Evaluation of Toxic Amyloid β42 Oligomers in Rat Primary Cerebral Cortex Cells and Human iPS-derived Neurons Treated with 10-Me-Aplog-1, a New PKC Activator

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. APP Expression Levels in Cultured Neuronal Cell Lines Treated with 1
2.2. Effects of 1 on Extracellular Aβ42/Aβ40 and Aβ Oligomerization in Rat Primary Cerebral Cortex Cells
2.3. Effects of 1 on Aβ Production and Degradation in Rat Primary Cerebral Cortex Cells
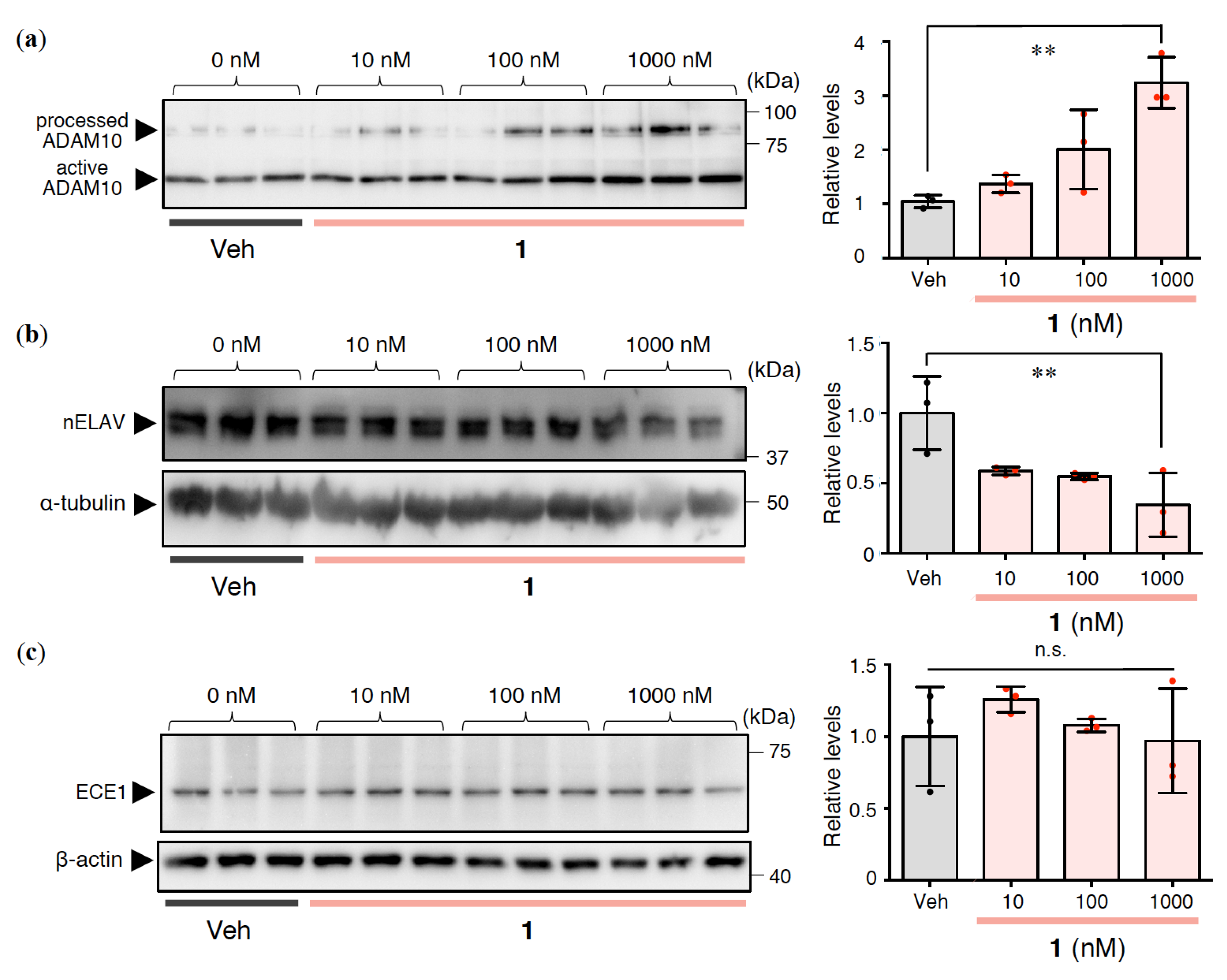
2.4. Effects of 1 on Intracellular Aβ Oligomerization in Rat Primary Cerebral Cortex Cells
2.5. Effects of 1 on the Cytotoxicity of Rat Primary Cerebral Cortex Cells
2.6. Effects of 1 on Aβ42/Aβ40, Aβ Oligomerization, and Neurotoxicity in Human iPS-Derived Neurons
3. Discussion
4. Materials and Methods
4.1. Rat Primary Cerebral Cortex Cells
4.2. ELISA
4.3. Western Blotting
4.4. MTT Assay
4.5. Generation and Characterization of Human iPS-Derived Neurons
4.6. Electrochemiluminescence Assays
4.7. ToxiLight Assay
4.8. Statistical Analyses
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid β-protein |
| AD | Alzheimer’s disease |
| ADAM10 | A disintegrin and metalloproteinase 10 |
| APP | Amyloid β precursor protein |
| BACE1 | β-site amyloid β precursor protein cleaving enzyme 1 |
| DMSO | Dimethyl sulfoxide |
| ECE1 | Endothelin converting enzyme 1 |
| ELISA | Enzyme-linked immunosorbent assay |
| E-MEM | Eagle’s minimal essential medium |
| FBS | Fetal bovine serum |
| HFIP | 1,1,1,3,3,3-hexafluoro-2-propanol |
| HRP | Horseradish peroxidase |
| HS | Horse serum |
| iPS | Induced pluripotent stem |
| MCI | Mild cognitive impairment |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | Phosphate-buffered saline |
| PKC | Protein kinase C |
| sAPP | Secreted amyloid β precursor protein |
| WB | Western blotting |
References
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.J.; Kellett, K.A.; Thinakaran, G.; Hooper, N.M. A Greek Tragedy: The growing complexity of Alzheimer amyloid precursor protein proteolysis. J. Biol. Chem. 2016, 291, 19235–19244. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid β-protein assembly and Alzheimer disease. J. Biol. Chem. 2009, 284, 4749–4753. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995, 9, 484–496. [Google Scholar] [CrossRef]
- Sun, M.K.; Alkon, D.L. The “memory kinases”: roles of PKC isoforms in signal processing and memory formation. Prog. Mol. Biol. Transl. Sci. 2014, 122, 31–59. [Google Scholar]
- Govoni, S.; Bergamaschi, S.; Racchi, M.; Battaini, F.; Binetti, G.; Bianchetti, A.; Trabucchi, M. Cytosol protein kinase C downregulation in fibroblasts from Alzheimer’s disease patients. Neurology 1993, 43, 2581–2586. [Google Scholar] [CrossRef]
- Yang, H.Q.; Pan, J.; Ba, M.W.; Sun, Z.K.; Ma, G.Z.; Lu, G.Q.; Xiao, Q.; Chen, S.D. New protein kinase C activator regulates amyloid precursor protein processing in vitro by increasing α-secretase activity. Eur. J. Neurosci. 2007, 26, 381–391. [Google Scholar] [CrossRef]
- Etcheberrigaray, R.; Tan, M.; Dewachter, I.; Kuiperi, C.; Van der Auwera, I.; Wera, S.; Qiao, L.; Bank, B.; Nelson, T.J.; Kozikowski, A.P.; et al. Therapeutic effects of PKC activators in Alzheimer’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2004, 101, 11141–11146. [Google Scholar] [CrossRef] [PubMed]
- Pascale, A.; Amadio, M.; Scapagnini, G.; Lanni, C.; Racchi, M.; Provenzani, A.; Govoni, S.; Alkon, D.L.; Quattrone, A. Neuronal ELAV proteins enhance mRNA stability by a PKCα-dependent pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 12065–12070. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Wang, D.; Yu, G.Q.; Zhu, G.; Kharazia, V.N.; Paredes, J.P.; Chang, W.S.; Deitchman, J.K.; Mucke, L.; Messing, R.O. PKCε increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8215–8220. [Google Scholar] [CrossRef] [PubMed]
- Hongpaisan, J.; Sun, M.K.; Alkon, D.L. PKCε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J. Neurosci. 2011, 31, 630–643. [Google Scholar] [CrossRef]
- Pacheco-Quinto, J.; Eckman, E.A. Endothelin-converting enzymes degrade intracellular β-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. J. Biol. Chem. 2013, 288, 5606–5615. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Khan, T.K.; Nelson, T.J.; Verma, V.A.; Wender, P.A.; Alkon, D.L. A cellular model of Alzheimer’s disease therapeutic efficacy: PKC activation reverses Aβ-induced biomarker abnormality on cultured fibroblasts. Neurobiol. Dis. 2009, 34, 332–339. [Google Scholar] [CrossRef]
- Alkon, D.L.; Sun, M.K.; Nelson, T.J. PKC signaling deficits: a mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci. 2007, 28, 51–60. [Google Scholar] [CrossRef]
- Wender, P.A.; Hardman, C.T.; Ho, S.; Jeffreys, M.S.; Maclaren, J.K.; Quiroz, R.V.; Ryckbosch, S.M.; Shimizu, A.J.; Sloane, J.L.; Stevens, M.C. Scalable synthesis of bryostatin 1 and analogs, adjuvant leads against latent HIV. Science 2017, 358, 218–223. [Google Scholar] [CrossRef]
- Trost, B.M.; Dong, G. Total synthesis of bryostatin 16 using atom-economical and chemoselective approaches. Nature 2008, 456, 485–488. [Google Scholar] [CrossRef]
- Kato, Y.; Scheuer, P.J. Aplysiatoxin and debromoaplysiatoxin, constituents of the marine mollusk Stylocheilus longicauda (Quoy and Gaimard, 1824). J. Am. Chem. Soc. 1974, 96, 2245–2246. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Yanagita, R.C.; Hamada, N.; Murakami, A.; Takahashi, H.; Saito, N.; Nagai, H.; Irie, K. A simple analogue of tumor-promoting aplysiatoxin is an antineoplastic agent rather than a tumor promoter: development of a synthetically accessible protein kinase C activator with bryostatin-like activity. Am. Chem. Soc. 2009, 131, 7573–7579. [Google Scholar] [CrossRef] [PubMed]
- Kikumori, M.; Yanagita, R.C.; Tokuda, H.; Suzuki, N.; Nagai, H.; Suenaga, K.; Irie, K. Structure-activity studies on the spiroketal moiety of a simplified analogue of debromoaplysiatoxin with antiproliferative activity. J. Med. Chem. 2012, 55, 5614–5626. [Google Scholar] [CrossRef]
- Blennow, K.; Mattsson, N.; Scholl, M.; Hansson, O.; Zetterberg, H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol. Sci. 2015, 36, 297–309. [Google Scholar] [CrossRef]
- Pike, K.E.; Savage, G.; Villemagne, V.L.; Ng, S.; Moss, S.A.; Maruff, P.; Mathis, C.A.; Klunk, W.E.; Masters, C.L.; Rowe, C.C. β-Amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain 2007, 130, 2837–2844. [Google Scholar] [CrossRef]
- Rowe, C.C.; Bourgeat, P.; Ellis, K.A.; Brown, B.; Lim, Y.Y.; Mulligan, R.; Jones, G.; Maruff, P.; Woodward, M.; Price, R.; et al. Predicting Alzheimer disease with β-amyloid imaging: results from the Australian imaging, biomarkers, and lifestyle study of ageing. Ann. Neurol. 2013, 74, 905–913. [Google Scholar] [CrossRef]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Figurski, M.; Coart, E.; Blennow, K.; Soares, H.; Simon, A.J.; Lewczuk, P.; Dean, R.A.; et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol. 2011, 121, 597–609. [Google Scholar] [CrossRef]
- Irie, K. New diagnostic method for Alzheimer’s disease based on the toxic conformation theory of amyloid β. Biosci. Biotechnol. Biochem. 2020, 84, 1–16. [Google Scholar] [CrossRef]
- Murakami, K.; Tokuda, M.; Suzuki, T.; Irie, Y.; Hanaki, M.; Izuo, N.; Monobe, Y.; Akagi, K.; Ishii, R.; Tatebe, H.; et al. Monoclonal antibody with conformational specificity for a toxic conformer of amyloid β42 and its application toward the Alzheimer’s disease diagnosis. Sci. Rep. 2016, 6, 29038. [Google Scholar] [CrossRef]
- Akiba, C.; Nakajima, M.; Miyajima, M.; Ogino, I.; Motoi, Y.; Kawamura, K.; Adachi, S.; Kondo, A.; Sugano, H.; Tokuda, T.; et al. Change of amyloid-β1-42 toxic conformer ratio after cerebrospinal fluid diversion predicts long-term cognitive outcome in patients with idiopathic normal pressure hydrocephalus. J. Alzheimers Dis. 2018, 63, 989–1002. [Google Scholar] [CrossRef]
- Masuda, Y.; Uemura, S.; Ohashi, R.; Nakanishi, A.; Takegoshi, K.; Shimizu, T.; Shirasawa, T.; Irie, K. Identification of physiological and toxic conformations in Aβ42 aggregates. ChemBioChem. 2009, 10, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Trejo, J.; Massamiri, T.; Deng, T.; Dewji, N.N.; Bayney, R.M.; Brown, J.H. A direct role for protein kinase C and the transcription factor Jun/AP-1 in the regulation of the Alzheimer’s β-amyloid precursor protein gene. J. Biol. Chem. 1994, 269, 21682–21690. [Google Scholar] [PubMed]
- Lahiri, D.K.; Nall, C. Promoter activity of the gene encoding the β-amyloid precursor protein is up-regulated by growth factors, phorbol ester, retinoic acid and interleukin-1. Brain Res. Mol. Brain Res. 1995, 32, 233–240. [Google Scholar] [CrossRef]
- Sarajarvi, T.; Jantti, M.; Paldanius, K.M.A.; Natunen, T.; Wu, J.C.; Makinen, P.; Tarvainen, I.; Tuominen, R.K.; Talman, V.; Hiltunen, M. Protein kinase C-activating isophthalate derivatives mitigate Alzheimer’s disease-related cellular alterations. Neuropharmacology 2018, 141, 76–88. [Google Scholar] [CrossRef]
- Sato, M.; Murakami, K.; Uno, M.; Nakagawa, Y.; Katayama, S.; Akagi, K.; Masuda, Y.; Takegoshi, K.; Irie, K. Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the Lys residues. J. Biol. Chem. 2013, 288, 23212–23224. [Google Scholar] [CrossRef]
- Yoshioka, T.; Murakami, K.; Ido, K.; Hanaki, M.; Yamaguchi, K.; Midorikawa, S.; Taniwaki, S.; Gunji, H.; Irie, K. Semisynthesis and structure-activity studies of uncarinic acid C isolated from Uncaria rhynchophylla as a specific inhibitor of the nucleation phase in amyloid β42 aggregation. J. Nat. Prod. 2016, 79, 2521–2529. [Google Scholar] [CrossRef]
- Amadio, M.; Pascale, A.; Wang, J.; Ho, L.; Quattrone, A.; Gandy, S.; Haroutunian, V.; Racchi, M.; Pasinetti, G.M. nELAV proteins alteration in Alzheimer’s disease brain: a novel putative target for amyloid-β reverberating on AβPP processing. J. Alzheimers Dis. 2009, 16, 409–419. [Google Scholar] [CrossRef]
- Kang, M.J.; Abdelmohsen, K.; Hutchison, E.R.; Mitchell, S.J.; Grammatikakis, I.; Guo, R.; Noh, J.H.; Martindale, J.L.; Yang, X.; Lee, E.K.; et al. HuD regulates coding and noncoding RNA to induce APP-->Aβ processing. Cell Rep. 2014, 7, 1401–1409. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Irie, Y.; Murakami, K.; Hanaki, M.; Hanaki, Y.; Suzuki, T.; Monobe, Y.; Takai, T.; Akagi, K.I.; Kawase, T.; Hirose, K.; et al. Synthetic models of quasi-stable amyloid β40 oligomers with significant neurotoxicity. ACS Chem. Neurosci. 2017, 8, 807–816. [Google Scholar] [CrossRef]
- Irie, Y.; Hanaki, M.; Murakami, K.; Imamoto, T.; Furuta, T.; Kawabata, T.; Kawase, T.; Hirose, K.; Monobe, Y.; Akagi, K.I.; et al. Synthesis and biochemical characterization of quasi-stable trimer models of full-length amyloid β40 with a toxic conformation. Chem. Commun. 2018, 55, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. iPSC-based compound screening and in vitro trials identify a synergistic anti-amyloid β combination for Alzheimer’s disease. Cell Rep. 2017, 21, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Crouch, S.P.; Kozlowski, R.; Slater, K.J.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. [Google Scholar] [CrossRef]
- Nelson, T.J.; Sun, M.K.; Lim, C.; Sen, A.; Khan, T.; Chirila, F.V.; Alkon, D.L. Bryostatin effects on cognitive function and PKCε in Alzheimer’s disease phase IIa and expanded access trials. J. Alzheimers Dis. 2017, 58, 521–535. [Google Scholar] [CrossRef]
- Aranda-Abreu, G.E.; Behar, L.; Chung, S.; Furneaux, H.; Ginzburg, I. Embryonic lethal abnormal vision-like RNA-binding proteins regulate neurite outgrowth and tau expression in PC12 cells. J. Neurosci. 1999, 19, 6907–6917. [Google Scholar] [CrossRef]
- Marchesi, N.; Amadio, M.; Colombrita, C.; Govoni, S.; Ratti, A.; Pascale, A. PKC activation counteracts ADAM10 deficit in HuD-silenced neuroblastoma cells. J. Alzheimers Dis. 2016, 54, 535–547. [Google Scholar] [CrossRef]
- Jarosz-Griffiths, H.H.; Corbett, N.J.; Rowland, H.A.; Fisher, K.; Jones, A.C.; Baron, J.; Howell, G.J.; Cowley, S.A.; Chintawar, S.; Cader, M.Z.; et al. Proteolytic shedding of the prion protein via activation of metallopeptidase ADAM10 reduces cellular binding and toxicity of amyloid-β oligomers. J. Biol. Chem. 2019, 294, 7085–7097. [Google Scholar] [CrossRef]
- Kaneko, N.; Yamamoto, R.; Sato, T.A.; Tanaka, K. Identification and quantification of amyloid β-related peptides in human plasma using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 2014, 90, 104–117. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Dore, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Hwang, S.S.; Chan, H.; Sorci, M.; Van Deventer, J.; Wittrup, D.; Belfort, G.; Walt, D. Detection of amyloid β oligomers toward early diagnosis of Alzheimer’s disease. Anal. Biochem. 2019, 566, 40–45. [Google Scholar] [CrossRef]
- Klaver, A.C.; Patrias, L.M.; Finke, J.M.; Loeffler, D.A. Specificity and sensitivity of the Aβ oligomer ELISA. J. Neurosci. Methods 2011, 195, 249–254. [Google Scholar] [CrossRef]
- Horikoshi, Y.; Mori, T.; Maeda, M.; Kinoshita, N.; Sato, K.; Yamaguchi, H. Aβ N-terminal-end specific antibody reduced β-amyloid in Alzheimer-model mice. Biochem. Biophys. Res. Commun. 2004, 325, 384–387. [Google Scholar] [CrossRef]
- Xia, W.; Yang, T.; Shankar, G.; Smith, I.M.; Shen, Y.; Walsh, D.M.; Selkoe, D.J. A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 2009, 66, 190–199. [Google Scholar] [CrossRef]
- Ohshima, Y.; Taguchi, K.; Mizuta, I.; Tanaka, M.; Tomiyama, T.; Kametani, F.; Yabe-Nishimura, C.; Mizuno, T.; Tokuda, T. Mutations in the β-amyloid precursor protein in familial Alzheimer’s disease increase Aβ oligomer production in cellular models. Heliyon 2018, 4, e00511. [Google Scholar] [CrossRef]
- Umeda, T.; Ramser, E.M.; Yamashita, M.; Nakajima, K.; Mori, H.; Silverman, M.A.; Tomiyama, T. Intracellular amyloid β oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol. Commun. 2015, 3, 51. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef]
- Guo, J.P.; Arai, T.; Miklossy, J.; McGeer, P.L. Aβ and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 1953–1958. [Google Scholar] [CrossRef]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef]
- Boyko, S.; Qi, X.; Chen, T.H.; Surewicz, K.; Surewicz, W.K. Liquid-liquid phase separation of tau protein: The crucial role of electrostatic interactions. J. Biol. Chem. 2019, 294, 11054–11059. [Google Scholar] [CrossRef]
- Vogler, T.O.; Wheeler, J.R.; Nguyen, E.D.; Hughes, M.P.; Britson, K.A.; Lester, E.; Rao, B.; Betta, N.D.; Whitney, O.N.; Ewachiw, T.E.; et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 2018, 563, 508–513. [Google Scholar] [CrossRef]
- Kume, T.; Kouchiyama, H.; Kaneko, S.; Maeda, T.; Akaike, A.; Shimohama, S.; Kihara, T.; Kimura, J.; Wada, K.; Koizumi, S. BDNF prevents NO mediated glutamate cytotoxicity in cultured cortical neurons. Brain Res 1997, 756, 200–204. [Google Scholar] [CrossRef]
- Izuo, N.; Kume, T.; Sato, M.; Murakami, K.; Irie, K.; Izumi, Y.; Akaike, A. Toxicity in rat primary neurons through the cellular oxidative stress induced by the turn formation at positions 22 and 23 of Aβ42. ACS Chem. Neurosci. 2012, 3, 674–681. [Google Scholar] [CrossRef]
- Nakagawa, M.; Taniguchi, Y.; Senda, S.; Takizawa, N.; Ichisaka, T.; Asano, K.; Morizane, A.; Doi, D.; Takahashi, J.; Nishizawa, M.; et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci. Rep. 2014, 4, 3594. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murakami, K.; Yoshimura, M.; Nakagawa, S.; Kume, T.; Kondo, T.; Inoue, H.; Irie, K. Evaluation of Toxic Amyloid β42 Oligomers in Rat Primary Cerebral Cortex Cells and Human iPS-derived Neurons Treated with 10-Me-Aplog-1, a New PKC Activator. Int. J. Mol. Sci. 2020, 21, 1179. https://doi.org/10.3390/ijms21041179
Murakami K, Yoshimura M, Nakagawa S, Kume T, Kondo T, Inoue H, Irie K. Evaluation of Toxic Amyloid β42 Oligomers in Rat Primary Cerebral Cortex Cells and Human iPS-derived Neurons Treated with 10-Me-Aplog-1, a New PKC Activator. International Journal of Molecular Sciences. 2020; 21(4):1179. https://doi.org/10.3390/ijms21041179
Chicago/Turabian StyleMurakami, Kazuma, Mayuko Yoshimura, Shota Nakagawa, Toshiaki Kume, Takayuki Kondo, Haruhisa Inoue, and Kazuhiro Irie. 2020. "Evaluation of Toxic Amyloid β42 Oligomers in Rat Primary Cerebral Cortex Cells and Human iPS-derived Neurons Treated with 10-Me-Aplog-1, a New PKC Activator" International Journal of Molecular Sciences 21, no. 4: 1179. https://doi.org/10.3390/ijms21041179
APA StyleMurakami, K., Yoshimura, M., Nakagawa, S., Kume, T., Kondo, T., Inoue, H., & Irie, K. (2020). Evaluation of Toxic Amyloid β42 Oligomers in Rat Primary Cerebral Cortex Cells and Human iPS-derived Neurons Treated with 10-Me-Aplog-1, a New PKC Activator. International Journal of Molecular Sciences, 21(4), 1179. https://doi.org/10.3390/ijms21041179