Isoform-Specific Lysine Methylation of RORα2 by SETD7 Is Required for Association of the TIP60 Coactivator Complex in Prostate Cancer Progression


Abstract
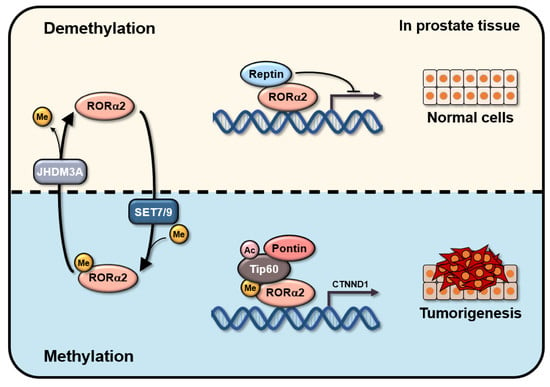
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
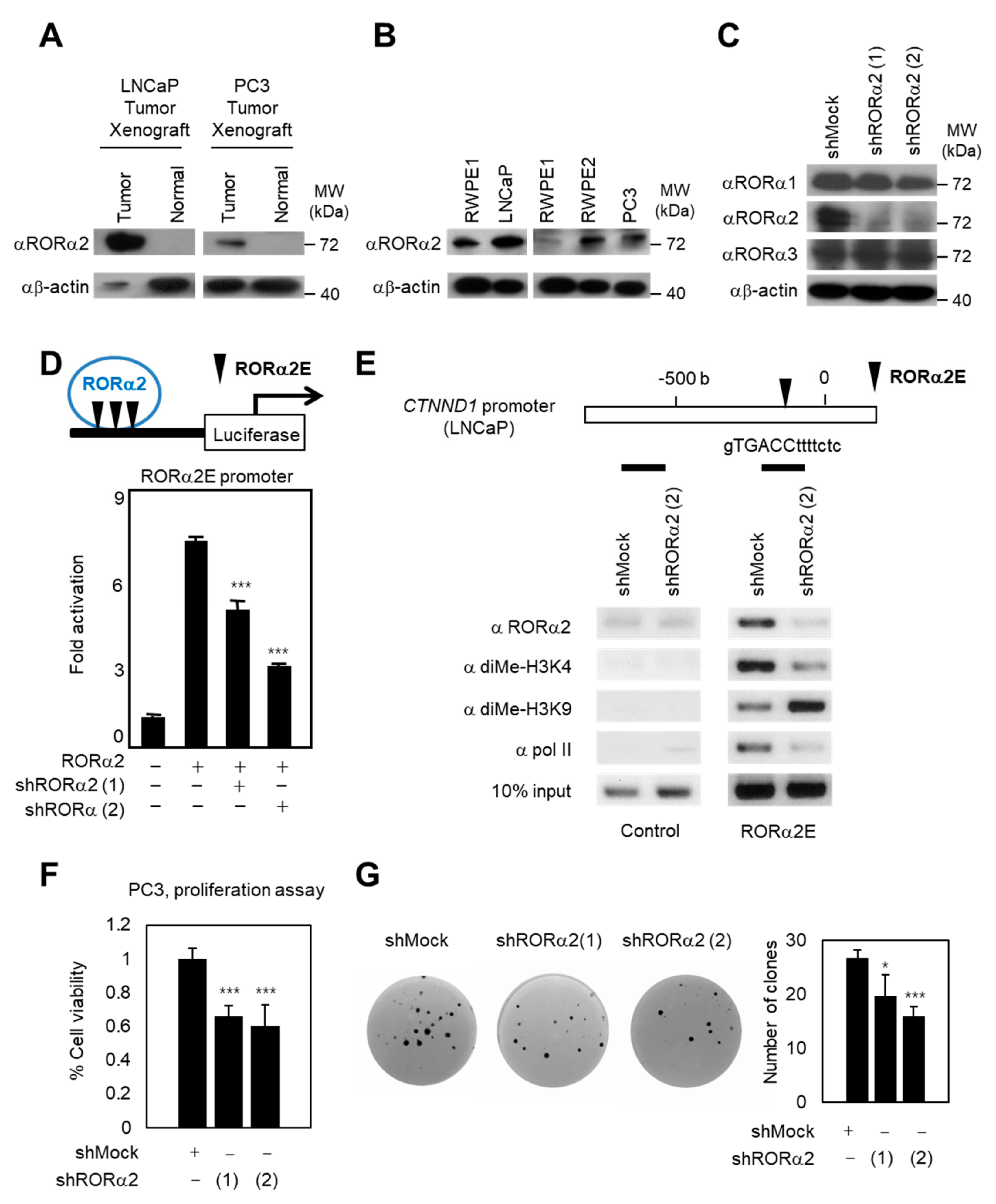
2.1. RORα2 Functions as a Selective Oncogene in PCa
2.2. RORα2 Selectively Binds to Pontin/Tip60 Coactivator Complex and Reptin Corepressor
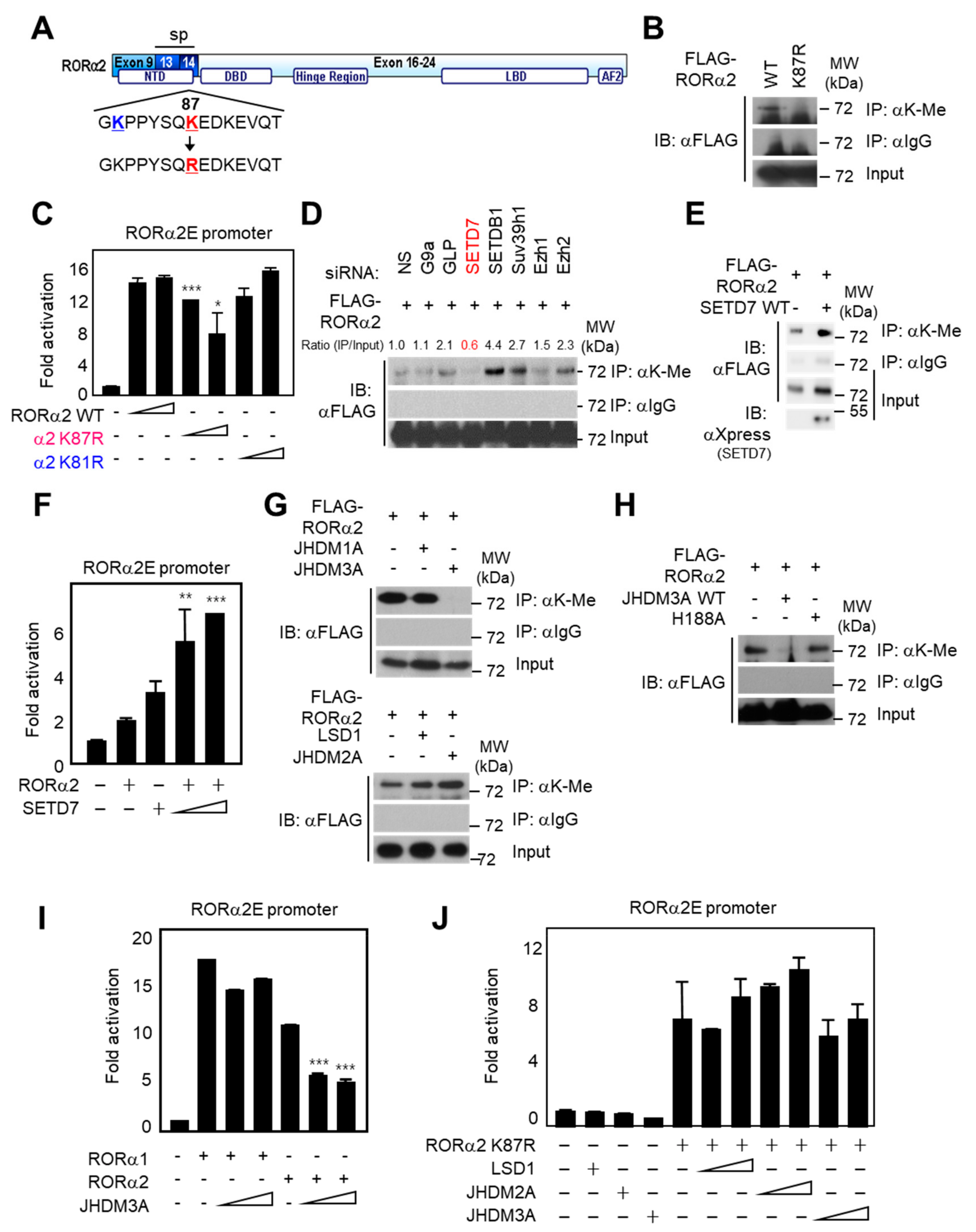
2.3. Lysine Methylation of RORα2 by SETD7 Is Crucial for Downstream Target Gene Activation
2.4. Methylation of RORα2 by SETD7 Alters the Binding Affinity of a Coactivator Complex and Increases Tumorigenesis in PCa
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. GST Pulldown Assays
4.3. Luciferase Reporter Assays
4.4. Purification and Identification of Binding Proteins for RORα2
4.5. LC–MS/MS and SEQUEST Analyses
4.6. Chromatin Immunoprecipitation (ChIP)
4.7. Cell Proliferation Assay
4.8. RNA Interference by shRNA of RORα2
4.9. Clonogenic and Tumorigenicity Assay
4.10. Real-Time Q-PCR
4.11. Plasmid Construction
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RORα | Retinoid acid-related orphan receptor α |
| ONR | Orphan nuclear receptor |
| NTD | N-terminal domain |
| PCa | Prostate cancer |
| DBD | DNA-binding domain |
| LBD | Ligand-binding domain |
References
- Blumberg, B.; Evans, R.M. Orphan nuclear receptors - new ligands and new possibilities. Genes Dev. 1998, 12, 3149–3155. [Google Scholar] [CrossRef]
- Steinmayr, M.; André, E.; Conquet, F.; Rondi-Reig, L.; Delhaye-Bouchaud, N.; Auclair, N.; Daniel, H.; Crepel, F.; Mariani, J.; Sotelo, C.; et al. Staggerer phenotype in retinoid-related orphan receptor α-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3960–3965. [Google Scholar] [CrossRef] [PubMed]
- Atkins, G.B.; Hu, X.; Guenther, M.G.; Rachez, C.; Freedman, L.P.; Lazar, M.A. Coactivators for the orphan nuclear receptor RORα. Mol. Endocrinol. 1999, 13, 1550–1557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jetten, A.M.; Kurebayashi, S.; Ueda, E. The ROR nuclear orphan receptor subfamily: Critical regulators of multiple biological processes. Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 205–247. [Google Scholar] [PubMed]
- Delerive, P.; Monté, D.; Dubois, G.; Trottein, F.; Fruchart-Najib, J.; Mariani, J.; Fruchart, J.C.; Staels, B. The orphan nuclear receptor ROR alpha is a negative regulator of the inflammatory response. EMBO Rep. 2001, 2, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Moraitis, A.N.; Giguère, V. The Co-repressor Hairless Protects RORalpha Orphan Nuclear Receptor from Proteasome-mediated Degradation. J. Biol. Chem. 2003, 278, 52511–52518. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.; Nixon, S.J.; Parton, R.G.; Muscat, G.E. RORalpha regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: Caveolin-3 and CPT-1 are direct targets of ROR. J. Biol. Chem. 2004, 279, 36828–36840. [Google Scholar] [CrossRef]
- Migita, H.; Satozawa, N.; Lin, J.H.; Morser, J.; Kawai, K. RORalpha1 and RORalpha4 suppress TNF-alpha-induced VCAM-1 and ICAM-1 expression in human endothelial cells. FEBS Lett. 2004, 557, 269–274. [Google Scholar] [CrossRef][Green Version]
- Moretti, R.M.; Montagnani, M.M.; Sala, A.; Motta, M.; Limonta, P. Activation of the orphan nuclear receptor RORalpha counteracts the proliferative effect of fatty acids on prostate cancer cells: Crucial role of 5-lipoxygenase. Int. J. Cancer 2004, 112, 87–93. [Google Scholar] [CrossRef]
- Kim, K.; Boo, K.; Yu, Y.S.; Oh, S.K.; Kim, H.; Jeon, Y.; Bhin, J.; Hwang, D.; Kim, K.I.; Lee, J.S.; et al. RORalpha controls hepatic lipid homeostasis via negative regulation of PPARgamma transcriptional network. Nat. Commun. 2017, 8, 162. [Google Scholar] [CrossRef]
- Oh, S.K.; Kim, D.; Kim, K.; Boo, K.; Yu, Y.S.; Kim, I.S.; Jeon, Y.; Im, S.K.; Lee, S.H.; Lee, J.M.; et al. RORalpha is crucial for attenuated inflammatory response to maintain intestinal homeostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 21140–21149. [Google Scholar] [CrossRef]
- Kim, K.; Lee, J.M.; Yu, Y.S.; Kim, H.; Nam, H.J.; Moon, H.-G.; Noh, D.-Y.; Kim, K.I.; Fang, S.; Baek, S.H. RORα2 requires LSD1 to enhance tumor progression in breast cancer. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, J.S.; Kim, H.; Kim, K.; Park, H.; Kim, J.Y.; Lee, S.H.; Kim, I.S.; Kim, J.; Lee, M.; et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 2012, 48, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, J.M.; Lee, G.; Bhin, J.; Oh, S.K.; Kim, K.; Pyo, K.E.; Lee, J.S.; Yim, H.Y.; Kim, K.I.; et al. DNA Damage-Induced RORα Is Crucial for p53 Stabilization and Increased Apoptosis. Mol. Cell 2011, 44, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Kim, I.S.; Kim, H.; Lee, J.S.; Kim, K.; Yim, H.Y.; Jeong, J.; Kim, J.H.; Kim, J.Y.; Lee, H.; et al. RORalpha attenuates Wnt/beta-catenin signaling by PKCalpha-dependent phosphorylation in colon cancer. Mol. Cell 2010, 37, 183–195. [Google Scholar] [CrossRef]
- Huang, J.; Dorsey, J.; Chuikov, S.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S.L. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef]
- Gaughan, L.; Stockley, J.; Wang, N.; McCracken, S.R.C.; Treumann, A.; Armstrong, K.; Shaheen, F.; Watt, K.; McEwan, I.J.; Wang, C.; et al. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011, 39, 1266–1279. [Google Scholar] [CrossRef]
- Shah, N.; Brown, M. The Sly Oncogene: FOXA1 Mutations in Prostate Cancer. Cancer Cell 2019, 36, 119–121. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Aparicio, A.M.; Shen, L.; Tapia, E.L.; Lu, J.F.; Chen, H.C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef]
- Burkhardt, L.; Fuchs, S.; Krohn, A.; Masser, S.; Mader, M.; Kluth, M.; Bachmann, F.; Huland, H.; Steuber, T.; Graefen, M.; et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res. 2013, 73, 2795–2805. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Fujimoto, N.; Kashiwagi, E.; Eto, M. The Role of Nuclear Receptors in Prostate Cancer. Cells 2019, 8, 602. [Google Scholar] [CrossRef] [PubMed]
- Cariello, M.; Ducheix, S.; Maqdasy, S.; Baron, S.; Moschetta, A.; Lobaccaro, J.A. LXRs, SHP, and FXR in Prostate Cancer: Enemies or Menage a Quatre With AR? Nucl Recept Signal. 2018, 15, 1550762918801070. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.B.; de Matos Oliveira, F.; Neves, A.F.; Araujo, G.R.; Marangoni, K.; Goulart, L.R.; Araujo, T.G. Association of vitamin D receptor variants with clinical parameters in prostate cancer. Springerplus 2016, 5, 364. [Google Scholar] [CrossRef]
- Kroon, J.; Puhr, M.; Buijs, J.T.; van der Horst, G.; Hemmer, D.M.; Marijt, K.A.; Hwang, M.S.; Masood, M.; Grimm, S.; Storm, G.; et al. Glucocorticoid receptor antagonism reverts docetaxel resistance in human prostate cancer. Endocr. Relat. Cancer 2016, 23, 35–45. [Google Scholar] [CrossRef]
- Park, S.C.; Park, I.G.; Kim, H.; Lee, J.M. N-Terminal Domain Mediated Regulation of RORalpha1 Inhibits Invasive Growth in Prostate Cancer. Int J. Mol. Sci. 2019, 20, 1684. [Google Scholar]
- Levy, D. Lysine methylation signaling of non-histone proteins in the nucleus. Cell. Mol. Life Sci. CMLS 2019, 76, 2873–2883. [Google Scholar]
- Rathert, P.; Dhayalan, A.; Murakami, M.; Zhang, X.; Tamas, R.; Jurkowska, R.; Komatsu, Y.; Shinkai, Y.; Cheng, X.; Jeltsch, A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat. Chem. Biol. 2008, 4, 344–346. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.M.; Nam, H.J.; Choi, H.J.; Yang, J.W.; Lee, J.S.; Kim, M.H.; Kim, S.I.; Chung, C.H.; Kim, K.I.; et al. SUMOylation of pontin chromatin-remodeling complex reveals a signal integration code in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 20793–20798. [Google Scholar] [CrossRef]
- Crea, F.; Sun, L.; Mai, A.; Chiang, Y.T.; Farrar, W.L.; Danesi, R.; Helgason, C.D. The emerging role of histone lysine demethylases in prostate cancer. Mol. Cancer 2012, 11, 52. [Google Scholar] [CrossRef]
- Carlson, S.M.; Gozani, O. Nonhistone Lysine Methylation in the Regulation of Cancer Pathways. Cold Spring Harbor Perspectives In Medicine 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Biggar, K.K.; Li, S.S. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 2015, 15, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Schrecengost, R.; Knudsen, K.E. Molecular pathogenesis and progression of prostate cancer. Semin. Oncol. 2013, 40, 244–258. [Google Scholar] [CrossRef]
- Liu, Q.; Geng, H.; Xue, C.; Beer, T.M.; Qian, D.Z. Functional regulation of hypoxia inducible factor-1alpha by SET9 lysine methyltransferase. Biochim. Biophys. Acta 2015, 1853, 881–891. [Google Scholar] [CrossRef]
- Ko, S.; Ahn, J.; Song, C.S.; Kim, S.; Knapczyk-Stwora, K.; Chatterjee, B. Lysine methylation and functional modulation of androgen receptor by Set9 methyltransferase. Mol. Endocrinol 2011, 25, 433–444. [Google Scholar] [CrossRef]
- Chu, C.H.; Wang, L.Y.; Hsu, K.C.; Chen, C.C.; Cheng, H.H.; Wang, S.M.; Wu, C.M.; Chen, T.J.; Li, L.T.; Liu, R.; et al. KDM4B as a target for prostate cancer: Structural analysis and selective inhibition by a novel inhibitor. J. Med. Chem 2014, 57, 5975–5985. [Google Scholar] [CrossRef]
- Guerra-Calderas, L.; Gonzalez-Barrios, R.; Herrera, L.A.; Cantu de Leon, D.; Soto-Reyes, E. The role of the histone demethylase KDM4A in cancer. Cancer Genet. 2015, 208, 215–224. [Google Scholar] [CrossRef]
- Shiota, M.; Yokomizo, A.; Masubuchi, D.; Tada, Y.; Inokuchi, J.; Eto, M.; Uchiumi, T.; Fujimoto, N.; Naito, S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 2010, 70, 540–554. [Google Scholar] [CrossRef]
- Matias, P.M.; Baek, S.H.; Bandeiras, T.M.; Dutta, A.; Houry, W.A.; Llorca, O.; Rosenbaum, J. The AAA+ proteins Pontin and Reptin enter adult age: From understanding their basic biology to the identification of selective inhibitors. Front. Mol. Biosci. 2015, 2, 17. [Google Scholar] [CrossRef]
- Huber, O.; Menard, L.; Haurie, V.; Nicou, A.; Taras, D.; Rosenbaum, J. Pontin and reptin, two related ATPases with multiple roles in cancer. Cancer Res. 2008, 68, 6873–6876. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.M.; Montagnani Marelli, M.; Motta, M.; Limonta, P. Role of the Orphan Nuclear Receptor Ror Alpha in the Control of the Metastatic Behavior of Androgen-Independent Prostate Cancer Cells. Oncol. Rep. 2002, 9, 1139–1143. [Google Scholar] [PubMed]
- Varier, R.A.; Timmers, H.T. Histone lysine methylation and demethylation pathways in cancer. Biochim. Biophys Acta 2011, 1815, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Dou, Z.; Sammons, M.A.; Levine, A.J.; Berger, S.L. Lysine methylation represses p53 activity in teratocarcinoma cancer cells. Proc. Natl. Acad. Sci. USA 2016, 113, 9822–9827. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Jung, M. New lysine methyltransferase drug targets in cancer. Nat. Biotechnol. 2012, 30, 622–623. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Trojer, P. Targeting histone lysine methylation in cancer. Pharmacol. Therap. 2015, 150, 1–22. [Google Scholar] [CrossRef]
- Li, N.; Dhar, S.S.; Chen, T.Y.; Kan, P.Y.; Wei, Y.; Kim, J.H.; Chan, C.H.; Lin, H.K.; Hung, M.C.; Lee, M.G. JARID1D Is a Suppressor and Prognostic Marker of Prostate Cancer Invasion and Metastasis. Cancer Res. 2016, 76, 831–843. [Google Scholar] [CrossRef]
- Roth, G.S.; Casanova, A.G.; Lemonnier, N.; Reynoird, N. Lysine methylation signaling in pancreatic cancer. Curr. Opin. Oncol. 2018, 30, 30–37. [Google Scholar] [CrossRef]
- Metzger, E.; Wang, S.; Urban, S.; Willmann, D.; Schmidt, A.; Offermann, A.; Allen, A.; Sum, M.; Obier, N.; Cottard, F.; et al. KMT9 monomethylates histone H4 lysine 12 and controls proliferation of prostate cancer cells. Nat. Struct. Mol. Biol. 2019, 26, 361–371. [Google Scholar] [CrossRef]
- Baek, S.H.; Ohgi, K.A.; Rose, D.W.; Koo, E.H.; Glass, C.K.; Rosenfeld, M.G. Exchange of N-CoR Corepressor and Tip60 coactivator Complexes Links Gene Expression by NF-κB and beta-amyloid precursor protein. Cell 2002, 110, 55–67. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, H.J.; Kim, B.; Kim, M.H.; Lee, J.M.; Kim, I.S.; Lee, M.H.; Choi, S.J.; Kim, K.I.; Kim, S.I.; et al. Roles of SUMOylation of a reptin chromatin remodeling complex in cancer metastasis. Nat. Cell. Biol. 2006, 8, 631–639. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, H.; Chu, J.W.; Park, S.C.; Im, H.; Park, I.-G.; Kim, H.; Lee, J.M. Isoform-Specific Lysine Methylation of RORα2 by SETD7 Is Required for Association of the TIP60 Coactivator Complex in Prostate Cancer Progression. Int. J. Mol. Sci. 2020, 21, 1622. https://doi.org/10.3390/ijms21051622
Song H, Chu JW, Park SC, Im H, Park I-G, Kim H, Lee JM. Isoform-Specific Lysine Methylation of RORα2 by SETD7 Is Required for Association of the TIP60 Coactivator Complex in Prostate Cancer Progression. International Journal of Molecular Sciences. 2020; 21(5):1622. https://doi.org/10.3390/ijms21051622
Chicago/Turabian StyleSong, Hyerin, Jung Woong Chu, Su Chan Park, Hyuntae Im, Il-Geun Park, Hyunkyung Kim, and Ji Min Lee. 2020. "Isoform-Specific Lysine Methylation of RORα2 by SETD7 Is Required for Association of the TIP60 Coactivator Complex in Prostate Cancer Progression" International Journal of Molecular Sciences 21, no. 5: 1622. https://doi.org/10.3390/ijms21051622
APA StyleSong, H., Chu, J. W., Park, S. C., Im, H., Park, I.-G., Kim, H., & Lee, J. M. (2020). Isoform-Specific Lysine Methylation of RORα2 by SETD7 Is Required for Association of the TIP60 Coactivator Complex in Prostate Cancer Progression. International Journal of Molecular Sciences, 21(5), 1622. https://doi.org/10.3390/ijms21051622