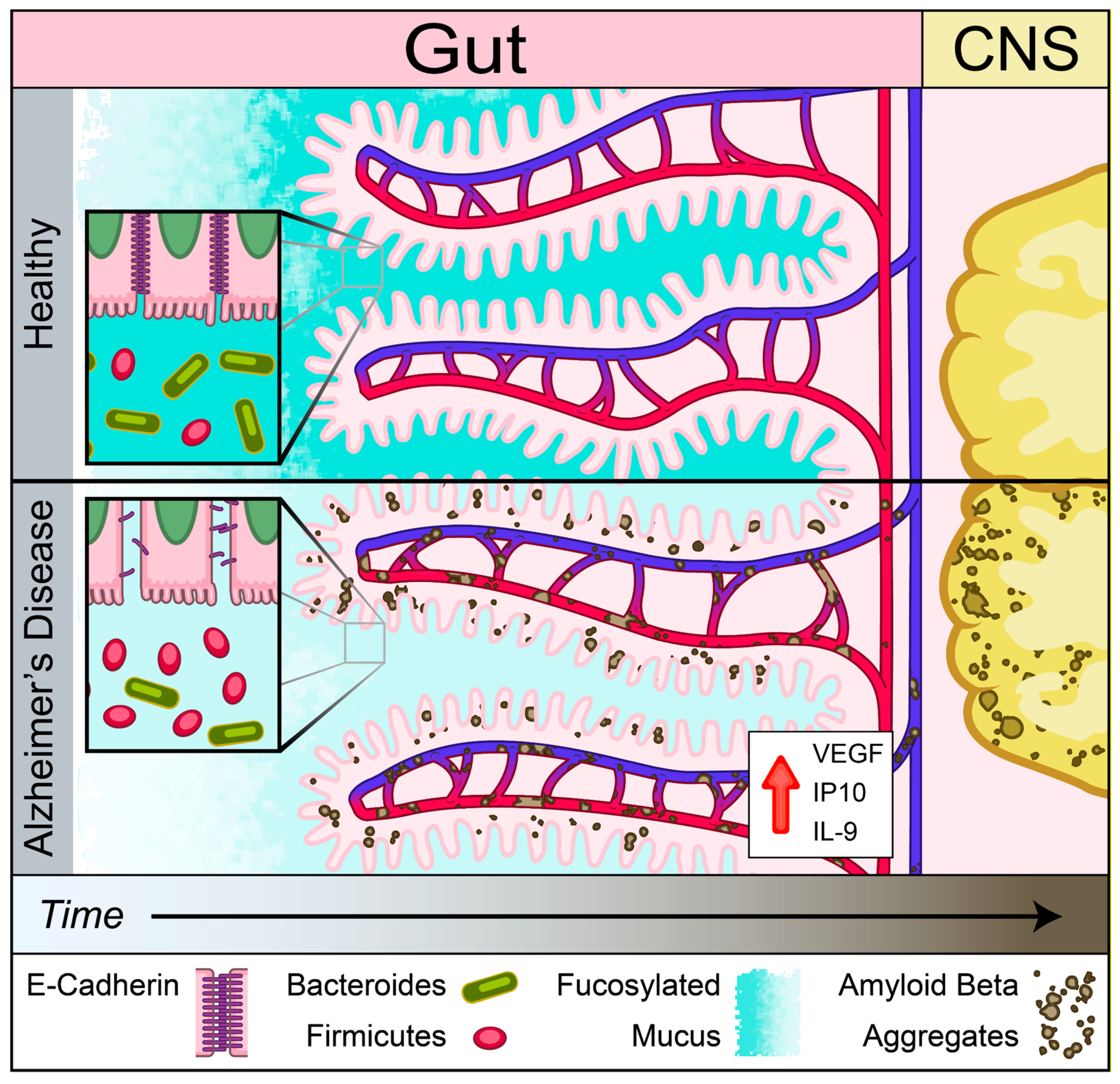
3. Discussion
4. Conclusions
5. Methods
5.1. Mice
5.2. Immunohistochemistry (IHC) and Microscopy
5.3. Fluorescent In Situ Hybridization (FISH)
5.4. Sample Collection and 16 S rRNA Sequencing
5.5. Cytokine Measurements
5.6. mRNA Gene Expression
5.7. Vitamin B12 Measurement by Mass Spectrometry
5.8. Two-Photon Microscopy to Visualize Amyloid-β in the Brain and Gut
5.9. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chung, C.G.; Lee, H.; Lee, S.B. Mechanisms of protein toxicity in neurodegenerative diseases. Cell. Mol. Life Sci. 2018, 75, 3159–3180. [Google Scholar] [CrossRef]
- Akiyama, H. Inflammatory response in Alzheimer’s disease. Tohoku J. Exp. Med. 1994, 174, 295–303. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Doty, K.R.; Guillot-Sestier, M.-V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.A.; Troyanova, N.I.; Kovalenko, E.I.; Sapozhnikov, A.M. Similarity and Differences in Inflammation-Related Characteristics of the Peripheral Immune System of Patients with Parkinson’s and Alzheimer’s Diseases. Int. J. Mol. Sci. 2017, 18. [Google Scholar]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. Gut instincts: Microbiota as a key regulator of brain development, ageing and neurodegeneration. J. Physiol. 2017, 595, 489–503. [Google Scholar] [CrossRef]
- Jang, S.-E.; Lim, S.-M.; Jeong, J.-J.; Jang, H.-M.; Lee, H.-J.; Han, M.J.; Kim, D.-H. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol. 2018, 11, 369–379. [Google Scholar] [CrossRef]
- Petrof, E.O.; Claud, E.C.; Gloor, G.B.; Allen-Vercoe, E. Microbial ecosystems therapeutics: A new paradigm in medicine? Benef. Microbes 2013, 4, 53–65. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; DI Sarno, R.; Giorgio, V.; Miele, L. Gut microbiota, obesity and metabolic disorders. Minerva Gastroenterol. Dietol. 2017, 63, 337–344. [Google Scholar] [PubMed]
- Ding, H.T.; Taur, Y.; Walkup, J.T. Gut Microbiota and Autism: Key Concepts and Findings. J. Autism Dev. Disord. 2017, 47, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Yang, N.J.; Chiu, I.M. Bacterial Signaling to the Nervous System through Toxins and Metabolites. J. Mol. Biol. 2017, 429, 587–605. [Google Scholar] [CrossRef]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Invest. 2015, 125, 926–938. [Google Scholar] [CrossRef]
- Pistollato, F.; Sumalla Cano, S.; Elio, I.; Masias Vergara, M.; Giampieri, F.; Battino, M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 2016, 74, 624–634. [Google Scholar] [CrossRef]
- Bell, J.S.; Spencer, J.I.; Yates, R.L.; Yee, S.A.; Jacobs, B.M.; DeLuca, G.C. From Nose to Gut - The Role of the Microbiome in Neurological Disease. Neuropathol. Appl. Neurobiol. 2018. [Google Scholar]
- Smith, P.A. The tantalizing links between gut microbes and the brain. Nature 2015, 526, 312–314. [Google Scholar] [CrossRef]
- Bercik, P.; Denou, E.; Collins, J.; Jackson, W.; Lu, J.; Jury, J.; Deng, Y.; Blennerhassett, P.; Macri, J.; McCoy, K.D.; et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 2011, 141, 599–609, 609.e1–3. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimers. Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Dodiya, H.B.; Kuntz, T.; Shaik, S.M.; Baufeld, C.; Leibowitz, J.; Zhang, X.X.; Gottel, N.; Zhang, X.X.; Butovsky, O.; Gilbert, J.A.; et al. Sex-specific effects of microbiome perturbations on cerebral Aβ amyloidosis and microglia phenotypes. J. Exp. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13, e1006654. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Bacteroides fragilis Lipopolysaccharide and Inflammatory Signaling in Alzheimer’s Disease. Front. Microbiol. 2016, 7, 1544. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Guan, N.L.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.-C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. North Am. 2017, 46, 77–89. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Bauer, B.; Abner, E.L.; Ashkenazy-Frolinger, T.; Hartz, A.M.S. A Comprehensive Behavioral Test Battery to Assess Learning and Memory in 129S6/Tg2576 Mice. PLoS ONE 2016, 11, e0147733. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.I.; Nielsen, R.; Birn, H. From bowel to kidneys: The role of cubilin in physiology and disease. Nephrol. Dial. Transplant 2013, 28, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.A.; Gama Sosa, M.A.; De Gasperi, R. Transgenic mouse models of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Kokjohn, T.A.; Roher, A.E. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: Understanding the paradigms, limitations, and contributions. Alzheimers. Dement. 2009, 5, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Crawford, F.; Mullan, M.; Kokjohn, T.A.; Emmerling, M.R.; Weller, R.O.; Roher, A.E. Elevated A beta and apolipoprotein E in A betaPP transgenic mice and its relationship to amyloid accumulation in Alzheimer’s disease. Mol. Med. 2000, 6, 430–439. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Larsson, J.M.H.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4659–4665. [Google Scholar] [CrossRef]
- Ganesh, B.P.; Klopfleisch, R.; Loh, G.; Blaut, M. Commensal Akkermansia muciniphila exacerbates gut inflammation in Salmonella Typhimurium-infected gnotobiotic mice. PLoS ONE 2013, 8, e74963. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Hansson, G.C. Mucus and the goblet cell. Dig. Dis. 2013, 31, 305–309. [Google Scholar] [CrossRef]
- Ganesh, B.P.; Hall, A.; Ayyaswamy, S.; Nelson, J.W.; Fultz, R.; Major, A.; Haag, A.; Esparza, M.; Lugo, M.; Venable, S.; et al. Diacylglycerol kinase synthesized by commensal Lactobacillus reuteri diminishes protein kinase C phosphorylation and histamine-mediated signaling in the mammalian intestinal epithelium. Mucosal Immunol. 2018, 11, 380–393. [Google Scholar] [CrossRef]
- Gómez-Escudero, J.; Moreno, V.; Martín-Alonso, M.; Hernández-Riquer, M.V.; Feinberg, T.; Colmenar, Á.; Calvo, E.; Camafeita, E.; Martínez, F.; Oudhoff, M.J.; et al. E-cadherin cleavage by MT2-MMP regulates apical junctional signaling and epithelial homeostasis in the intestine. J. Cell Sci. 2017, 130, 4013–4027. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef]
- Li, J. Molecular regulators of nerve conduction - Lessons from inherited neuropathies and rodent genetic models. Exp. Neurol. 2015, 267, 209–218. [Google Scholar] [CrossRef]
- Denninger, A.R.; Breglio, A.; Maheras, K.J.; LeDuc, G.; Cristiglio, V.; Demé, B.; Gow, A.; Kirschner, D.A. Claudin-11 Tight Junctions in Myelin Are a Barrier to Diffusion and Lack Strong Adhesive Properties. Biophys. J. 2015, 109, 1387–1397. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef]
- Knudsen, K.; Fedorova, T.D.; Hansen, A.K.; Sommerauer, M.; Otto, M.; Svendsen, K.B.; Nahimi, A.; Stokholm, M.G.; Pavese, N.; Beier, C.P.; et al. In-vivo staging of pathology in REM sleep behaviour disorder: A multimodality imaging case-control study. Lancet. Neurol. 2018, 17, 618–628. [Google Scholar] [CrossRef]
- Elmi, A.; Nasher, F.; Jagatia, H.; Gundogdu, O.; Bajaj-Elliott, M.; Wren, B.; Dorrell, N. Campylobacter jejuni outer membrane vesicle-associated proteolytic activity promotes bacterial invasion by mediating cleavage of intestinal epithelial cell E-cadherin and occludin. Cell. Microbiol. 2016, 18, 561–572. [Google Scholar] [CrossRef]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef]
- Torres-Fuentes, C.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. The microbiota-gut-brain axis in obesity. Lancet. Gastroenterol. Hepatol. 2017, 2, 747–756. [Google Scholar] [CrossRef]
- Khanna, S.; Pardi, D.S.; Kelly, C.R.; Kraft, C.S.; Dhere, T.; Henn, M.R.; Lombardo, M.-J.; Vulic, M.; Ohsumi, T.; Winkler, J.; et al. A Novel Microbiome Therapeutic Increases Gut Microbial Diversity and Prevents Recurrent Clostridium difficile Infection. J. Infect. Dis. 2016, 214, 173–181. [Google Scholar] [CrossRef]
- Bostanciklioğlu, M. The Role of Gut Microbiota in Pathogenesis of Alzheimer’s Disease. J. Appl. Microbiol. 2019. [Google Scholar] [CrossRef]
- Liu, P.; Wu, L.; Peng, G.; Han, Y.; Tang, R.; Ge, J.; Zhang, L.; Jia, L.; Yue, S.; Zhou, K.; et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain. Behav. Immun. 2019. [Google Scholar] [CrossRef]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef]
- Chen, J.; Huang, C.; Wang, J.; Zhou, H.; Lu, Y.; Lou, L.; Zheng, J.; Tian, L.; Wang, X.; Cao, Z.; et al. Dysbiosis of intestinal microbiota and decrease in paneth cell antimicrobial peptide level during acute necrotizing pancreatitis in rats. PLoS ONE 2017, 12, e0176583. [Google Scholar] [CrossRef]
- Kuang, Y.-S.; Lu, J.-H.; Li, S.-H.; Li, J.-H.; Yuan, M.-Y.; He, J.-R.; Chen, N.-N.; Xiao, W.-Q.; Shen, S.-Y.; Qiu, L.; et al. Connections between the human gut microbiome and gestational diabetes mellitus. Gigascience 2017, 6, 1–12. [Google Scholar] [CrossRef]
- Blander, J.M.; Longman, R.S.; Iliev, I.D.; Sonnenberg, G.F.; Artis, D. Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 2017, 18, 851–860. [Google Scholar] [CrossRef]
- Tanuma, N.; Sakuma, H.; Sasaki, A.; Matsumoto, Y. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006, 112, 195–204. [Google Scholar] [CrossRef]
- Britschgi, M.; Wyss-Coray, T. Immune cells may fend off Alzheimer disease. Nat. Med. 2007, 13, 408–409. [Google Scholar] [CrossRef]
- Vakilian, A.; Khorramdelazad, H.; Heidari, P.; Sheikh Rezaei, Z.; Hassanshahi, G. CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem. Int. 2017, 103, 1–7. [Google Scholar] [CrossRef]
- Steenwinckel, V.; Louahed, J.; Lemaire, M.M.; Sommereyns, C.; Warnier, G.; McKenzie, A.; Brombacher, F.; Van Snick, J.; Renauld, J.-C. IL-9 promotes IL-13-dependent paneth cell hyperplasia and up-regulation of innate immunity mediators in intestinal mucosa. J. Immunol. 2009, 182, 4737–4743. [Google Scholar] [CrossRef] [PubMed]
- Hufford, M.M.; Kaplan, M.H. A gut reaction to IL-9. Nat. Immunol. 2014, 15, 599–600. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.; Miners, J.S.; Allen, S.; Love, S. VEGFR1 and VEGFR2 in Alzheimer’s Disease. J. Alzheimers. Dis. 2018, 61, 741–752. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef]
- Galimberti, D.; Schoonenboom, N.; Scheltens, P.; Fenoglio, C.; Bouwman, F.; Venturelli, E.; Guidi, I.; Blankenstein, M.A.; Bresolin, N.; Scarpini, E. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch. Neurol. 2006, 63, 538–543. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef]
- Dean, D.C.; Hurley, S.A.; Kecskemeti, S.R.; O’Grady, J.P.; Canda, C.; Davenport-Sis, N.J.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Asthana, S.; et al. Association of Amyloid Pathology With Myelin Alteration in Preclinical Alzheimer Disease. JAMA Neurol. 2017, 74, 41–49. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Morita, K.; Sasaki, H.; Fujimoto, K.; Furuse, M.; Tsukita, S. Claudin-11/OSP-based tight junctions of myelin sheaths in brain and Sertoli cells in testis. J. Cell Biol. 1999, 145, 579–588. [Google Scholar] [CrossRef]
- Morrison, B.M.; Lee, Y.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of axons. Trends Cell Biol. 2013, 23, 644–651. [Google Scholar] [CrossRef]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Chow, E.; Mottahedeh, J.; Prins, M.; Ridder, W.; Nusinowitz, S.; Bronstein, J.M. Disrupted compaction of CNS myelin in an OSP/Claudin-11 and PLP/DM20 double knockout mouse. Mol. Cell. Neurosci. 2005, 29, 405–413. [Google Scholar] [CrossRef]
- Gow, A.; Southwood, C.M.; Li, J.S.; Pariali, M.; Riordan, G.P.; Brodie, S.E.; Danias, J.; Bronstein, J.M.; Kachar, B.; Lazzarini, R.A. CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell 1999, 99, 649–659. [Google Scholar] [CrossRef]
- Minter, M.R.; Hinterleitner, R.; Meisel, M.; Zhang, C.; Leone, V.; Zhang, X.; Oyler-Castrillo, P.; Zhang, X.; Musch, M.W.; Shen, X.; et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1ΔE9 murine model of Alzheimer’s disease. Sci. Rep. 2017, 7, 10411. [Google Scholar] [CrossRef]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef]
- Lagos, R.; Baeza, M.; Corsini, G.; Hetz, C.; Strahsburger, E.; Castillo, J.A.; Vergara, C.; Monasterio, O. Structure, organization and characterization of the gene cluster involved in the production of microcin E492, a channel-forming bacteriocin. Mol. Microbiol. 2001, 42, 229–243. [Google Scholar] [CrossRef]
- Romero, D.; Aguilar, C.; Losick, R.; Kolter, R. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc. Natl. Acad. Sci. USA 2010, 107, 2230–2234. [Google Scholar] [CrossRef]
- Fang, H.; Kang, J.; Zhang, D. Microbial production of vitamin B12: A review and future perspectives. Microb. Cell Fact. 2017, 16, 15. [Google Scholar] [CrossRef]
- Tian, D.; Shi, X.; Zhao, Y.; Peng, X.; Zou, L.; Xu, L.; Ma, Y.; Wen, Y.; Faulhaber-Walter, R.; Chen, L. The effect of A1 adenosine receptor in diabetic megalin loss with caspase-1/IL18 signaling. Diabetes. Metab. Syndr. Obes. 2019, 12, 1583–1596. [Google Scholar] [CrossRef]
- Miller, A.; Korem, M.; Almog, R.; Galboiz, Y. Vitamin B12, demyelination, remyelination and repair in multiple sclerosis. J. Neurol. Sci. 2005, 233, 93–97. [Google Scholar] [CrossRef]
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.-L.; Brito, A.; Guéant, J.-L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.-H.; et al. Vitamin B12 deficiency. Nat. Rev. Dis. Prim. 2017, 3, 17040. [Google Scholar] [CrossRef]
- Gröber, U.; Kisters, K.; Schmidt, J. Neuroenhancement with vitamin B12-underestimated neurological significance. Nutrients 2013, 5, 5031–5045. [Google Scholar] [CrossRef]
- Briani, C.; Dalla Torre, C.; Citton, V.; Manara, R.; Pompanin, S.; Binotto, G.; Adami, F. Cobalamin deficiency: Clinical picture and radiological findings. Nutrients 2013, 5, 4521–4539. [Google Scholar] [CrossRef]
- Scott, M.R.; Köhler, R.; Foster, D.; Prusiner, S.B. Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci. 1992, 1, 986–997. [Google Scholar] [CrossRef]
- Nelson, M.C.; Morrison, H.G.; Benjamino, J.; Grim, S.L.; Graf, J. Analysis, optimization and verification of Illumina-generated 16S rRNA gene amplicon surveys. PLoS ONE 2014, 9, e94249. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Putluri, N.; Shojaie, A.; Vasu, V.T.; Vareed, S.K.; Nalluri, S.; Putluri, V.; Thangjam, G.S.; Panzitt, K.; Tallman, C.T.; Butler, C.; et al. Metabolomic profiling reveals potential markers and bioprocesses altered in bladder cancer progression. Cancer Res. 2011, 71, 7376–7386. [Google Scholar] [CrossRef]
- Vantaku, V.; Donepudi, S.R.; Piyarathna, D.W.B.; Amara, C.S.; Ambati, C.R.; Tang, W.; Putluri, V.; Chandrashekar, D.S.; Varambally, S.; Terris, M.K.; et al. Large-scale profiling of serum metabolites in African American and European American patients with bladder cancer reveals metabolic pathways associated with patient survival. Cancer 2019, 125, 921–932. [Google Scholar] [CrossRef]
- Vantaku, V.; Dong, J.; Ambati, C.R.; Perera, D.; Donepudi, S.R.; Amara, C.S.; Putluri, V.; Ravi, S.S.; Robertson, M.J.; Piyarathna, D.W.B.; et al. Multi-omics Integration Analysis Robustly Predicts High-Grade Patient Survival and Identifies CPT1B Effect on Fatty Acid Metabolism in Bladder Cancer. Clin. Cancer Res. 2019, 25, 3689–3701. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honarpisheh, P.; Reynolds, C.R.; Blasco Conesa, M.P.; Moruno Manchon, J.F.; Putluri, N.; Bhattacharjee, M.B.; Urayama, A.; McCullough, L.D.; Ganesh, B.P. Dysregulated Gut Homeostasis Observed Prior to the Accumulation of the Brain Amyloid-β in Tg2576 Mice. Int. J. Mol. Sci. 2020, 21, 1711. https://doi.org/10.3390/ijms21051711
Honarpisheh P, Reynolds CR, Blasco Conesa MP, Moruno Manchon JF, Putluri N, Bhattacharjee MB, Urayama A, McCullough LD, Ganesh BP. Dysregulated Gut Homeostasis Observed Prior to the Accumulation of the Brain Amyloid-β in Tg2576 Mice. International Journal of Molecular Sciences. 2020; 21(5):1711. https://doi.org/10.3390/ijms21051711
Chicago/Turabian StyleHonarpisheh, Pedram, Caroline R. Reynolds, Maria P. Blasco Conesa, Jose F. Moruno Manchon, Nagireddy Putluri, Meenakshi B. Bhattacharjee, Akihiko Urayama, Louise D. McCullough, and Bhanu P. Ganesh. 2020. "Dysregulated Gut Homeostasis Observed Prior to the Accumulation of the Brain Amyloid-β in Tg2576 Mice" International Journal of Molecular Sciences 21, no. 5: 1711. https://doi.org/10.3390/ijms21051711
APA StyleHonarpisheh, P., Reynolds, C. R., Blasco Conesa, M. P., Moruno Manchon, J. F., Putluri, N., Bhattacharjee, M. B., Urayama, A., McCullough, L. D., & Ganesh, B. P. (2020). Dysregulated Gut Homeostasis Observed Prior to the Accumulation of the Brain Amyloid-β in Tg2576 Mice. International Journal of Molecular Sciences, 21(5), 1711. https://doi.org/10.3390/ijms21051711