Treatment of Neonatal Hypoxic-Ischemic Encephalopathy with Erythropoietin Alone, and Erythropoietin Combined with Hypothermia: History, Current Status, and Future Research


Abstract
1. Introduction
- Early identification, within 2–6 h of birth, of those at high risk. A high risk of HIE is likely in infants with fetal bradycardia (<100 beats/minute), an Apgar score of five or less at 5 minutes [8], a cord blood pH of 7 or less, and/or a base deficit of 16 or more [9]. The Apgar score enables a quick and accurate assessment of the respiratory, cardio-circulatory, and neurological condition of the newborn. The Apgar score at 10 minutes correlates with poor outcomes following HIE [10].
- Adequate perfusion of the brain through supportive care. The supportive care can involve the provision of oxygen, volume expanders, ionotropes, diurectics, and antibiotics (see also Section 5).
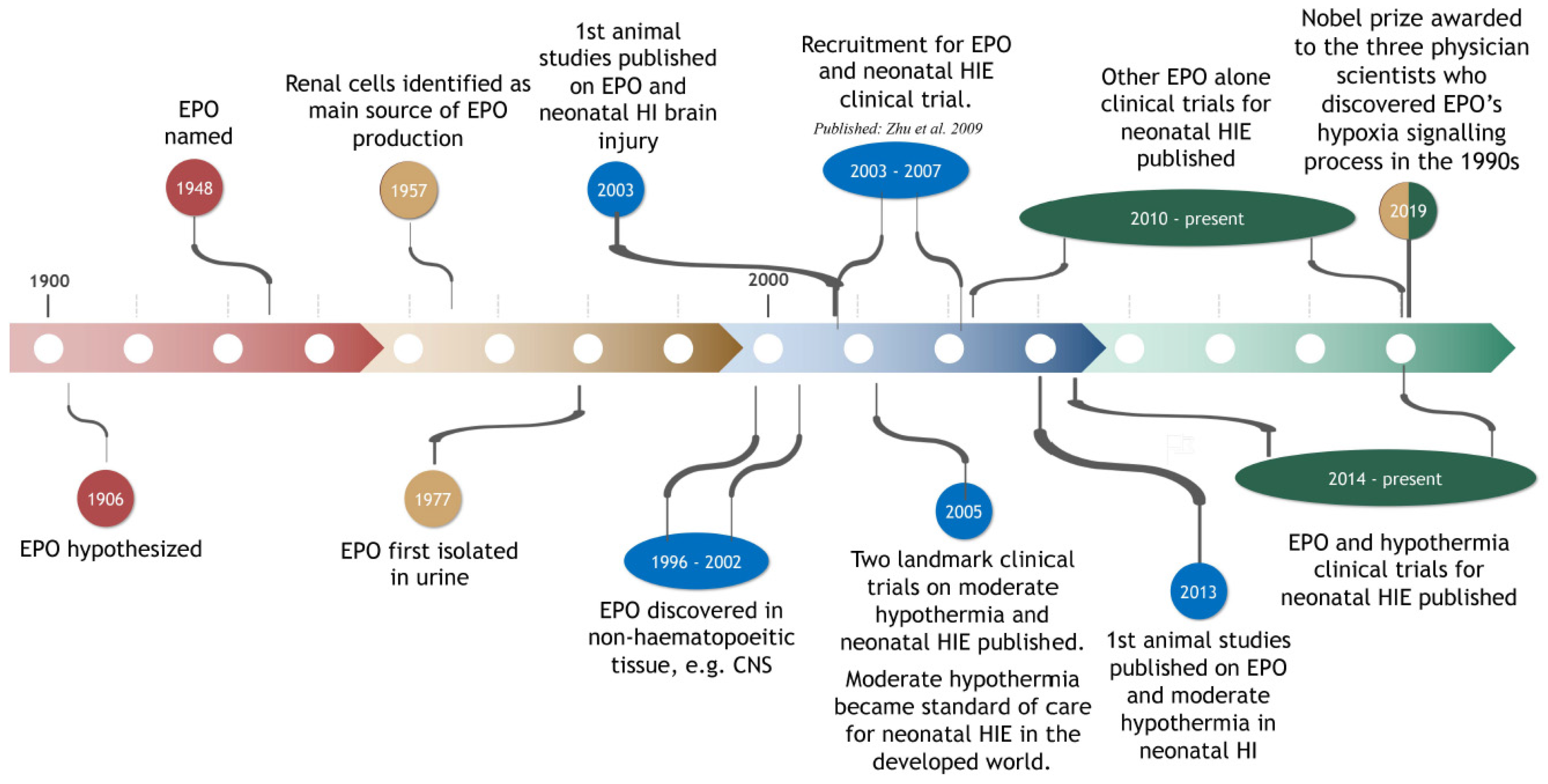
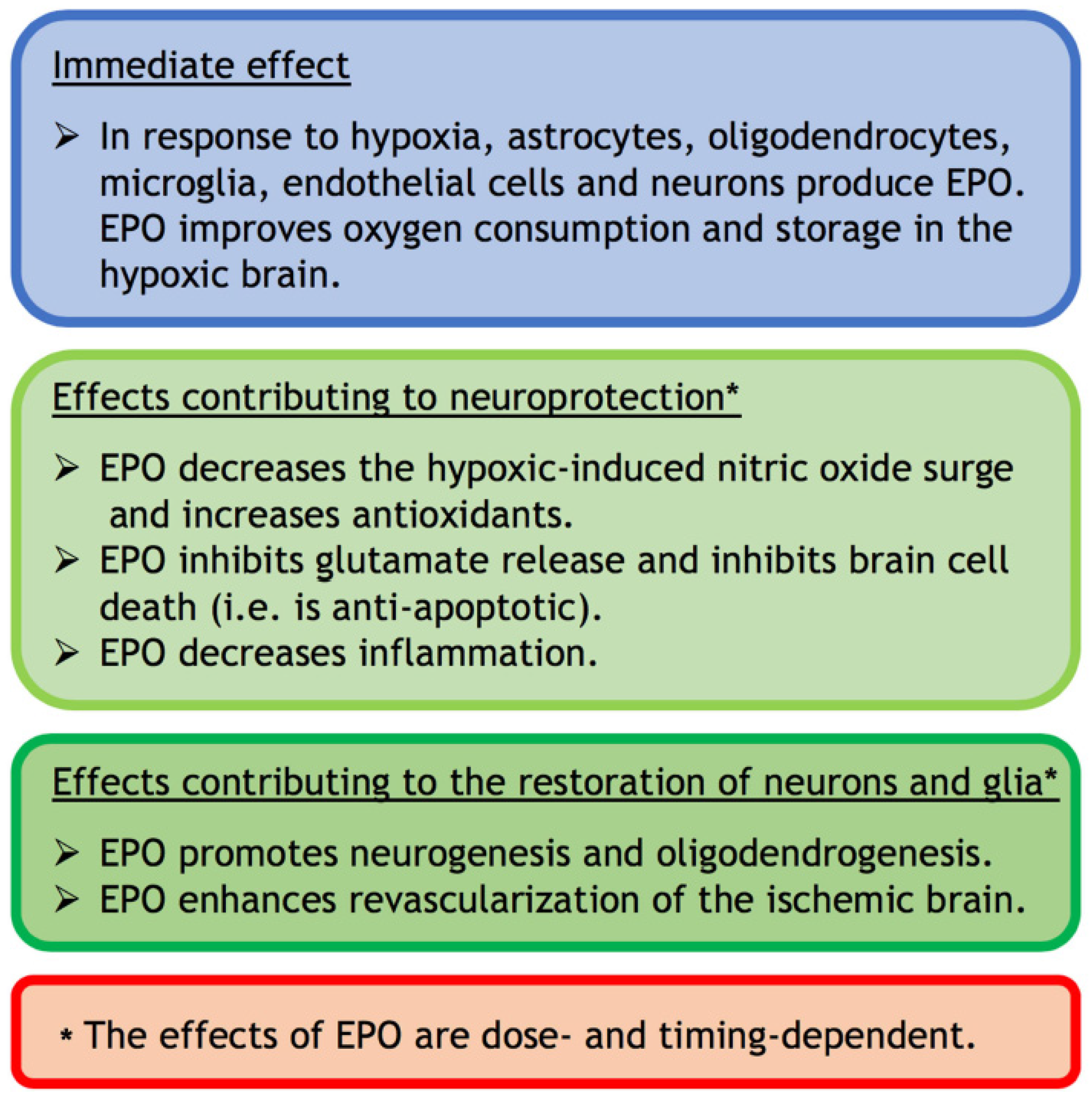
2. EPO: Brief History and Synopsis of Functions
3. Animal Studies on Neonatal Hypoxia-Ischemia: Effect of EPO Alone
4. Animal Studies on Neonatal Hypoxia-Ischemia: Effect of EPO in Combination with Moderate Hypothermia
5. Clinical Studies on Neonatal Hypoxic-Ischemic Encephalopathy: Effect of EPO Alone
6. Clinical Studies on Neonatal Hypoxic-Ischemic Encephalopathy: Effect of EPO Combined with Moderate Hypothermia
7. Effects of EPO in Clinical Trials for Preterm Infants
8. Potential for Adverse Effects of EPO in Neonatal HIE
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martinello, K.; Hart, A.R.; Yap, S.; Mitra, S.; Robertson, N.J. Management and investigation of neonatal encephalopathy; 2017 update. Arch. Dis. Child. Fetal Neonatal Ed. 2017, 102, F346–F358. [Google Scholar] [CrossRef] [PubMed]
- Kurinczuk, J.J.; White-Koning, M.; Badawi, N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum. Dev. 2010, 86, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E.; Ferriero, D.M. Pharmacologic neuroprotective strategies in neonatal brain injury. Clin. Perinatol. 2014, 41, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Black, R.E.; Cousens, S.; Johnson, H.L.; Lawn, J.E.; Rudan, I.; Bassani, D.G.; Jha, P.; Campbell, H.; Fischer Walker, C.; Cibulskis, R.; et al. For the child Health Epidemiology Reference Group of WHO and UNICEF. Global, regional, and national causes of child mortality in 2008: A systematic analysis. Lancet 2010, 375, 1969–1987. [Google Scholar] [CrossRef]
- Akamatsu, T.; Sugiyama, T.; Aoki, Y.; Kawabata, K.; Shimizu, M.; Okazaki, K.; Kondo, M.; Takahashi, K.; Yokoyama, Y.; Takahashi, N.; et al. A pilot study of soluble form of LOX-1 as a novel biomarker for neonatal hypoxic-ischemic encephalopathy. J. Pediatr. 2019, 206, 49–55. [Google Scholar]
- Sarnat, H.B.; Sarnat, M.S. Neonatal encephalopathy following fetal distress. Arch. Neurol. 1976, 33, 696–705. [Google Scholar] [CrossRef]
- Thompson, C.M.; Puterman, A.S.; Linley, L.L.; Hann, M.; van der Elst, C.W.; Molten, C.D.; Malan, A.F. The value of a scoring system for hypoxic ischaemic encephalopathy in predicting neurodevelopmental outcome. Acta Paediatr. 1997, 86, 757–761. [Google Scholar] [CrossRef]
- Apgar, V. A proposal for a new method of evaluation of the newborn infant. Curr. Res. Anesth. Analg. 1953, 32, 260–267. [Google Scholar] [CrossRef]
- Perlman, J.M. Summary proceedings from the neurology group on hypoxic-ischemic encephalopathy. Pediatrics 2006, 117, S28–S33. [Google Scholar] [CrossRef]
- Natarajan, G.; Shankaran, S.; Laptook, A.R.; Pappas, A.; Bann, C.M.; McDonald, S.A.; Das, A.; Higgins, R.D.; Hintz, S.R.; Vohr, B.R. Apgar scores at 10 min and outcomes at 6–7 years following hypoxic-ischaemic encephalopathy. Arch. Dis. Child. Fetal Neonatal Ed. 2013, 98, F473–F479. [Google Scholar] [CrossRef]
- Amer, A.R.; Oorschot, D.E. Xenon combined with hypothermia in perinatal hypoxic-ischemic encephalopathy: A noble gas, a noble mission. Pediatr. Neurol. 2018, 84, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Perlman, J.M.; Wyllie, J.; Kattwinkel, J.; Atkins, D.L.; Chameides, L.; Goldsmith, J.P.; Guinsburg, R.; Hazinski, M.F.; Morley, C.; Richmond, S.; et al. and Neonatal Resuscitation Chapter Collaborators. Part II: Neonatal resuscitation: 2010 international consensus on cardiopulmonary resuscitation and emergency cardiovascular care science with treatment recommendations. Circulation 2010, 122, S516–S538. [Google Scholar] [CrossRef] [PubMed]
- Rossouw, G.; Irlam, J.; Horn, A.R. Therapeutic hypothermia for hypoxic ischaemic encephalopathy using low-technology methods: A systematic review and meta-analysis. Acta Paediatr. 2015, 104, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Sabir, H.; Osredkar, D.; Maes, E.; Wood, T.; Thoresen, M. Xenon combined with therapeutic hypothermia is not neuroprotective after severe hypoxia-ischemia in neonatal rats. PLoS ONE 2016, 1, e0156759. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, S.; Laptook, A.R.; Pappas, A.; McDonald, S.A.; Das, A.; Tyson, J.E.; Poindexter, B.B.; Schibler, K.; Bell, E.F.; Heyne, R.J.; et al. Effect of depth and duration of cooling on deaths in the NICU among neonates with hypoxic ischemic encephalopathy: A randomized clinical trial. JAMA 2014, 312, 2629–2639. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.E.; Berg, M.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Ver. 2013. [Google Scholar] [CrossRef]
- Tagin, M.A.; Woolcott, C.G.; Vincer, M.J.; Whyte, R.K.; Stinson, D.A. Hypothermia for neonatal hypoxic ischemic encephalopathy: An updated systematic review and meta-analysis. Arch. Pediatr. Adolesc. Med. 2012, 166, 558–566. [Google Scholar] [CrossRef]
- Hobson, A.; Baines, J.; Weiss, M.D. Beyond hypothermia: Alternative therapies for hypoxic ischemic encephalopathy. Open Pharmacol. J. 2013, 7, 26–40. [Google Scholar] [CrossRef]
- Gunn, A.J.; Groenendaal, F. Delayed neuroporotection in the era of hypothermia: What can we add? J. Clin. Neonatol. 2016, 5, 3–7. [Google Scholar] [CrossRef]
- Rao, R.; Trivedi, S.; Vesoulis, Z.; Liao, S.M.; Smyser, C.D.; Mathur, A.M. Safety and short-term outcomes of therapeutic hypothermia in preterm neonates 34-35 weeks gestational age with hypoxic-ischemic encephalopathy. J. Pediatr. 2017, 183, 37–42. [Google Scholar] [CrossRef]
- McPherson, R.J.; Juul, S.E. Erythropoietin (Epo) for infants with hypoxic-ischemic encephalopathy (HIE). Curr. Opin. Pediatr. 2010, 22, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Krumdieck, N. Erythropoietic substance in the serum of anemic animals. Proc. Soc. Exp. Biol. Med. 1943, 54, 14–17. [Google Scholar] [CrossRef]
- Ostrowski, D.; Heinrich, R. Alternative erythropoietin receptors in the nervous system. J. Clin. Med. 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, A. Nobel Prize 2019 pays tribute to translational physiology on oxygen sensing. Pflugers Arch.—Eur. J. Physiol. 2019, 471, 1341–1342. [Google Scholar] [CrossRef]
- Carnot, P.; Deflandre, C. Sur l’activite hemopoietique de serum au cours de la regeneration du sang. Acad. Sci. 1906, 143, 384–386. [Google Scholar]
- Fisher, J.W. Erythropoietin: Physiology and pharmacology update. Exp. Biol. Med. 2003, 228, 1–14. [Google Scholar] [CrossRef]
- Sytkowski, A.J. Does erythropoietin have a dark side? Epo signaling and cancer cells. Sci. STKE 2007, 395, pe38. [Google Scholar] [CrossRef]
- Bonsdorff, E.; Jalavisto, E. A humoral mechanism in anoxic erythrocytosis. Acta Physiol. Rev. 1948, 16, 150–170. [Google Scholar] [CrossRef]
- Miyake, T.; Kung, C.K.H.; Goldwasser, E. Purification of human erythropoietin. J. Biol. Chem. 1977, 252, 5558–5564. [Google Scholar]
- Ng, T.; Marx, G.; Littlewood, T.; Macdougall, I. Recombinant erythropoietin in clinical practice. Postgrad. Med. J. 2003, 79, 367–376. [Google Scholar] [CrossRef][Green Version]
- McGraw, K.; List, A. Erythropoietin receptor signaling and lipid rafts. Vitam. Horm. 2017, 105, 79–100. [Google Scholar] [PubMed]
- Jacobson, L.O.; Goldwasser, E.; Fried, W.; Plzak, L. Role of the kidney in erythropoiesis. Nature 1957, 179, 633–634. [Google Scholar] [CrossRef] [PubMed]
- Erslev, A.J. In vitro production of erythropoietin by kidneys perfused with a serum-free solution. Blood 1974, 44, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Marti, H.H. Erythropoietin and the hypoxic brain. J. Exp. Biol. 2004, 207, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Nagao, M.; Takahata, K.; Konishi, Y.; Gallyas, F., Jr.; Tabira, T.; Sasaki, R. Functional erythropoietin receptor of the cells with neural characteristics. Comparison with receptor properties of erythroid cells. J. Biol. Chem. 1993, 268, 11208–11216. [Google Scholar]
- Li, Y.; Juul, S.E.; Morris-Wiman, J.A.; Calhoun, D.A.; Christensen, R.D. Erythropietin receptors are expressed in the central nervous system of mid-trimester human fetuses. Pediatr. Res. 1996, 40, 376–380. [Google Scholar] [CrossRef][Green Version]
- Morishita, E.; Masuda, S.; Nagao, M.; Yasuda, Y.; Sasaki, R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 1997, 76, 105–116. [Google Scholar] [CrossRef]
- Juul, S.E.; Anderson, D.K.; Li, Y.; Christensen, R.D. Erythropoietin and erythropoietin receptor in the developing human central nervous system. Pediatr. Res. 1998, 43, 40–49. [Google Scholar] [CrossRef]
- Kirkeby, A.; van Beek, J.; Nielsen, J.; Leist, M.; Helboe, L. Functional immunochemical characterization of different antibodies against the erythropoietin receptor. J. Neurosci. Methods 2007, 164, 50–58. [Google Scholar] [CrossRef]
- Ott, C.; Martens, H.; Hassouna, I.; Oliveira, B.; Erck, C.; Zafeiriou, M.-P.; Peteri, U.-K.; Hesse, D.; Gerhart, S.; Altas, B.; et al. Widespread expression of erythropoietin receptor in brain and its induction by injury. Mol. Med. 2015, 21, 803–815. [Google Scholar] [CrossRef]
- Masuda, S.; Okano, M.; Yamagishi, K.; Nagao, M.; Ueda, M.; Sasaki, R. A novel site of erythropoietin production. Oxygen-dependent production in cultured rat astrocytes. J. Biol. Chem. 1994, 269, 9488–9493. [Google Scholar]
- Van der Kooij, M.A.; Groenendaal, F.; Kavelaars, A.; Heijnen, C.J.; van Bel, F. Neuroprotective properties and mechanisms of erythropoietin in in vitro and in vivo experimental models for hypoxia/ischemia. Brain Res. Rev. 2008, 59, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for oxygen sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Landau ubiquitylation complex by oxygen-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Liu, Y.; Christou, H.; Moritia, T.; Laughner, E.; Semenza, G.L.; Kourembanas, S. Carbon monoxide and nitric oxide suppress the hypoxic induction of vascular endothelial growth factor gene via the 5′ enhancer. J. Biol. Chem. 1998, 273, 15257–15262. [Google Scholar] [CrossRef]
- Kumral, A.; Baskin, H.; Gokmen, N.; Yilmaz, O.; Genc, K.; Genc, S.; Tatli, M.M.; Duman, N.; Ozer, E.; Ozkan, H. Selective inhibition of nitric oxide in hypoxic-ischemic brain model in newborn rats: Is it an explanation for the protective role of erythropoietin? Biol. Neonate 2004, 85, 51–54. [Google Scholar] [CrossRef]
- Roberts, J.D.; Polaner, D.M.; Lang, P.; Zapol, W.M. Inhaled nitric oxide in persistent pulmonary hypertension of the newborn. Lancet 1992, 340, 818–819. [Google Scholar] [CrossRef]
- Kinsella, J.P.; Neish, S.R.; Shaffer, E.; Abman, S.H. Low-dose inhalational nitric oxide in persistent pulmonary hypertension of the newborn. Lancet 1992, 340, 819–820. [Google Scholar] [CrossRef]
- Schelshorn, D.W.; Schneider, A.; Kuschinsky, W.; Weber, D.; Kruger, C.; Dittgen, T.; Burgers, H.F.; Sabouri, F.; Gassler, N.; Bach, A.; et al. Expression of haemoglobin in rodent neurons. J. Cereb. Blood Flow Metabol. 2009, 29, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Ferriero, D.M. Oxidant mechanisms in neonatal hypoxia-ischemia. Dev. Neurosci. 2001, 23, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Kumral, A.; Gonenc, S.; Acikgoz, O.; Sonmez, A.; Genc, K.; Yilmaz, O.; Gokmen, N.; Duman, N.; Ozkan, H. Erythropoietin increases glutathione peroxidase enzyme activity and decreases lipid peroxidation levels in hypoxic-ischemic brain injury in neonatal rats. Biol. Neonate 2005, 87, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Juul, S. Neuroprotective role of erythropoietin in neonates. J. Matern. Fetal Neonatal Med. 2012, 25, 105–107. [Google Scholar] [CrossRef]
- Genc, S.; Akhisaroglu, M.; Kuralay, F.; Genc, K. Erythropoietin restores glutathione peroxidase activity in 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced neurotoxicity in C57BL mice and stimulates murine astroglial glutathione peroxidase production in vitro. Neurosci. Lett. 2002, 321, 73–76. [Google Scholar] [CrossRef]
- Juul, S. Erythropoietin in the central nervous system, and its use to prevent hypoxic-ischemic brain damage. Acta Paediatr. Suppl. 2002, 91, 36–42. [Google Scholar] [CrossRef]
- Xiong, T.; Qu, Y.; Mu, D.; Ferriero, D. Erythropoietin for neonatal brain injury: Opportunity and challenge. Int. J. Dev. Neurosci. 2011, 29, 583–591. [Google Scholar] [CrossRef]
- Vairano, M.; Dello Russo, C.; Pozzoli, G.; Battaglia, A.; Scambia, G.; Tringali, G.; Aloe-Spiriti, M.A.; Preziosi, P.; Navarra, P. Erythropoietin exerts anti-apoptotic effects on rat microglial cells in vitro. Eur. J. Neurosci. 2002, 16, 584–592. [Google Scholar] [CrossRef]
- Jantzie, L.L.; Miller, R.H.; Robinson, S. Erythropoietin signalling promotes oligodendrocyte development following prenatal systemic hypoxic-ischemic brain injury. Pediatr. Res. 2013, 74, 658–667. [Google Scholar] [CrossRef]
- Kumral, A.; Uysal, N.; Tugyan, K.; Sonmez, A.; Yilmaz, O.; Gokmen, N.; Kiray, M.; Genc, S.; Duman, N.; Koroglu, T.F.; et al. Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hypoxia-ischemia in rats. Behav. Brain Res. 2004, 153, 77–86. [Google Scholar] [CrossRef]
- Kawakami, M.; Sekihuchi, M.; Sato, K.; Kozaki, S.; Takahashi, M. Erythropoietin receptor-mediated inhibition of exocytotic glutamate release confers neuroprotection during chemical ischemia. J. Biol. Chem. 2001, 276, 39469–39475. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, R.C.; Perlman, J.M. Interventions for perinatal hypoxic-ischemic encephalopathy. Pediatrics 1997, 100, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E.; Pet, G.C. Erythropoietin and neonatal neuroprotection. Clin. Perinatol. 2015, 42, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Buemi, M.; Alafaci, C.; Sfacteria, A.; Passalacqua, M.; Sturiale, A.; Calapai, G.; De Vico, G.; Piedimonte, G.; Salpietro, F.M.; et al. Beneficial effects of systemic administration of recombinant human erythropoietin in rabbits subjected to subarachnoid hemorrhage. Proc. Natl. Acad. Sci. USA 2002, 99, 5627–5631. [Google Scholar] [CrossRef]
- Sola, A.; Wen, T.-C.; Hamrick, S.E.G.; Ferriero, D.M. Potential for protection and repair following injury to the developing brain: A role for erythropoietin? Pediatr. Res. 2005, 57, 110–117. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Towfighi, J.; Vannucci, S.J. Secondary energy failure after cerebral hypoxia-ischemia in the immature rat. J. Cereb. Blood Flow Metabol. 2004, 24, 1090–1097. [Google Scholar] [CrossRef]
- Inoue, N.; Takeuchi, M.; Ohashi, H.; Suzuki, T. The production of recombinant human erythropoietin. Biotechnol. Annu. Rev. 1995, 1, 297–313. [Google Scholar]
- Sirén, A.-L.; Faßhauser, T.; Bartels, C.; Ehrenreich, H. Therapeutic potential of erythropoietin and its structural or functional varients in the nervous system. Neurotherapeutics 2009, 6, 108–127. [Google Scholar] [CrossRef]
- Smith, R.E.; Jaiyesimi, I.A.; Meza, L.A.; Tchekmedyian, N.S.; Chan, D.; Griffith, H.; Brosman, S.; Bukowski, R.; Murdock, M.; Rarick, M.; et al. Novel erythropoiesis stimulating protein (NESP) for the treatment of anaemia of chronic disease associated with cancer. Br. J. Cancer 2001, 84, 24–30. [Google Scholar] [CrossRef]
- Leist, M.; Ghezzi, P.; Grasso, G.; Bianchi, R.; Villa, P.; Fratelli, M.; Savino, C.; Bianchi, M.; Nielsen, J.; Gerwien, J.; et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 2004, 305, 239–242. [Google Scholar] [CrossRef]
- Matsushita, H.; Johnston, M.V.; Lange, M.S.; Wilson, M.A. Protective effect of erythropoietin in neonatal hypoxic ischemia in mice. Neuroreport 2003, 14, 1757–1761. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aydin, A.; Genc, K.; Akhisaroglu, M.; Yorukoglu, K.; Gokmen, N.; Gonullu, E. Erythropoietin exerts neuroprotective effect in neonatal rat model of hypoxic-ischemic brain injury. Brain Dev. 2003, 27, 494–498. [Google Scholar] [CrossRef]
- Kumral, A.; Ozer, E.; Yilmaz, O.; Akhisaroglu, M.; Gokmen, N.; Duman, N.; Ulukus, C.; Genc, S.; Ozkan, H. Neuroprotective effect of erythropoietin on hypoxic-ischemic brain injury in neonatal rats. Biol. Neonate 2003, 83, 224–228. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Connor, J.R.; Mauger, D.T.; Palmer, C.; Smith, M.B.; Towfighi, J.; Vannucci, S.J. Rat model of perinatal hypoxic-ischemic brain damage. J. Neurosci. Res. 1999, 55, 158–163. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.E.; Vannucci, R.C.; Brierly, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Bona, E.; Gilland, E.; Puka-Sundvall, M. Hypoxia-ischaemia model in the 7-day-old rat: Possibilities and shortcomings. Acta Paediatr. Suppl. 1997, 422, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Koehler, R.C.; Yang, Z.-J.; Lee, J.K.; Martin, L.J. Perinatal hypoxic-ischemic brain injury in larger animal models: Relevance to human neonatal encephalopathy. J. Cereb. Blood Flow Metabol. 2018, 38, 2092–2111. [Google Scholar] [CrossRef]
- Brines, M.L.; Ghezzi, P.; Keenan, S.; Agnello, D.; de Lanerolle, N.C.; Cerami, C.; Itri, L.M.; Cerami, A. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. USA 2000, 97, 10526–10531. [Google Scholar] [CrossRef]
- Spandou, E.; Papoutsopoulou, S.; Soubasi, V.; Karkavelas, G.; Simeonidou, C.; Kremenopoulos, G.; Guiba-Tziampiri, O. Hypoxia-ischemia affects erythropoietin and erythropoietin receptor expression pattern in the neonatal rat brain. Brain Res. 2004, 1021, 167–172. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, C.; Polk, P.; Nanda, A.; Zhang, J.H. Mechanisms of erythropoietin-induced brain protection in neonatal hypoxia-ischemia rat model. J. Cereb. Blood Flow Metabol. 2004, 24, 259–270. [Google Scholar] [CrossRef] [PubMed]
- McClure, M.M.; Threlkeld, S.W.; Fitch, R.H. Auditory processing and learning/memory following erythropoietin administration in neonatally hypoxic-ischemic injured rats. Brain Res. 2007, 1132, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.L.; Hill, C.A.; Rosenkrantz, T.S.; Fitch, R.H. Evaluation of the therapeutic benefit of delayed administration of erythropoietin following early hypoxic-ischemic injury in rodents. Dev. Neurosci. 2012, 34, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Spandou, E.; Papadopoulou, Z.; Soubasi, V.; Karkavelas, G.; Simeonidou, C.; Pazaiti, A.; Guiba-Tziampiri, O. Erythropoietin prevents long-term sensorimotor deficits and brain injury following neonatal hypoxia-ischemia in rats. Brain Res. 2005, 1045, 22–30. [Google Scholar] [CrossRef]
- Demers, E.J.; McPherson, R.J.; Juul, S.E. Erythropoietin protects dopaminergic neurons and improves neurobehavioral outcomes in juvenile rats after neonatal hypoxia-ischemia. Pediatr. Res. 2005, 58, 297–301. [Google Scholar] [CrossRef]
- Van der Kooij, M.A.; Groenendaal, F.; Kavelaars, A.; Heijnen, C.J.; van Bel, F. Combination of deferoxamine and erythropoietin: Therapy for hypoxia-ischemia-induced brain injury in the neonatal rat? Neurosci. Lett. 2009, 451, 109–113. [Google Scholar] [CrossRef]
- Fan, X.; Heijnen, C.J.; van der kooij, M.A.; Groenendaal, F.; van Bel, F. Beneficial effect of erythropoietin on sensorimotor function and white matter after hypoxia-ischemia in neonatal mice. Pediatr. Res. 2011, 69, 56–61. [Google Scholar] [CrossRef]
- Wen, T.C.; Rogido, M.; Peng, H.; Genetta, T.; Moore, J.; Sola, A. Gender differences in long-term beneficial effects of erythropoietin given after neonatal stroke in postnatal day-7 rats. Neuroscience 2006, 139, 803–811. [Google Scholar] [CrossRef]
- Sola, A.; Rogido, M.; Lee, B.H.; Genetta, T.; Wen, T.-C. Erythropoietin after focal cerebral ischemia activates the janus kinase-signal transducer and activator of transcription signaling pathway and improves brain injury in postnatal day 7 rats. Pediatr. Res. 2005, 57, 481–487. [Google Scholar] [CrossRef]
- Chang, Y.S.; Mu, D.; Wendland, M.; Sheldon, R.A.; Vexler, Z.S.; McQuillen, P.S.; Ferriero, D.M. Erythropoietin improves functional and histological outcome in neonatal stroke. Pediatr. Res. 2005, 58, 106–111. [Google Scholar] [CrossRef]
- Gonzalez, F.F.; Abel, R.; Almli, C.R.; Mu, D.; Wendland, M.; Ferriero, D.M. Erythropoietin sustains cognitive function and brain volume after neonatal stroke. Dev. Neurosci. 2009, 31, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.F.; Larpthaveesarp, A.; McQuillen, P.; Derugin, N.; Wendland, M.; Spadafora, R.; Ferriero, D.M. Erythropoietin increases neurogenesis and oligodendrogliosis of subventricular zone precursor cells after neonatal stroke. Stroke 2013, 44, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Kellert, B.A.; McPherson, R.J.; Juul, S.E. A comparison of high-dose recombinant erythropoietin treatment regimens in brain-injured neonatal rats. Pediatr. Res. 2007, 61, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Iwai, M.; Cao, G.; Yin, W.; Stetler, R.A.; Liu, J.; Chen, J. Erythropoietin promotes neuronal replacement through revascularization and neurogenesis after neonatal hypoxia/ischemia in rats. Stroke 2007, 38, 2795–2803. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Calvert, J.W.; Zhang, J.H. Neonatal hypoxia/ischemia is associated with decreased inflammatory mediators after erythropoietin administration. Stroke 2005, 36, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Iwai, M.; Stetler, R.A.; Xing, J.; Hu, X.; Gao, Y.; Zhang, W.; Chen, J.; Cao, G. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke 2010, 41, 1032–1037. [Google Scholar] [CrossRef]
- Chen, H.; Spagnoli, F.; Burris, M.; Rolland, W.B.; Fajilan, A.; Dou, H.; Tang, J.; Zhang, J. Nanoerythropoietin is 10-times more effective than regular erythropoietin in neuroprotection in a neonatal rat model of hypoxia and ischemia. Stroke 2012, 43, 884–887. [Google Scholar] [CrossRef]
- Ifudu, O.; Uribarri, J.; Rajwani, I.; Vlacich, V.; Reydel, K.; Delosreyes, G.; Friedman, E.A. Gender modulates responsiveness to recombinant erythropoietin. Am. J. Kidney Dis. 2001, 38, 518–522. [Google Scholar] [CrossRef]
- Zeng, J.; Yankowitz, S.M.; Widemness, J.A.; Strauss, R.G. Etiology of differences in hematocrit between males and females: Sequence-based polymorphisms in erythropoietin and its receptor. J. Gend. Specif. Med. 2001, 4, 35–40. [Google Scholar]
- Charriaut-Marlangue, C.; Besson, V.C.; Baud, O. Sexually dimorphic outcomes after neonatal stroke and hypoxia-ischemia. Int. J. Mol. Sci. 2018, 19, 61. [Google Scholar] [CrossRef]
- Dou, H.; Destache, C.J.; Morehead, J.R.; Mosley, R.L.; Boska, M.D.; Kingsley, J.; Gorantia, S.; Poluektova, L.; Nelson, J.A.; Chaubal, M.; et al. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood 2006, 108, 2827–2835. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.J.; Gunn, T.R.; Gunning, M.I.; Williams, C.E.; Gluckman, P.D. Neuroporetection with prolonged head cooling started before postischemic seizures in fetal sheep. Pediatrics 1998, 102, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Wyatt, J.S.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Polin, A.; Robertson, C.M.; Thoresen, M.; Whitelaw, A.; et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: Multicenter randomized trial. Lancet 2005, 365, 663–670. [Google Scholar] [CrossRef]
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, R.A.; Tyson, J.E.; McDonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D.; et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 2005, 353, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; van Bel, F.; van der Kooij, M.A.; Heijnen, C.J.; Groenendaal, F. Hypothermia and erythropoietin for neuroprotection after neonatal brain damage. Pediatr. Res. 2013, 73, 18–23. [Google Scholar] [CrossRef]
- Fang, A.Y.; Gonzalez, F.F.; Sheldon, R.A.; Ferriero, D.M. Effects of combination therapy using hypothermia and erythropoietin in a rat model of neonatal hypoxia-ischemia. Pediatr. Res. 2013, 73, 12–17. [Google Scholar] [CrossRef]
- Traudt, C.M.; McPherson, R.J.; Bauer, L.A.; Richards, T.L.; Burbacher, T.M.; McAdams, R.M.; Juul, S.E. Concurrent erythropoietin and hypothermia treatment improve outcomes in a term nonhuman primate model of perinatal asphyxia. Dev. Neurosci. 2013, 35, 491–503. [Google Scholar] [CrossRef]
- McAdams, R.M.; Fleiss, B.; Traudt, C.; Schwendimann, L.; Snyder, J.M.; Haynes, R.L.; Natarayan, N.; Gressens, P.; Juul, S.E. Long-term neuropathological changes associated with cerebral palsy in a nonhuman primate model of hypoxic-ischemic encephalopathy. Dev. Neurosci. 2017, 39, 124–140. [Google Scholar] [CrossRef]
- Halperin, D.S.; Wacker, P.; Lacourt, G.; Felix, M.; Babel, J.-F.; Aapro, M.; Wyss, M. Effects of recombinant human erythropoietin in infants with the anemia of prematurity: A pilot study. J. Pediatr. 1990, 116, 779–786. [Google Scholar] [CrossRef]
- Ananthan, A.; Balasubramanian, H.; Rao, S.; Patole, S. Clinical outcomes related to the gastrointestinal trophic effects of erythropoietin in preterm neonates: A systematic review and meta-analysis. Adv. Nutr. 2018, 9, 238–246. [Google Scholar] [CrossRef]
- Aher, S.M.; Ohlsson, A. Early versus late erythropoietin for preventing red blood cell transfusion in preterm and/or low birth weight infants. Cochrane Database Syst. Rev. 2006, 3. [Google Scholar] [CrossRef]
- Ohlsson, A.; Aher, S.M. Early erythropoietin for preventing red blood cell transfusion in preterm and/or low birth weight infants. Cochrane Database Syst. Rev. 2006, 3. [Google Scholar] [CrossRef]
- Natalucci, G.; Latal, B.; Koller, B.; Ruegger, C.; Sick, B.; Held, L.; Bucher, H.U.; Fauchere, J.-C. Effect of early prophylactic high-dose recombinant human erythropoietin in very preterm infants on neurodevelopmental outcome at 2 years. A randomized clinical trial. JAMA 2016, 315, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Kang, W.; Xu, F.; Cheng, X.; Zhang, Z.; Jia, L.; Ji, L.; Guo, X.; Xiong, H.; Simbruner, G.; et al. Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrics 2009, 124, e218–e226. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.-Y.; Wu, S.-J.; Wang, Q.-L.; Yang, L.-H.; Ren, P.-S.; Qiao, B.-J.; Wang, Z.-Y.; Li, J.-H.; Gu, X.-L.; Li, L.-X. Effect of erythropoietin combined with hypothermia on serum tau protein levels and neurodevelopmental outcome in neonates with hypoxic-ischemic encephalopathy. Neural Regen. Res. 2017, 12, 1655–1663. [Google Scholar] [PubMed]
- Elmahdy, H.; El-Mashad, A.-R.; El-Bahrawy, H.; El-Gohary, T.; El-Barbary, A.; Aly, H. Human recombinant erythropoietin in asphyxia neonatorum: Pilot trial. Pediatrics 2010, 125, e1135–e1142. [Google Scholar] [CrossRef] [PubMed]
- Avasiloaiei, A.; Dimitriu, C.; Moscalu, M.; Paduraru, L.; Stamatin, M. High-dose phenobarbital or erythropoietin for the treatment of perinatal asphyxia in term newborns. Pediatr. Int. 2013, 55, 589–593. [Google Scholar] [CrossRef]
- El Shimi, M.S.; Awad, H.A.; Hassanein, S.M.; Gad, G.I.; Imam, S.S.; Shaaban, H.; El Maraghy, O. Single dose recombinant erythropoietin versus moderate hypothermia for neonatal hypoxic ischemic encephalopathy in low resource settings. J. Matern. Fetal Neonatal Med. 2014, 27, 1295–1300. [Google Scholar] [CrossRef]
- Malla, R.R.; Asimi, R.; Teli, M.A.; Shaheen, F.; Bhat, M.A. Erythropoietin monotherapy in perinatal asphyxia with moderate to severe encephalopathy: A randomized placebo-controlled trial. J. Perinatol. 2017, 37, 596–601. [Google Scholar] [CrossRef]
- Frankenburg, W.K.; Doods, J.; Arhcer, P.; Shapiro, H.; Bresnick, B. The Denver II: A major revision and restandardization of the Denver Developmental Screening Test. Pediatrics 1992, 89, 91–97. [Google Scholar]
- Garg, B.; Sharma, D.; Bansal, A. Systematic review seeking erythropoietin role for neuroprotection in neonates with hypoxic ischemic encephalopathy: Presently where do we stand. J. Matern. Fetal Neonatal Med. 2018, 31, 3214–3224. [Google Scholar] [CrossRef] [PubMed]
- Baserga, M.C.; Beachy, J.C.; Roberts, J.K.; Ward, R.M.; DiGeronimo, R.J.; Walsh, W.F.; Ohls, R.K.; Anderson, J.; Mayock, D.E.; Juul, S.E.; et al. Darbepoetin administration to neonates undergoing cooling for encephalopathy: A safety and pharmacokinetic trial. Pediatr. Res. 2015, 78, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Valera, I.T.; Vázquez, M.D.; González, M.D.; Jaraba, M.P.; Benitez, M.V.R.; Morano, C.C.; Laso, E.L.; Cabanas, J.M.G.; Quiles, M.J.P. Erythropoietin with hypothermia improves outcomes in neonatal hypoxic ischemic encephalopathy. J. Clin. Neonatol. 2015, 4, 244–249. [Google Scholar]
- Rogers, E.E.; Bonifacio, S.L.; Glass, H.C.; Juul, S.E.; Chang, T.; Mayock, D.E.; Durand, D.J.; Song, D.; Barkovich, A.J.; Ballard, R.A.; et al. Erythropoietin and hypothermia for hypoxic-ischemic encephalopathy. Pediatr. Neurol. 2014, 51, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Mulkey, S.B.; Ramakrishnaiah, R.H.; Mckinstry, R.C.; Chang, T.; Mathur, A.M.; Mayock, D.E.; Van Meurs, K.P.; Schaefer, G.B.; Luo, C.; Bai, S.; et al. Erythropoietin and brain magnetic resonance imaging findings in hypoxic-ischemic encephalopathy: Volume of acute brain injury and 1-year neurodevelopmental outcome. J. Pediatr. 2017, 186, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Mathir, A.M.; Chang, T.; McKinstry, R.C.; Mulkey, S.B.; Mayock, D.E.; Van Meurs, K.P.; Rogers, E.E.; Gonzalez, F.F.; Comstock, B.A.; et al. High-dose erythropoietin and hypothermia for hypoxic-ischemic encephalopathy: A phase II trial. Pediatrics 2016, 137. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Bauer, L.A.; Ballard, R.A.; Ferriero, D.M.; Glidden, D.V.; Mayock, D.E.; Chang, T.; Durand, D.J.; Song, D.; Bonifacio, S.L.; et al. Erythropoietin for neuroprotection in neonatal encephalopathy: Safety and pharmacokinetics. Pediatrics 2012, 130, 683–691. [Google Scholar] [CrossRef]
- Statler, P.A.; McPherson, R.J.; Bauer, L.A.; Kellert, B.A.; Juul, S.E. Pharmacokinetics of high-dose recombinant erythropoietin in plasma and brain of neonatal rats. Pediatr. Res. 2007, 61, 671–675. [Google Scholar] [CrossRef]
- Wu, Y.W.; Goodman, A.M.; Chang, T.; Mulkey, S.B.; Gonzalez, F.F.; Mayock, D.E.; Juul, S.E.; Mathur, A.M.; Van Meurs, K.; McKinstry, R.C.; et al. Placental pathology and neonatal brain MRI in a randomized trial of erythropoietin for hypoxic-ischemic encephalopathy. Pediatr. Res. 2019. [Google Scholar] [CrossRef]
- Juul, S.E.; Comstock, B.A.; Heagerty, P.J.; Mayock, D.E.; Goodman, A.M.; Hauge, S.; Gonzalez, F.; Wu, Y.W. High-dose erythropoietin for asphyxia and encephalopathy (HEAL): A randomized controlled trial—background, aims, and study protocol. Neonatology 2018, 113, 331–338. [Google Scholar] [CrossRef]
- Liley, H. Preventing adverse outcomes of neonatal hypoxic ischaemic encephalopathy with erythropoietin: A phase III randomised placebo controlled multi-centre clinical trial. ClinicalTrials.gov Identifier 2017, NCT03079167. [Google Scholar]
- Rangarajan, V.R.; Juul, S.E. Erythropoietin: Emerging role of erythropoietin in neonatal neuroprotection. Pediatr. Neurol. 2014, 51, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Storring, P.L.; Tiplady, R.J.; Gaines Das, R.E.; Stenning, B.E.; Lamikanra, A.; Rafferty, B.; Lee, J. Epoetin alfa and beta differ in their erythropoietin isoform compositions and biological properties. Br. J. Haematol. 1998, 100, 79–89. [Google Scholar] [PubMed]
- Wang, S.-Y. Effect of mild hypothermia combined with VitC and EPO therapy on target organ damage in children with neonatal asphyxia. J. Hainan Med. Univ. 2017, 23, 117–120. [Google Scholar]
- Nonomura, M.; Harada, S.; Asada, Y.; Matsumura, H.; Iwami, H.; Tanaka, Y.; Ichiba, H. Combination therapy with erythropoietin, magnesium sulfate and hypothermia for hypoxic-ischemic encephalopathy: An open-label pilot study to assess the safety and feasibility. BMC Pediatr. 2019, 19, 13. [Google Scholar] [CrossRef]
- McDonald, J.W.; Silverstein, F.S.; Johnston, M.V. Magnesium reduces N-methyl-D-asparate (NMDA)-mediated brain injury in perinatal rats. Neurosci. Lett. 1990, 109, 234–238. [Google Scholar] [CrossRef]
- Robertson, N.J.; Tan, S.; Groenendaal, F.; van Bel, F.; Juul, S.E.; Bennet, L.; Derrick, M.; Back, S.A.; Valdez, R.C.; Northington, F.; et al. Which neuroprotective agents are ready for bench to bedside translation in the newborn infant? J. Pediatr. 2012, 160, 544–552. [Google Scholar] [CrossRef]
- Mazur, M.; Miller, R.M.; Robinson, S. Postnatal erythropoietin treatment mitigates neural cell loss after systemic prenatal hypoxic-ischemic injury. J. Neurosurg. Pediatr. 2010, 6, 206–221. [Google Scholar] [CrossRef]
- Polglase, G.R.; Barton, S.K.; Melville, J.M.; Zahra, V.; Wallace, M.J.; Siew, M.L.; Tolcos, M.; Moss, T.J.M. Prophylactic erythropoietin exacerbates ventilation-induced lung inflammation and injury in preterm lambs. J. Physiol. 2014, 592, 1993–2002. [Google Scholar] [CrossRef]
- Barton, S.K.; McDougall, A.R.A.; Melville, J.M.; Moss, T.J.M.; Zahra, V.; Lim, T.; Crossley, K.J.; Polglase, G.R.; Tolcos, M. Differential short-term regional effects of early high dose erythropoietin on white matter in preterm lambs after mechanical ventilation. J. Physiol. 2016, 594, 1437–1449. [Google Scholar] [CrossRef]
- Chan, K.Y.Y.; LaRosa, D.A.; Tolcos, M.; Li, A.; Zahra, V.; Polglase, G.R.; Barton, S.K. Optimizing the dose of erythropoietin required to prevent acute ventilation-induced cerebral white matter injury in preterm lambs. Dev. Neurosci. 2017, 39, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Allison, B.J.; LaRosa, D.A.; Barton, S.K.; Hooper, S.; Zahra, V.; Tolcos, M.; Chan, K.Y.Y.; Barbuto, J.; Inocencio, I.M.; Moss, T.J.M.; et al. Dose-dependent exacerbation of ventilation-induced lung injury by erythropoietin in preterm newborn lambs. J. Appl. Physiol. 2019, 126, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, A.P.; Voss, W.; Wachtendor, M.; Jungmann, T. Erythropoietin improves neurodevelopmental outcome of extremely preterm infants. Ann. Neurol. 2010, 67, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Ohls, R.K.; Kamath-Rayne, B.D.; Christensen, R.D.; Wiedmeier, S.E.; Rosenberg, A.; Fuller, J.; Lacy, C.B.; Roohi, M.; Lambert, D.K.; Burnett, J.J.; et al. Cognitive outcomes of preterm infants randomized to darbepoetin, erythropoietin, or placebo. Pediatrics 2014, 133, 1023–1030. [Google Scholar] [CrossRef]
- Leuchter, R.H.-V.; Gui, L.; Poncet, A.; Hagmann, C.; Lodygensky, G.A.; Martin, E.; Koller, B.; Darque, A.; Bucher, H.U.; Huppi, P.S. Association between early administration of high-dose erythropoietin in preterm infants and brain MRI abnormality at term-equivalent age. JAMA 2014, 312, 817–824. [Google Scholar] [CrossRef]
- Fischer, H.S.; Reibel, N.J.; Bührer, C.; Dame, C.P. Prophylactic early erythropoietin for neuroprotection in preterm infants: A meta-analysis. Pediatrics 2017, 139, e20164317. [Google Scholar] [CrossRef]
- Juul, S.E.; Comstock, B.A.; Wadhawan, R.; Mayock, D.E.; Courtney, S.E.; Robinson, T.; Ahmad, K.A.; Bendel-Stenzel, E.; Baserga, M.; LaGamma, E.F.; et al. A randomized trial of erythropoietin for neuroprotection in preterm infants. N. Engl. J. Med. 2020, 382, 233–243. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study | Animal Model, Age and Species | Gender | Dose | Type of EPO | Timing of Administration | Route of Administration | Key Findings: Short-Term Findings in Italics |
|---|---|---|---|---|---|---|---|
| Matsushita et al. 2003 [71] | Rice-Vannucci, PN7 mice | Unknown | 1 U/g or 5 U/g | Unknown | 1 h before hypoxia | Intraperitoneal (i.p.) | Decreased infarct volume or cerebral injury score at 24 h and 7 days post-injury |
| Aydin et al. 2003 [72] | Rice-Vannucci, PN 7 rat | Unknown | 20 U | r-Hu-EPO | Immediate post-treatment | Intracerebroventricular (ICV) | EPO decreased brain infarct volume at 7 days post-injury |
| Kumral et al. 2003 [73] | Rice-Vannucci, PN7 rat | Unknown | 1000 U/kg | r-Hu-EPO | Immediate post-treatment | i.p. | EPO decreased brain infarct volume at 3 days post-injury |
| Kumral et al. 2004 [48] | Rice-Vannucci, PN7 rat | Unknown | 1000 U/kg | r-Hu-EPO | Immediate post-treatment | i.p. | Decreased overproduction of cerebral nitric oxide |
| Kumral et al. 2005 [53] | Rice-Vannucci, PN7 rat | Unknown | 1000 U/kg | r-Hu-EPO | Immediate post-treatment | i.p. | Decreased lipid peroxidation; Increased activity of antioxidant enzyme |
| Kumral et al. 2004 [60] | Rice-Vannucci, PN7 rat | Unknown | 1000 U/kg | r-Hu-EPO | Immediate post-treatment | i.p. | 20 weeks post-injury: Improved memory and cerebral volume |
| McClure et al. 2007 [82] | Rice-Vannucci, PN7 rat | Male | 300 or 1000 U/kg | Unknown | Immediate post-treatment | i.p. | 2–3 months post-injury: Improved memory, rapid auditory processing |
| Alexander et al. 2012 [83] | Rice-Vannucci, PN 7 rat | Male | 1000 U/kg | Unknown | Immediate post-treatment, or delayed treatment at 1 h or 3 h | i.p. | Immediate, but not delayed, treatment with EPO has therapeutic benefit for auditory processing |
| Spandou et al. 2005 [84] | Rice-Vannucci, PN 7 rat | Unknown | 2000 U/kg | Unknown | Immediate post-treatment | i.p. | 6 weeks post-injury: Improved sensorimotor function; Reduced brain damage |
| Demers et al. 2005 [85] | Rice-Vannucci, PN 7 rat, 1.5 hypoxia | Unknown | 2500 U/kg | rEPO | Repeated daily, 3 days post-injury | Subcutaneous (s.c.) | 3 weeks post-injury: Prevented rotation, reduced sensory neglect. No effect on overall brain injury score. |
| van der Kooij et al. 2009 [86] | Rice-Vannucci, PN 7 rat, 1.5 hypoxia | Unknown | 1000 U/kg | r-Hu-EPO, epoetin-alfa | Repeated daily, 0 h, 24 h, 48 h post-injury | i.p. | No effect on cerebral white or grey matter damage |
| Fan et al. 2011 [87] | Rice-Vannucci, PN 9 mice, 45 min hypoxia | Male and female | 5000 U/kg | Unknown | Repeated daily, 0 h, 24 h, 48 h post-injury | i.p. | At 72 h, Increased progenitor cells in subventricular zone and dentate gyrus in females but not males |
| Wen et al. 2006 [88] | Neonatal stroke, PN7 rat | Male and female | 1000 U/kg | Unknown | Repeated daily, 15 min, 24 h, 48 h post-injury | i.p. | Long-term neuroprotection-more beneficial in females |
| Sola et al. 2005 [89] | Neonatal stroke, PN7 rat | Unknown | 100 or 1000, 5000 U/kg | r-Hu-EPO | Repeated, after 15 min and on days 1 and 2 post-stroke | i.p. | Reduced cerebral infarct volume at 3 days post-stroke |
| Chang et al. 2005 [90] | Neonatal stroke, PN10 rat | Unknown | 5000 U/kg | r-Hu-EPO | Immediately after hypoxia | i.p. | Short-term improvements in sensorimotor outcomes |
| Gonzalez et al. 2009 [91] | Neonatal stroke, PN10 rat | Unknown | 1000 U/kg | r-Hu-EPO | Repeated: 0 h, 24 h, 7 days post-injury | i.p. | No long-term difference in memory at 3-months compared to shams |
| Gonzalez et al. 2013 [92] | Neonatal stroke, PN7 rat | Unknown | 1000 U/kg | r-Hu-EPO | Repeated: 0 h, 24 h, 7 days post-injury | i.p. | Increased neurogenesis and oligodendrogenesis after EPO |
| Kellert et al. 2007 [93] | Rice-Vannucci, PN 7 rat | Unknown | 5000 or 30,000 U/kg | r-Hu-EPO, epoetin-alfa | Repeated: 1, 3 or 7 daily injections, started immediately after hypoxia | s.c. | Maximal benefit against brain injury with 3 doses of 5000 U/kg or 1 dose of 30,000 U/kg |
| Iwai et al. 2007 [94] | Rice-Vannucci, PN 7 rat | Unknown | 1000 U/kg | r-Hu-EPO | Repeated: 20 min, 2, 4 & 6 days post-injury | i.p. | EPO decreased cerebral infarct volume 1 week later, and improved motor outcomes 4 weeks later |
| Sun et al. 2005 [95] | Rice-Vannucci, PN 7 rat | Unknown | 5000 U/kg | r-Hu-EPO | Repeated: 24 h, 48 h, 72 h post-injury | i.p. | EPO neuroprotective and decreased the inflammatory response |
| Iwai et al. 2010 [96] | Rice-Vannucci, PN 7 rat | Unknown | 1000 U/kg | r-Hu-EPO | Repeated: 2, and for 4 days until 13 days post-injury | i.p. | EPO did not prevent brain volume loss, but improved oligodendro-genesis and sensorimotor function at day 14 post-injury |
| Chen et al. 2012 [97] | Rice-Vannucci, PN 10 rat | Unknown | 300 U/kg | Nano-EPO | Repeated: 1 h, 48 h, 48 h post-injury | i.p. | Similar outcomes to 5000 U/kg: EPO neuroprotective at 72 h post-injury, and improved functional deficit at 21 days post-injury |
| Study | Animal Model, Age and Species | Gender | Dose | Type of EPO | Timing of Administration | Route of Administration | Key Findings |
|---|---|---|---|---|---|---|---|
| Fan et al. 2013 [105] | Rice-Vannucci, PN7 rat | Males and females | 5000 U/kg | EPREX | Immediately after 3 h of hypothermia (4 °C decrease) | Intraperitoneal (i.p.) | Combined treatment had only a borderline (p = 0.07) neuroprotective effect at 6 weeks-of-age |
| Fang et al. 2013 [106] | Rice-Vannucci, PN 7 rat | Unknown | 1000 U/kg | r-Hu-EPO | EPO post-treatment at 0 h, 24 h and 7 days; hypothermia at 1–9 h post-hypoxia | i.p. | No adverse effects of combined treatment at 2- and 6-weeks post-injury |
| Traudt et al. 2013 [107] | Non-human primate model of perinatal asphyxia | Unknown | 1000, 2500 or 3500 U/kg | r-Hu-EPO | EPO post-treatment at 0.5 h, 24 h, 48 h and 7 days; hypothermia at 3–75 h post-hypoxia | Intravenous (i.v.); Either at 3500 U/kg for the first dose and then 2500 U/kg thereafter or at 1000 U/kg for all four doses | Hypothermia combined with 4 doses of EPO significantly decreased the risk of cerebral palsy at 9 months-of-age |
| McAdams et al. 2017 [108] | Non-human primate model of perinatal asphyxia | Unknown | 1000, 2500 or 3500 U/kg | r-Hu-EPO | EPO post-treatment at 0.5 h, 24 h, 48 h and 7 days; hypothermia at 3–75 h post-hypoxia | i.v. | Hypothermia combined with 4 doses of EPO significantly decreased neuropathology at 9 months-of-age |
| Study | Number of Term Newborn Infants | Gender | Dose | Type of EPO | Timing of Administration | Route of Administration | Key Findings |
|---|---|---|---|---|---|---|---|
| Zhu et al. 2009 [114] | 167, born between August 2003 and January 2007, with either moderate or severe hypoxic-ischemic encephalopathy (HIE) | Males and females | 300 or 500 U/kg (n = 83) or con-ventional (n = 84) | r-Hu-EPO | 1–48 h after birth for 1st dose; then every other day for 2 weeks | Subcutaneously (s.c.) for 1st dose; Intravenous (i.v.) thereafter | When 18-months-old, improved long-term outcomes after EPO treatment in the infants with moderate HIE, but not in those with severe HIE |
| Elmahdy et al. 2010 [116] | 45, 3 groups: normal (n = 15), HIE with conventional treatment * (n = 15), or HIE with EPO treatment (n = 15) | Males and females | 2500 U/kg | r-Hu-EPO | 4–6 h after birth for 1st dose; then daily for 4 days | s.c. | When 2-weeks-old for HIE infants, EPO decreased nitric oxide concentration and breakthrough seizures compared to conventional treatment. When 6-month-old for HIE infants, EPO decreased neurologic and developmental abnormalities. |
| Avasiloaiei et al. 2013 [117] | 67, 3 HIE groups treated with EPO & supportive care ** (n = 22), or phenobarbital (n = 22) or supportive care alone (n = 23) | Unknown | 1000 IU/kg | EPO | EPO post-treatment during the 1st 3 days | s.c. | When 18-months-old, neurodevelop-mental delay was lower in both the EPO and phenobarbital treatment groups, although the differences were not statistically analyzed |
| El Shimi et al. 2014 [118] | 45, HIE/EPO (n = 15), HIE/hypothermia (n = 15), normal (n = 15) | Males and females | 1500 U/kg | r-Hu-EPO | Single dose on postnatal day 1 | s.c. | When 3-months-old, no significant differences in neuromuscular function nor brain MRI score |
| Malla et al. 2017 [119] | 100, HIE/EPO (n = 50), HIE/placebo (saline, n = 50) | Males and females | 500 U/kg | r-Hu-EPO | 1st dose within 6 h of birth; then every other day for a total of 5 doses | i.v. | When 19-months-old, the EPO-treated group had a lower risk of cerebral palsy. EPO also decreased death |
| Study | Number of Term Newborn Infants | Gender | Dose | Type of EPO | Timing of Administration | Route of Administration | Key Findings |
|---|---|---|---|---|---|---|---|
| Baserga et al. 2015 [122] | 30, with hypoxic-ischemic encephalopathy (HIE), 3 groups, placebo (n = 10), EPO (low dose, n = 10), EPO (high dose, n = 10) | Males and females | 2 or 10 U/kg | darbe-poietin | EPO within 12 h of birth, and a 2nd dose 7 days later; Hypothermia started within 11 h of birth and given for 72 h | Intravenous (i.v.) | HT combined with EPO was safe. Weekly administration of darbepoietin was sufficient |
| Valera et al. 2015 [123] | 15, HIE and treated with EPO and moderate hypothermia | Males and females | 400 U/kg | r-Hu-EPO | Every 48 h for 2 weeks, commencing within 3 h of birth, along with hypothermia for 72 h | i.v. | When 18-months-old, 80% survival with no neurodevelopmental disability. Unfortunately, no control group |
| Rogers et al. 2014 [124] | 24, HIE and treated with EPO and moderate hypothermia; 250 U/kg EPO (n = 3), 500 (n = 6), 1000 (n = 7), 2500 (n = 8) | Males and females | 250 to 2500 U/kg | r-Hu-EPO | EPO at 24 h after birth, and then every 48 h; Hypothermia started within 6 h of birth and given for 72 h | i.v. | When 8-34-months-old, neurodevelopmental delay was lower compared to treatment with hypothermia alone. However, study statistically underpowered to detect a statistical difference |
| Mulkey et al. 2017 [125] | 44, HIE; In addition to treatment with moderate hypothermia, treated with EPO (n = 20) or saline (n = 24) | Males and females | 1000 U/kg | r-Hu-EPO | EPO on postnatal day 1 (at <24 h), 2&3 (n = 11), plus day 5 (n = 8), plus day 7 (n = 1); Hypothermia started within 6 h of birth and given for 72 h | i.v. | Statistically significant lower volume of acute brain injury in the EPO-hypothermia-treated group compared with the saline-hypothermia group |
| Wu et al. 2016 [126] | 50, HIE; In addition to treatment with moderate hypothermia, treated with EPO (n = 24) or saline (n = 26) | Males and females | 1000 U/kg | r-Hu-EPO | EPO on postnatal day 1 (at <24 h), 2, 3, 5 and 7; Hypothermia started within 6 h of birth and given for 72 h | i.v. | Significantly less brain injury at 5 days-of-age, and better 12-month motor outcomes, in the EPO-hypothermia-treated group compared with the saline-hypothermia group |
| Juul et al. 2018 [130] | Recruiting 500, HIE; In addition to treatment with moderate hypothermia, treated with EPO or saline | Males and females | 1000 U/kg | r-Hu-EPO | EPO on postnatal day 1 (at <24 h), 2, 3, 5 and 7; Hypothermia started within 6 h of birth and given for 72 h | i.v. | Assessment up to 24 months-of-age. Hypothesize that EPO in combination with hypothermia reduces mortality and neurodevelopmental disability |
| Patkai et al. 2014 [132] | Recruiting 120, HIE; In addition to treatment with moderate hypothermia, treated with EPO or saline | Males and females | 1000–1500 U/kg | beta r-Hu-EPO | EPO on postnatal day 1 (at <12 h), 2 and 3 (each 24 h after previous dose); Hypothermia started within 6 h of birth and given for 72 h | i.v. | Assessment up to 24 months-of-age. Hypothesize that EPO in combination with hypothermia increases survival and reduces neurological sequelae |
| Wang 2017 [134] | 68, with HIE, 2 groups, moderate hypothermia, EPO & Vitamin C (n = 34), EPO & Vitamin C (n = 34) | Males and females | 500 U/kg | r-Hu-EPO | EPO given 3 times per week (start time unknown); Vitamin C given once per day; Hypothermia given for 72 h | i.v. for EPO and Vitamin C (250 mg/kg) | Mild hypothermia, EPO & Vitamin C combined more effective. This was achieved through decreased apoptosis and oxygen free radicals, and increased antioxidant capacity |
| Nonomura et al. 2019 [135] | 9, with severe HIE, moderate hypothermia, EPO & magnesium sulfate (Mg) | Males and females | 300 U/kg | Epoietin alfa | EPO & Mg given within 6 h of birth, then every other day for 2 weeks. Hypothermia started within 6 h of birth, for 72 h | i.v. for EPO and Mg (250 mg/kg) | No deaths and all 9 neonates did not have any serious adverse effects |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oorschot, D.E.; Sizemore, R.J.; Amer, A.R. Treatment of Neonatal Hypoxic-Ischemic Encephalopathy with Erythropoietin Alone, and Erythropoietin Combined with Hypothermia: History, Current Status, and Future Research. Int. J. Mol. Sci. 2020, 21, 1487. https://doi.org/10.3390/ijms21041487
Oorschot DE, Sizemore RJ, Amer AR. Treatment of Neonatal Hypoxic-Ischemic Encephalopathy with Erythropoietin Alone, and Erythropoietin Combined with Hypothermia: History, Current Status, and Future Research. International Journal of Molecular Sciences. 2020; 21(4):1487. https://doi.org/10.3390/ijms21041487
Chicago/Turabian StyleOorschot, Dorothy E., Rachel J. Sizemore, and Ashraf R. Amer. 2020. "Treatment of Neonatal Hypoxic-Ischemic Encephalopathy with Erythropoietin Alone, and Erythropoietin Combined with Hypothermia: History, Current Status, and Future Research" International Journal of Molecular Sciences 21, no. 4: 1487. https://doi.org/10.3390/ijms21041487
APA StyleOorschot, D. E., Sizemore, R. J., & Amer, A. R. (2020). Treatment of Neonatal Hypoxic-Ischemic Encephalopathy with Erythropoietin Alone, and Erythropoietin Combined with Hypothermia: History, Current Status, and Future Research. International Journal of Molecular Sciences, 21(4), 1487. https://doi.org/10.3390/ijms21041487