Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
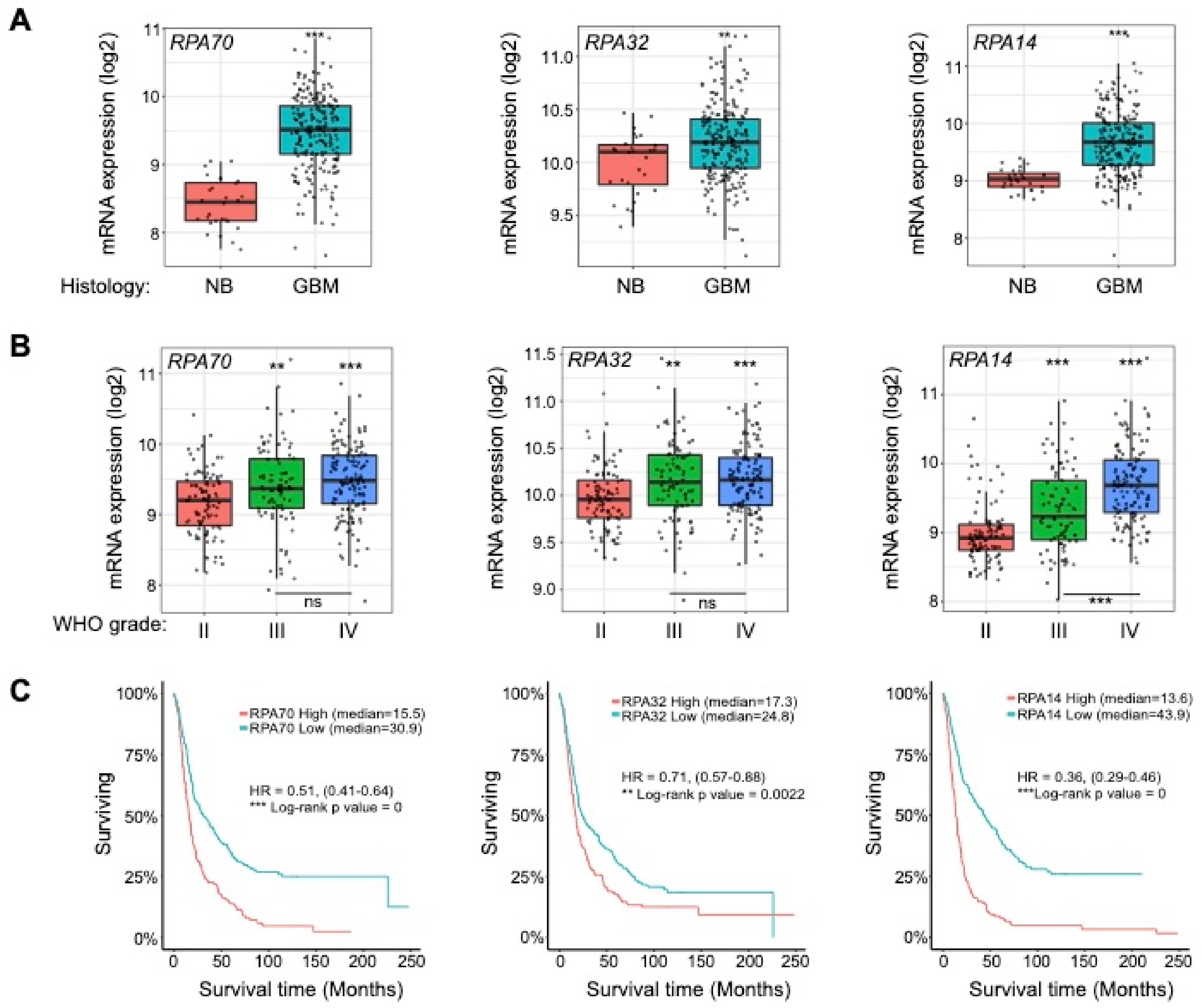
2.1. RPA is Overexpressed in High-Grade Gliomas and Informs Patient Survival
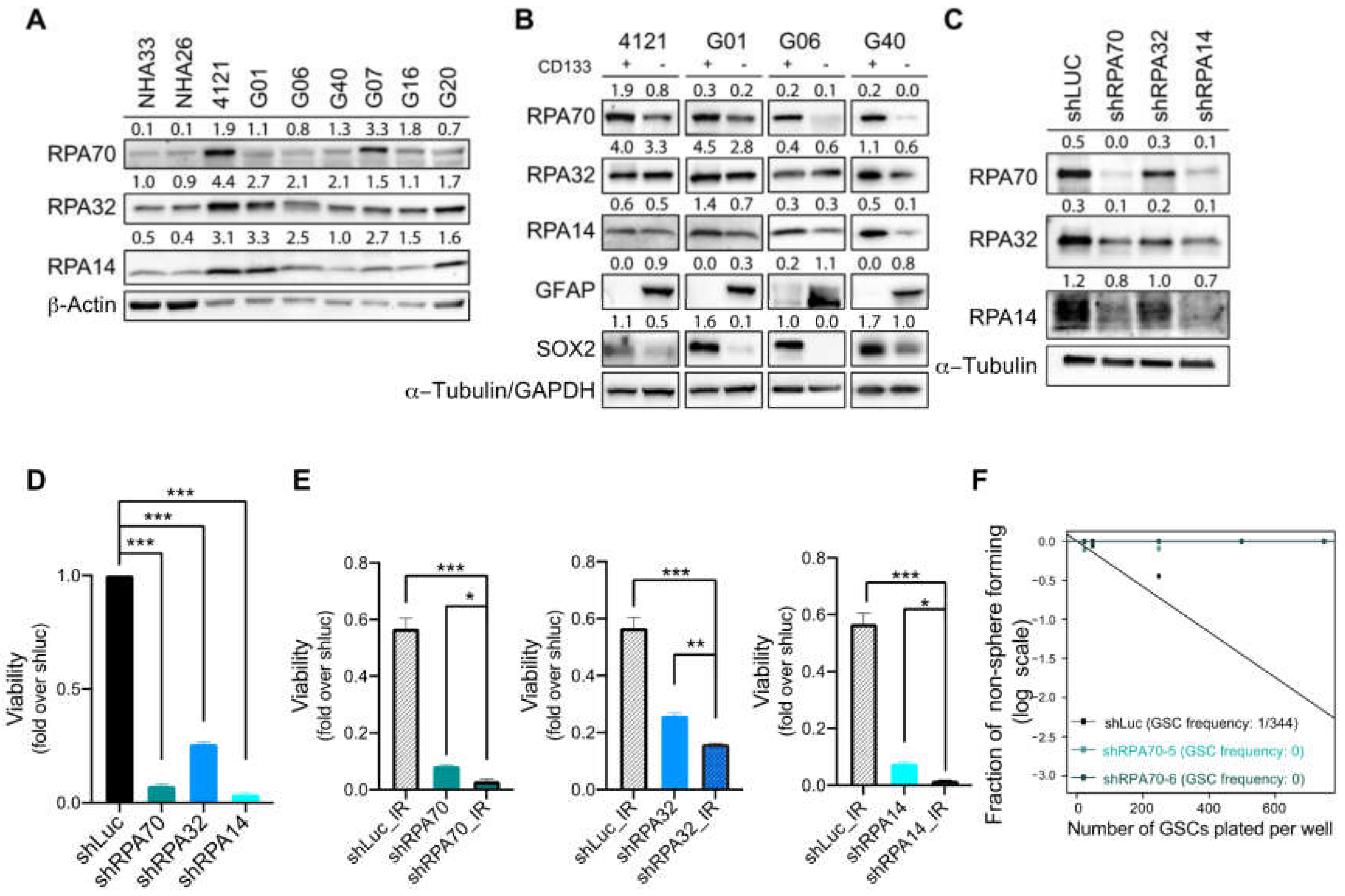
2.2. RPA Expression is Crucial for the Maintenance of Glioblastoma Cancer Stem-Like Cells
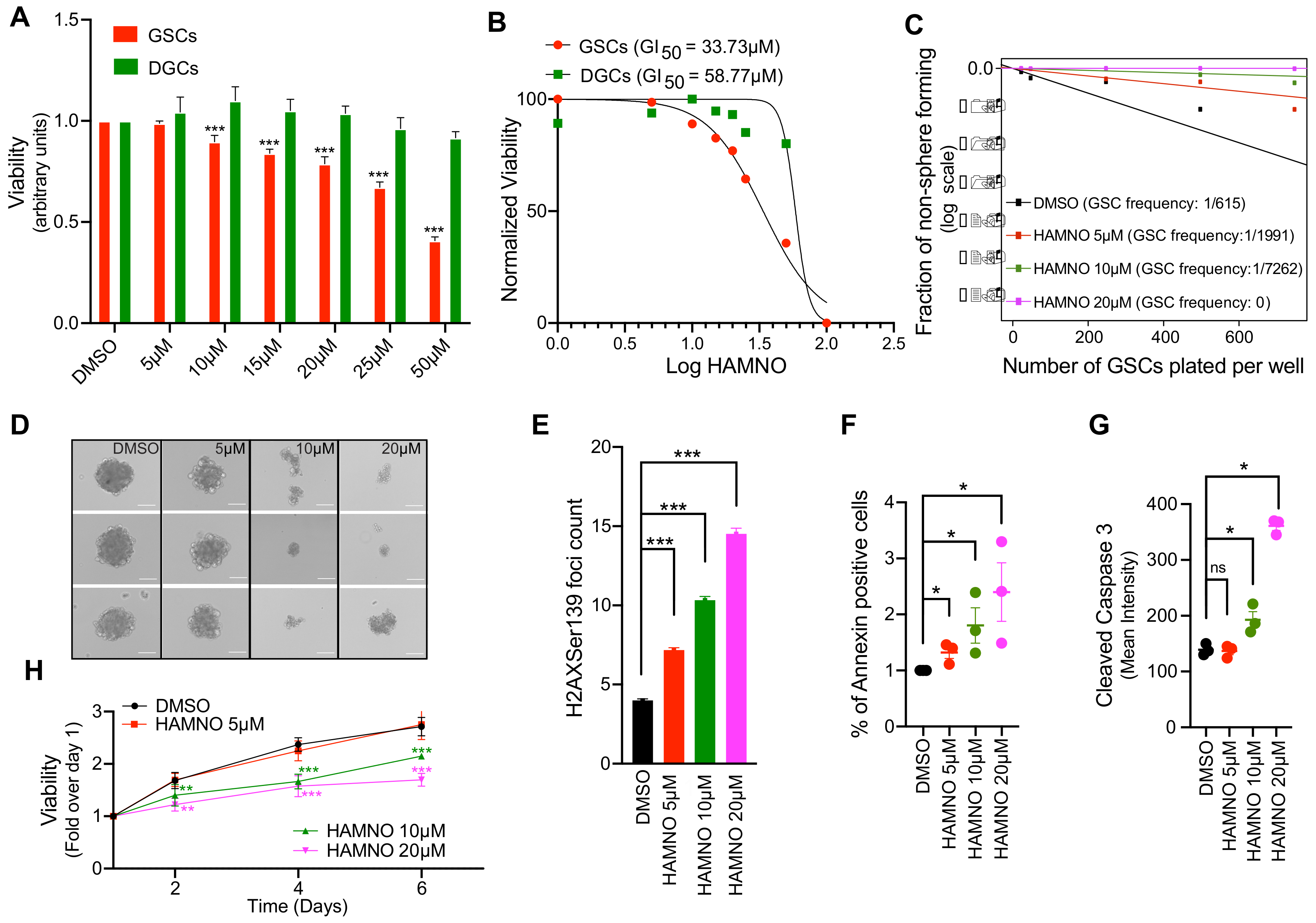
2.3. RPA70-Specific Inhibitor HAMNO Targets Glioblastoma Cancer Stem-Like Cells
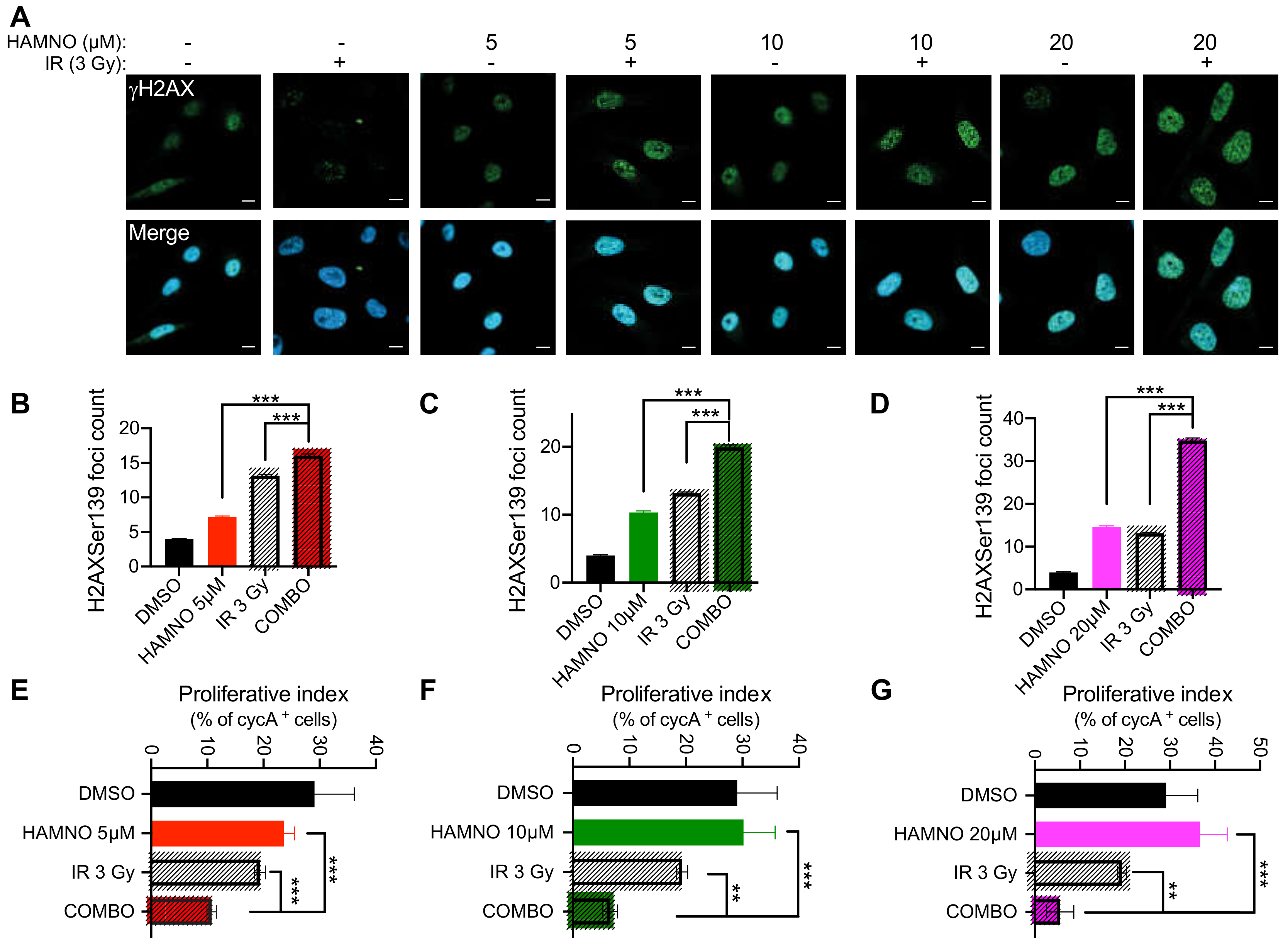
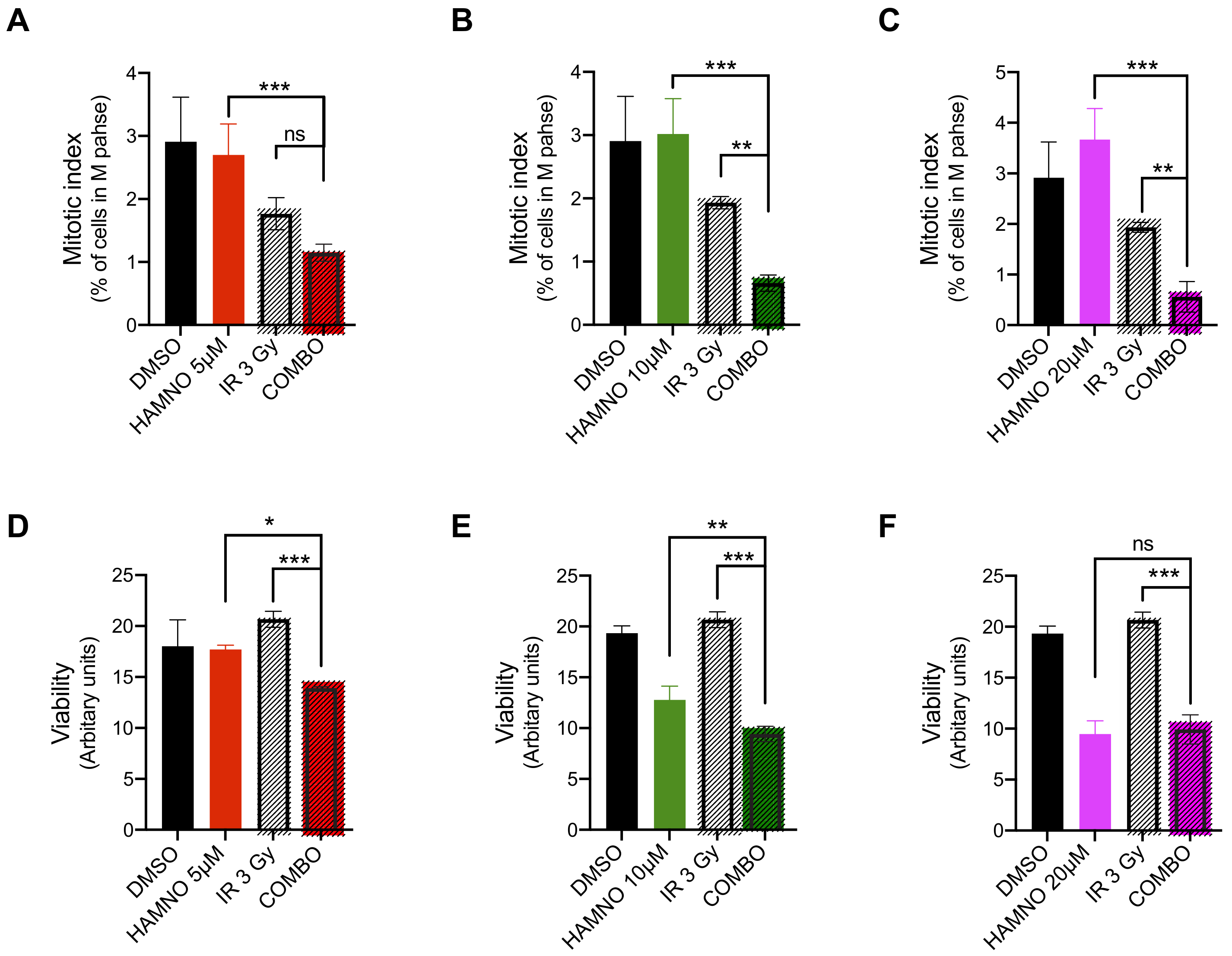
2.4. HAMNO Treatment Sensitizes Glioblastoma Cancer Stem-Like Cells to Ionizing Radiation
3. Discussion
4. Methodology
4.1. Tumor Dissociation, Cell Culture and MACS Sorting
4.2. Flow Cytometry Analysis of Apoptosis
4.3. Drug Preparation and Irradiation
4.4. Proliferation Assay
4.5. Lentiviral Particle Preparation
4.6. Cell Viability
4.7. Neurosphere Formation and Extreme Limiting Dilution Assay (ELDA)
4.8. Immunoblotting
4.9. Immunofluorescence and Image Analysis
4.10. In Silico Analysis of Public Data Sets
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276. [Google Scholar] [CrossRef]
- Ubhi, T.; Brown, G.W. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res. 2019, 79, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wold, M.S. Replication protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. BioEssays 2014, 36, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, J. Replication protein A and more: Single-stranded DNA-binding proteins in eukaryotic cells. Acta Biochim Biophys Sin. (Shanghai) 2016, 48, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 8, 509–520. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Binger, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells--much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef]
- Tamura, K.; Aoyagi, M.; Wakimoto, H.; Ando, N.; Nariai, T.; Yamamoto, M.; Ohno, K. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J. Neurosurg. 2010, 113, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Linkous, A.G.; Yazlovitskaya, E.M. Novel radiosensitizing anticancer therapeutics. Anticancer Res. 2012, 32, 2487–2499. [Google Scholar] [PubMed]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef]
- Rasmussen, R.D.; Gajjar, M.K.; Jensen, K.E.; Hamerlik, P. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol. Oncol. 2016, 10, 751–763. [Google Scholar] [CrossRef]
- Choudhary, S.K.; Li, R. BRCA1 modulates ionizing radiation-induced nuclear focus formation by the replication protein A p34 subunit. J. Cell Biochem. 2002, 84, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; Ionescu, D.; Ingles, C.J. Interaction between BRCA2 and replication protein A is compromised by a cancer-predisposing mutation in BRCA2. Oncogene 2003, 22, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.G.; Liu, S.; Wang, L.; Mosel, A.; Peng, A.; Oakley, G.G. RPA inhibition increases replication stress and suppresses tumor growth. Cancer Res. 2014, 74, 5165–5172. [Google Scholar] [CrossRef]
- Venere, M.; Hamerlik, P.; Wu, Q.; Rasmussen, R.D.; Song, L.A.; Vasanji, A.; Tenley, N.; Flavahan, W.A.; Hjelmeland, A.B.; Bartek, J.; et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 258–269. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, Y.; Wu, X.; Shell, S.M. Functions of human replication protein A (RPA): From DNA replication to DNA damage and stress responses. J. Cell Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef]
- Tomkiel, J.E.; Alansari, H.; Tang, N.; Virgin, J.B.; Yang, X.; VandeVord, P.; Karvonen, R.L.; Granda, J.L.; Kraut, M.J.; Ensley, J.F.; et al. Autoimmunity to the M(r) 32,000 subunit of replication protein A in breast cancer. Clin. Cancer Res. 2002, 8, 752–758. [Google Scholar] [PubMed]
- Dai, Z.; Wang, S.; Zhang, W.; Yang, Y. Elevated Expression of RPA3 Is Involved in Gastric Cancer Tumorigenesis and Associated with Poor Patient Survival. Dig. Dis Sci. 2017, 62, 2369–2375. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Zheng, J.; Zhou, B.; Pan, L. Replication Protein A 3 Is Associated with Hepatocellular Carcinoma Tumorigenesis and Poor Patient Survival. Dig. Dis. 2018, 36, 26–32. [Google Scholar] [CrossRef]
- Levidou, G.; Gakiopoulou, H.; Kavantzas, N.; Saetta, A.A.; Karlou, M.; Pavlopoulos, P.; Thymara, I.; Diamantopoulou, K.; Patsouris, E.; Korkolopoulou, P. Prognostic significance of replication protein A (RPA) expression levels in bladder urothelial carcinoma. BJU Int. 2011, 108, E59–E65. [Google Scholar] [CrossRef] [PubMed]
- Belanger, F.; Fortier, E.; Dube, M.; Lemay, J.F.; Buisson, R.; Masson, J.Y.; Elsherbiny, A.; Costantino, S.; Carmona, E.; Mes-Masson, A.M.; et al. Replication Protein A Availability during DNA Replication Stress Is a Major Determinant of Cisplatin Resistance in Ovarian Cancer Cells. Cancer Res. 2018, 78, 5561–5573. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, T.; Chen, H.; Li, H.; Zheng, S. Oncogene RPA1 promotes proliferation of hepatocellular carcinoma via CDK4/Cyclin-D pathway. Biochem. Biophys. Res. Commun. 2018, 498, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef]
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Møllgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek, J., Jr.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016, 7, 13398. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol Methods. 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedersen, H.; Anne Adanma Obara, E.; Elbæk, K.J.; Vitting-Serup, K.; Hamerlik, P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. Int. J. Mol. Sci. 2020, 21, 1588. https://doi.org/10.3390/ijms21051588
Pedersen H, Anne Adanma Obara E, Elbæk KJ, Vitting-Serup K, Hamerlik P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. International Journal of Molecular Sciences. 2020; 21(5):1588. https://doi.org/10.3390/ijms21051588
Chicago/Turabian StylePedersen, Henriette, Elisabeth Anne Adanma Obara, Kirstine Juul Elbæk, Kristoffer Vitting-Serup, and Petra Hamerlik. 2020. "Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells" International Journal of Molecular Sciences 21, no. 5: 1588. https://doi.org/10.3390/ijms21051588
APA StylePedersen, H., Anne Adanma Obara, E., Elbæk, K. J., Vitting-Serup, K., & Hamerlik, P. (2020). Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. International Journal of Molecular Sciences, 21(5), 1588. https://doi.org/10.3390/ijms21051588