Bioinformatics for Renal and Urinary Proteomics: Call for Aggrandization

 ,
, 
Abstract
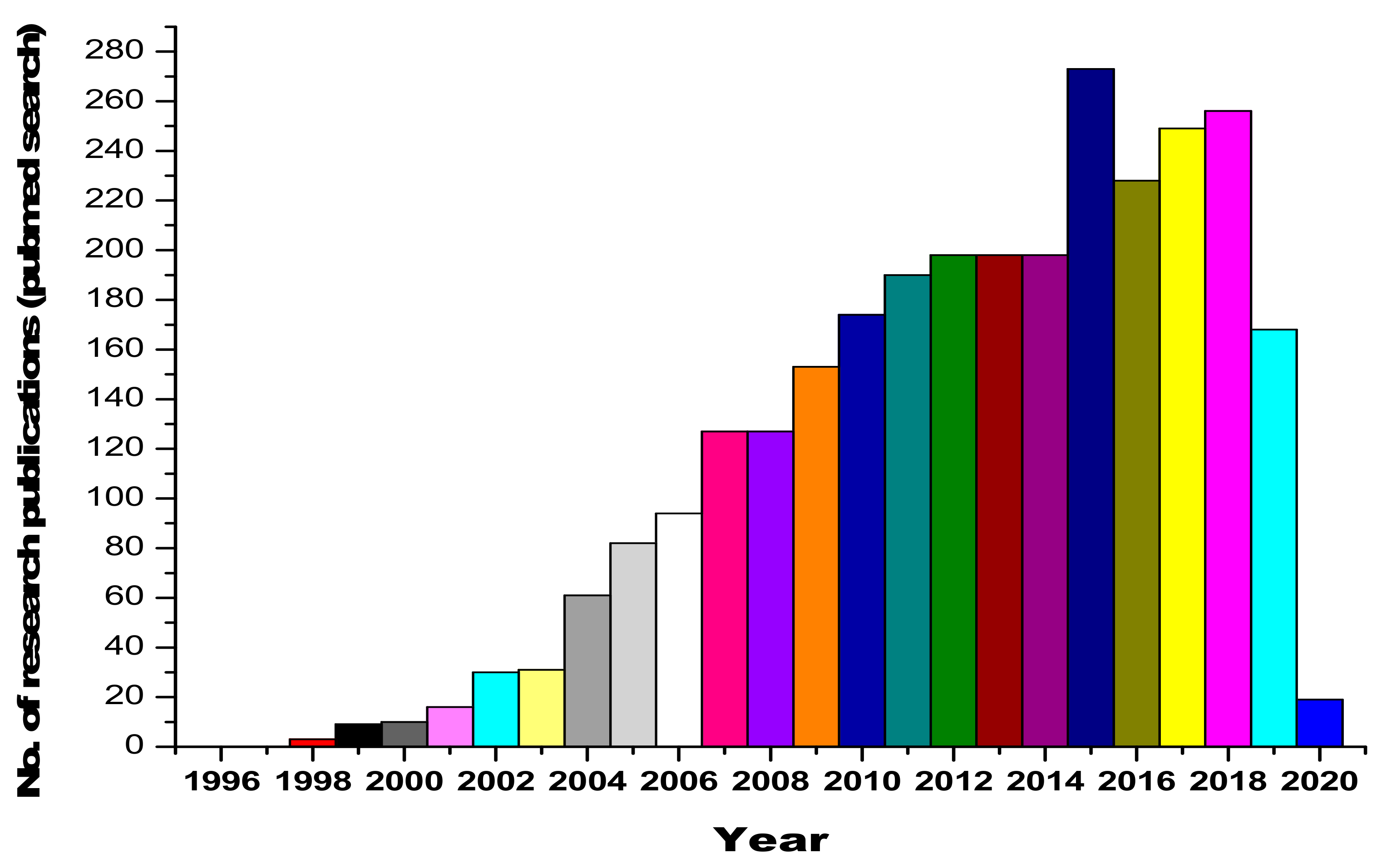
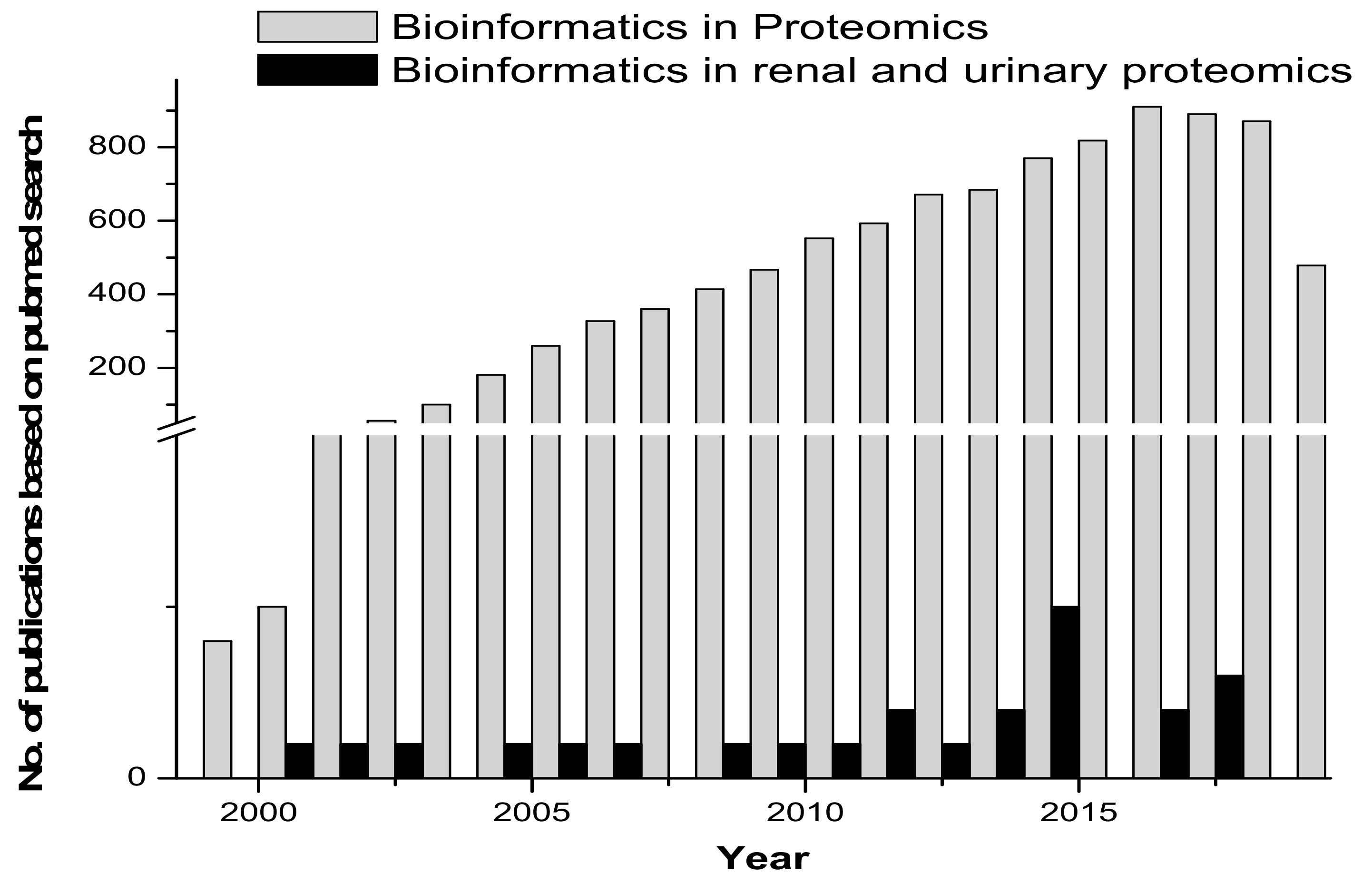
1. Introduction
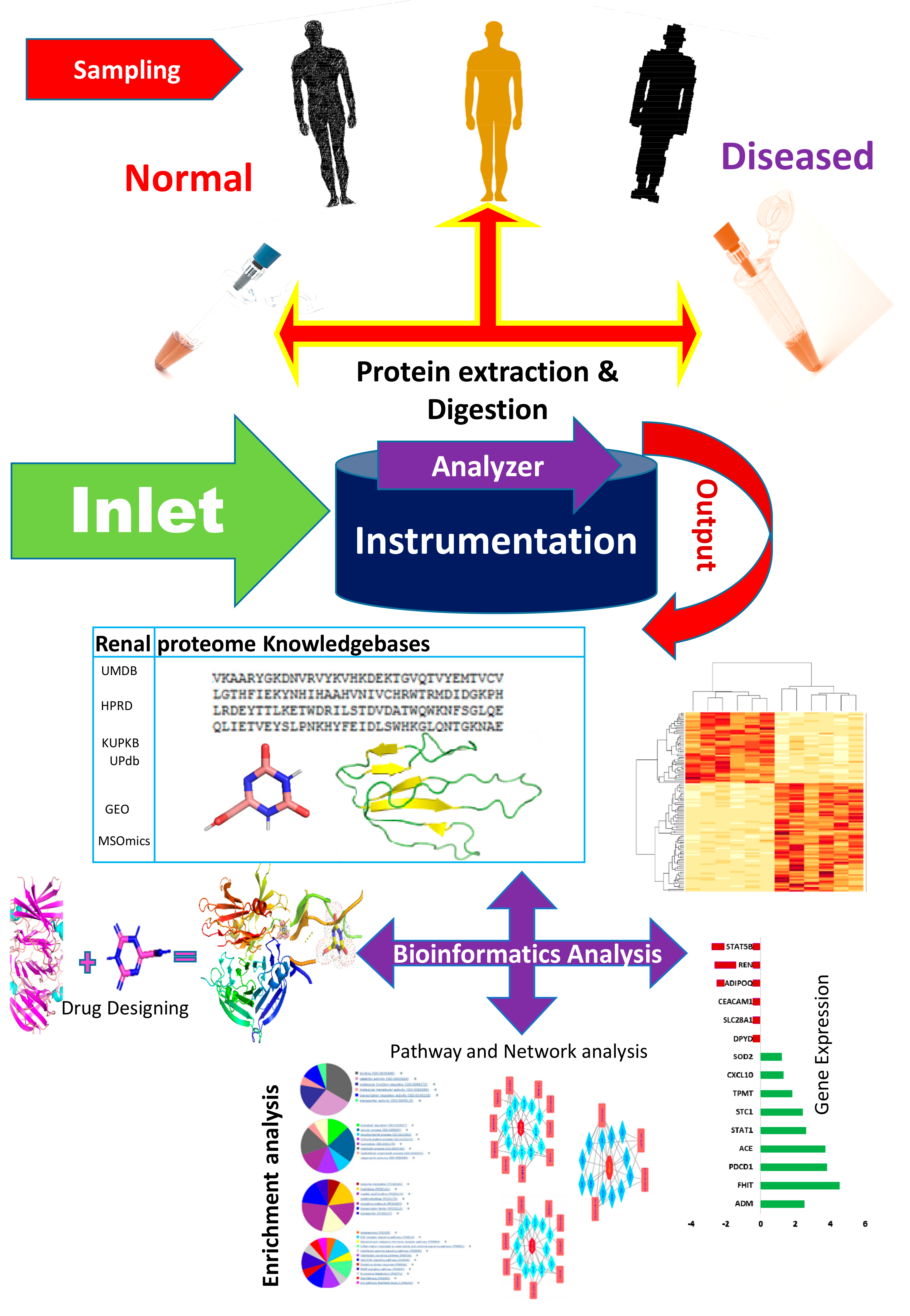
2. Bioinformatics Repositories for Proteomics
3. Consolidating Available Bioinformatics Tools for Renal and Urinary Proteomics
4. Future Perspectives on Bioinformatics Applications: Limitations
5. Conclusions
Funding
Conflicts of Interest
References
- Wilkins, M.R.; Sanchez, J.C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with proteome projects: Why all proteins expressed by a genome should be identified and how to do it. Biotechnol. Genet. Eng. Rev. 1996, 13, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. Proteome and proteomics: New technologies, new concepts, and new words. Electrophoresis 1998, 19, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.B.; Thongboonkerd, V. Overview of Proteomics. Contrib. Nephrol. 2004, 141, 1–10. [Google Scholar] [PubMed]
- Adachi, J.; Kumar, C.; Zhang, Y.; Olsen, J.V.; Mann, M. Tehuman urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome Biol. 2006, 7, R80. [Google Scholar] [CrossRef]
- Husi, H.; Stephens, N.; Cronshaw, A.; Stephens, N.A.; Wackerhage, H.; Greig, C.; Fearon, K.C.; Ross, J.A. Proteomic analysis of urinary upper gastrointestinal cancer markers. Proteom. Clin. Appl. 2011, 5, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Wasinger, V.C.; Zeng, M.; Yau, Y. Current status and advances in quantitative proteomic mass spectrometry. Int. J. Proteom. 2013. [Google Scholar] [CrossRef] [PubMed]
- Good, D.M.; Tongboonkerd, V.; Novak, J.; Bascands, J.L.; Schanstra, J.P.; Coon, J.J.; Dominiczak, A.; Mischak, H. Body fluid proteomics for biomarker discovery: Lessons from the past hold the key to success in the future. J. Proteome Res. 2007, 6, 4549–4555. [Google Scholar] [CrossRef]
- Cui, S.; Verroust, P.J.; Moestrup, S.K.; Christensen, E. Megalin/gp330 mediates uptake of albumin in renal proximal tubule. Ren. Fluid Electrolyte Physiol. 1996, 271, 900–907. [Google Scholar] [CrossRef]
- Pisitkun, T.; Shen, R.F.; Knepper, M.A. Identifcation and proteomic profling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373. [Google Scholar] [CrossRef]
- Castagna, A.; Cecconi, D.; Sennels, L.; Rappsilber, J.; Guerrier, L.; Fortis, F.; Boschetti, E.; Lomas, L.; Righetti, P.G. Exploring the hidden human urinary proteome via ligand library beads. J. Proteome Res. 2005, 4, 1917–1930. [Google Scholar] [CrossRef]
- Pieper, R.; Gatlin, C.L.; McGrath, A.M.; Makusky, A.J.; Mondal, M.; Seonarain, M.; Field, E.; Schatz, C.R.; Estock, M.A.; Ahmed, N.; et al. Characterization of the human urinary proteome: A method for high-resolution display of urinary proteins on two-dimensional electrophoresis gels with a yield of nearly 1400 distinct protein spots. Proteomics 2004, 4, 1159–1174. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, F.; Wu, S.; Wang, X.; Zheng, D.; Wang, J.; Gao, Y. Human urine proteome analysis by three separation approaches. Proteomics 2005, 5, 4994–5001. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, F.; Sun, W.; Wu, S.; Wang, X.; Zhang, L.; Zheng, D.; Wang, J.; Gao, Y. Concanavalin A-captured glycoproteins in healthy human urine. Mol. Cell. Proteom. 2006, 5, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Ozgo, M.; Skrzypczak, W.F.; Herosimczyk, A.; Mazur, A. Proteomika a fzjologiai patofzjologia nerek. MedWet 2007, 63, 1146–1150. [Google Scholar]
- Haraldsson, B.; Sorensson, J. Why do we not all have ¨proteinuria? An update of our current understanding of the glomerular barrier. News Physiol. Sci. 2004, 19, 7–10. [Google Scholar] [CrossRef]
- Maunsbach, A.B. Absorption of I125-labeled homologous albumin by rat kidney proximal tubule cells. A study of microperfused single proximal tubules by electron microscopic autoradiography and histochemistry. 1966. J. Am. Soc. Nephrol. 1997, 8, 323–351. [Google Scholar]
- Burne, M.J.; Osicka, T.M.; Comper, W.D. Fractional clearance of high molecular weight proteins in conscious rats using a continuous infusion method. Kidney Int. 1999, 55, 261–270. [Google Scholar] [CrossRef][Green Version]
- Batuman, V.; Verroust, P.J.; Navar, G.L.; Kaysen, J.H.; Goda, F.O.; Campbell, W.C.; Simon, E.; Pontillon, F.; Lyles, M.; Bruno, J.; et al. Myeloma light chains are ligands for cubilin (gp280). Ren. Physiol. 1998, 275, F246–F254. [Google Scholar] [CrossRef]
- Christensen, E.; Gburek, J. Protein reabsorption in renal proximal tubule—Function and dysfunction in kidney pathophysiology. Pediatr. Nephrol. 2004, 19, 714–721. [Google Scholar] [CrossRef]
- Ho, J.; Lucy, M.; Krokhin, O.; Hayglass, K.; Pascoe, E.; Darroch, G.; Rush, D.; Nickerson, P.; Rigatto, C.; Reslerova, M. Mass spectrometry-based proteomic analysis of urine in acute kidney injury following cardiopulmonary bypass: A nested case-control study. Am. J. Kidney Dis. 2009, 53, 584–595. [Google Scholar] [CrossRef]
- Orenes-Piñero, E.; Cortón, M.; González-Peramato, P.; Algaba, F.; Casal, I.; Serrano, A.; Sánchez-Carbayo, M. Searching urinary tumor markers for bladder cancer using a two-dimensional differential gel electrophoresis (2D-DIGE) approach. J. Proteome Res. 2007, 6, 4440–4448. [Google Scholar] [CrossRef] [PubMed]
- Zubiri, I.; Posada-Ayala, M.; Sanz-Maroto, A.; Calvo, E.; Martin-Lorenzo, M.; Gonzalez-Calero, L.; de la Cuesta, F.; Lopez, J.A.; Fernandez-Fernandez, B.; Ortiz, A.; et al. Diabetic nephropathy induces changes in the proteome of human urinary exosomes as revealed by label-free comparative analysis. J. Proteomics 2014, 96, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Tuijls, G.; van Wijck, K.; Grootjans, J.; Derikx, J.P.; van Bijnen, A.A.; Heineman, E.; Dejong, C.H.; Buurman, W.A.; Poeze, M. Early diagnosis of intestinal ischemia using urinary and plasma fatty acid binding proteins. Ann. Surg. 2011, 253, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zheng, C.X.; Peng, S.F.; Zhou, H.Y.; Su, Z.Y.; He, L.; Ai, T. Evaluation of urinary S100B protein level and lactate/creatinine ratio for early diagnosis and prognostic prediction of neonatal hypoxic-ischemic encephalopathy. Neonatology 2010, 97, 41–44. [Google Scholar] [CrossRef]
- Chugh, S.; Ouzounian, M.; Lu, Z.; Mohamed, S.; Li, W.; Bousette, N.; Liu, P.P.; Gramolini, A.O. Pilot study identifying myosin heavy chain 7, desmin, insulin-like growth factor 7, and annexin A2 as circulating biomarkers of human heart failure. Proteomics 2013, 13, 2324–2334. [Google Scholar] [CrossRef]
- Li, Q.R.; Fan, K.X.; Li, R.X.; Dai, J.; Wu, C.C.; Zhao, S.L.; Wu, J.R.; Shieh, C.H.; Zeng, R. A comprehensive and non-prefractionation on the protein level approach for the human urinary proteome: touching phosphorylation in urine. 2010, 24, 823–832. Rapid Commun. Mass Spectrom. 2010, 24, 823–832. [Google Scholar] [CrossRef]
- Zheng, J.; Liu, L.; Wang, J.; Jin, Q. Urinary proteomic and non-prefractionation quantitative phosphoproteomic analysis during pregnancy and non-pregnancy. BMC Genom. 2013, 14, 777. [Google Scholar] [CrossRef]
- Santucci, L.; Candiano, G.; Petretto, A.; Bruschi, M.; Lavarello, C.; Inglese, E.; Giorgio Righetti, P.; Marco Ghiggeri, G. From hundreds to thousands: Widening the normal human Urinome. J. Proteom. 2014, 112, 53–62. [Google Scholar]
- Desiere, F.; Deutsch, E.W.; King, N.L.; Nesvizhskii, A.I.; Mallick, P.; Eng, J.; Chen, S.; Eddes, J.; Loevenich, S.N.; Aebersold, R. PeptideAtlas project. Nucleic Acids Res. 2006, 34, D655–D658. [Google Scholar] [CrossRef]
- Tongboonkerd, V. Current status of renal and urinary proteomics: ready for routine clinical application. Nephrol. Dial. Transplant. 2010, 25, 11–16. [Google Scholar] [CrossRef]
- Kalantari, S.; Jafari, A.; Moradpoor, R.; Ghasemi, E.; Khalkhal, E. Human Urine Proteomics: Analytical Techniques and Clinical Applications in Renal Diseases. Int. J. Proteom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Spengler, S.J. Techview: Computers and biology. Bioinform. Inf. Agem Sci. 2000, 287, 1221–1223. [Google Scholar]
- Chen, C.; Huang, H.; Wu, C.H. Protein Bioinformatics Databases and Resources. Methods Mol. Biol. 2017, 1558, 3–39. [Google Scholar] [PubMed]
- wwPDB Consortium. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J. MPDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The Database of Disordered Proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef]
- Hoogland, C.; Mostaguir, K.; Sanchez, J.C.; Hochstrasser, D.F.; Appel, R.D. SWISS-2DPAGE, ten years later. Proteomics 2004, 4, 2352–2356. [Google Scholar] [CrossRef]
- Hoogland, C.; Mostaguir, K.; Appel, R.D.; Lisacek, F. The World-2DPAGE Constellation to promote and publish gel-based proteomics data through the ExPASy server. J. Proteom. 2008, 71, 245–248. [Google Scholar] [CrossRef]
- Yeats, C.; Maibaum, M.; Marsden, R.; Dibley, M.; Lee, D.; Addou, S.; Orengo, C.A. Gene3D: Modelling protein structure, function and evolution. Nucleic Acids Res. 2006, 34, D281–D284. [Google Scholar] [CrossRef]
- Pedruzzi, I.; Rivoire, C.; Auchincloss, A.H.; Coudert, E.; Keller, G.; de Castro, E.; Baratin, D.; Cuche, B.A.; Bougueleret, L.; Poux, S.; et al. HAMAP in 2015: Updates to the protein family classification and annotation system. Nucleic Acids Res. 2015, 43, D1064–D1070. [Google Scholar] [CrossRef]
- Mitchell, A.; Chang, H.Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Attwood, T.K.; Bradley, P.; Flower, D.R.; Gaulton, A.; Maudling, N.; Mitchell, A.; Moulton, G.; Nordle, A.; Paine, K.; Taylor, P.; et al. PRINTS; its automatic supplement, prePRINTS. Nucleic Acids Res. 2003, 31, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Servant, F.; Bru, C.; Carrère, S.; Courcelle, E.; Gouzy, J.; Peyruc, D.; Kahn, D. ProDom: Automated clustering of homologous domains. Brief. Bioinform. 2002, 3, 246–251. [Google Scholar] [CrossRef]
- Sigrist, C.J.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2013, 41, D344–D347. [Google Scholar] [CrossRef]
- Selengut, J.D.; Haft, D.H.; Davidsen, T.; Ganapathy, A.; Gwinn-Giglio, M.; Nelson, W.C.; Richter, A.R.; White, O. TIGRFAMs and Genome Properties: Tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res. 2007, 35, D260–D264. [Google Scholar] [CrossRef]
- Wilson, D.; Pethica, R.; Zhou, Y.; Talbot, C.; Vogel, C.; Madera, M.; Chothia, C.; Gough, J. SUPERFAMILY—Comparative Genomics, Datamining and Sophisticated Visualisation. Nucleic Acids Res. 2009, 37, D380–D386. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 4, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Adachi, J.J.; Olsen, J.V.; Shi, R.; de Souza, G.; Pasini, E.; Foster, L.J.; Macek, B.; Zougman, A.; Kumar, C.; et al. MAPU: Max-planck unifed database of organellar, cellular, tissue and body fluid proteomes. Nucleic Acids Res. 2007, 35, D771–D779. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, S.J.; Peng, M.; Li, H.; Liu, B.S.; Wang, C.; Wu, J.R.; Li, Y.X.; Zeng, R. Sys-BodyFluid: A systematical database for human body fluid proteome research. Nucleic Acids Res. 2009, 37, D907–D912. [Google Scholar] [CrossRef]
- Zhao, M.; Li, M.; Yang, Y.; Guo, Z.; Sun, Y.; Shao, C.; Li, M.; Sun, W.; Gao, Y. A comprehensive analysis and annotation of human normal urinary proteome. Sci. Rep. 2017, 7, 3024. [Google Scholar] [CrossRef]
- Siwy, J.; Mullen, W.; Golovko, I.; Franke, J.; Zurbig, P. Human urinary peptide database for multiple disease biomarker discovery. Proteom. Clin. Appl. 2011, 5, 367–374. [Google Scholar] [CrossRef]
- Ling, X.B.; Sigdel, T.K.; Lau, K.; Ying, L.; Lau, I.; Schilling, J.; Sarwal, M.M. Integrative urinary peptidomics in renal transplantation identifies biomarkers for acute rejection. J. Am. Soc. Nephrol. 2010, 21, 646–653. [Google Scholar] [CrossRef]
- Sigdel, T.K.; Kaushal, A.; Gritsenko, M.; Norbeck, A.D.; Qian, W.J.; Xiao, W.; Camp, D.G., 2nd; Smith, R.D.; Sarwal, M.M. Shotgun proteomics identifies proteins specific for acute renal transplant rejection. Proteom. Clin. Appl. 2010, 4, 32–47. [Google Scholar] [CrossRef]
- Sigdel, T.K.; Sarwal, M.M. The proteogenomic path towards biomarker discovery. Pediatr. Transpl. 2008, 2, 737–747. [Google Scholar] [CrossRef]
- Sigdel, T.K.; Nicora, C.D.; Hsieh, S.C.; Qian, W.J.; Sarwal, M.M. Optimization for peptide sample preparation for urine peptidomics. Clin. Proteom. 2014, 11, 7. [Google Scholar] [CrossRef]
- Sigdel, T.K.; Salomonis, N.; Nicora, C.D.; Ryu, S.; He, J.; Dinh, V.; Orton, D.J.; Moore, R.J.; Hsieh, S.C.; Dai, H. The identification of novel potential injury mechanisms and candidate biomarkers in renal allograft rejection by quantitative proteomics. Mol. Cell Proteom. 2014, 13, 621–631. [Google Scholar] [CrossRef]
- Nankivell, B.J.; Alexander, S.I. Rejection of the kidney allograft. N. Engl. J. Med. 2010, 363, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Bohl, D.L.; Brennan, D.C. BK virus nephropathy and kidney transplantation. Clin. J. Am. Soc. Nephrol. 2007, 2, S36–S46. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.T.; Nankivell, B.J.; Alexander, S.I. Chronic allograft nephropathy. Pediatr. Nephrol. 2009, 24, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Chen, W.; Lu, H.; Zhao, H.; Gao, S.; Liu, W.; Dong, X.; Guo, Z. Exploring the Therapeutic Mechanism of Desmodium styracifolium on Oxalate Crystal-Induced Kidney Injuries Using Comprehensive Approaches Based on Proteomics and Network Pharmacology Frontiers in Pharmacology. Front. Pharm. 2018, 9, 620. [Google Scholar] [CrossRef] [PubMed]
- Diller, D.J.; Merz, K.M. High Throughput Docking for Library Design and Library Prioritization. Proteins 2001, 43, 113–124. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef]
- Van, J.A.; Scholey, J.W.; Konvalinka, A. Insights into diabetic kidney disease using urinary proteomics and bioinformatics. J. Am. Soc. Nephrol. 2017, 28, 1050–1061. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Worachartcheewan, A.; Nantasenamat, C.; IsarankuraNa-Ayudhya, C.; Pidetcha, P.; Prachayasittikul, V. Identification of metabolic syndrome using decision tree analysis. Diabetes Res. Clin. Pract. 2010, 90, e15–e18. [Google Scholar] [CrossRef]
- Worachartcheewan, A.; Nantasenamat, C.; IsarankuraNa-Ayudhya, C.; Prachayasittikul, V. Quantitative population-health relationship (QPHR) for assessing metabolic syndrome. Excli J. 2013, 12, 569–583. [Google Scholar]
- Worachartcheewan, A.; Nantasenamat, C.; Prasertsrithong, P.; Amranan, J.; Monnor, T.; Chaisatit, T.; Nuchpramool, W.; Prachayasittikul, V. Machine learning approaches for discerning intercorrelation of hematological parameters and glucose level for identification of diabetes mellitus. EXCLI J. 2013, 12, 885–893. [Google Scholar] [PubMed]
- Worachartcheewan, A.; Shoombuatong, W.; Pidetcha, P.; Nopnithipat, W.; Prachayasittikul, V.; Nantasenamat, C. Predicting metabolic syndrome using the random forest method. Sci. World J. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Fang, X. Are you becoming a diabetic? A data mining approach. Int. Conf. Fuzzy Sys. Know Disc. 2009, 5, 18–22. [Google Scholar]
- Altelaar, A.F.M.; Munoz, J.; Heck, A.J.R. Nextgeneration proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2013, 14, 35–48. [Google Scholar] [CrossRef]
- Navarro, P.; Trevisan-Herraz, P.; Bonzon-Kulichenko, E.; Núñez, E.; Martínez-Acedo, P.; Pérez-Hernández, D.; Jorge, I.; Mesa, R.; Calvo, E.; Carrascal, M. General statistical framework for quantitative proteomics by stable isotope labeling. J. Proteome Res. 2014, 13, 1234–1247. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef]
- Oughtred, R.; Chatr-aryamontri, A.; Breitkreutz, B.J.; Chang, C.S.; Rust, J.M.; Theesfeld, C.L.; Heinicke, S.; Breitkreutz, A.; Chen, D.; Hirschman, J.; et al. Use of the BioGRID database for analysis of yeast protein and genetic interactions. Cold Spring Harb. Protoc. 2016, 2016, 1. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Pujol, A.; Mosca, R.; Farrés, J.; Aloy, P. Unveiling the role of network and systems biology in drug discovery. Trends Pharmacol. Sci. 2010, 31, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Guillén-Gómez, E.; Bardají-de-Quixano, B.; Ferrer, S.; Brotons, C.; Knepper, M.A.; Carrascal, M.; Abian, J.; Mas, J.M.; Calero, F.; Ballarín, J.A.; et al. Urinary Proteome Analysis Identified Neprilysin and VCAM as Proteins Involved in Diabetic Nephropathy. J. Diabetes Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The human urine metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef] [PubMed]
- Husi, H.; Barr, J.B.; Skipworth, R.J.; Stephens, N.A.; Greig, C.A.; Wackerhage, H.; Barron, R.; Fearon, K.C.; Ross, J.A. The Human Urinary Proteome Fingerprint Database UPdb. Int J. Proteom. 2013, 2013, 760208. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empiricalstatistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]
- Kentsis, A.; Monigatti, F.; Dorff, K.; Campagne, F.; Bachur, R.; Steen, H. Urine proteomics for profiling of human disease using high accuracy mass spectrometry. Proteom. Clin. Appl. 2009, 3, 1052–1061. [Google Scholar] [CrossRef]
- Marimuthu, A.; O’Meally, R.N.; Chaerkady, R.; Subbannayya, Y.; Nanjappa, V.; Kumar, P.; Kelkar, D.S.; Pinto, S.M.; Sharma, R.; Renuse, S.; et al. A comprehensive map of the human urinary proteome. Proteome Res. 2011, 10, 2734–2743. [Google Scholar] [CrossRef]
- Jupp, S.; Klein, J.; Schanstra, J.; Stevens, R. Developing a kidney and urinary pathway knowledge base. J. Biomed. Semant. 2011, 2, S7. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Sun, Z.; Campbell, D.; Kusebauch, U.; Chu, C.S.; Mendoza, L.; Shteynberg, D.; Omenn, G.S.; Moritz, R.L. State of the Human Proteome in 2014/2015 As Viewed through PeptideAtlas: Enhancing Accuracy and Coverage through the AtlasProphet. J. Proteome Res. 2015, 14, 3461–3473. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.H.; Yu, H.; Kim, K.W.; Kim, T.H.; Kim, Y.; Kim, S.I. Application of irregular and unbalanced data to predict diabetic nephropathy using visualization and feature selection methods. Artif Intell. Med. 2008, 42, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Neuhoff, N.V.; Kaiser, T.; Wittke, S.; Krebs, R.; Pitt, A.; Burchard, A.; Sundmacher, A.; Schlegelberger, B.; Kolch, W.; Mischak, H. Mass spectrometry for the detection of differentially expressed proteins: A comparison of surface-enhanced laser desorption/ionization and capillary electrophoresis/mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 149–156. [Google Scholar] [CrossRef]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted metabolomics strategies—Challenges and emerging directions. J. Am. Soc. Mass Spectrom 2016, 27, 1897–1905. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef] [PubMed]
- Kann, M.; Ettou, S.; Jung, Y.L.; Lenz, M.O.; Taglienti, M.E.; Park, P.J.; Schermer, B.; Benzing, T.; Kreidberg, J.A. Genome-Wide Analysis of Wilms’ Tumor 1-Controlled Gene Expression in Podocytes Reveals Key Regulatory Mechanisms. J. Am. Soc. Nephrol. 2015. [Google Scholar] [CrossRef]
- Karnovsky, A.; Weymouth, T.; Hull, T.; Tarcea, V.G.; Scardoni, G.; Laudanna, C.; Sartor, M.A.; Stringer, K.A.; Jagadish, H.V.; Burant, C.; et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 2012, 28, 373–380. [Google Scholar] [CrossRef]
- Basu, S.; Duren, W.; Evans, C.R.; Burant, C.; Michailidis, G.; Karnovsky, A. Sparse network modeling and Metscape-based visualization methods for the analysis of large-scale metabolomics data. Bioinformatics 2017, 33, 1545–1553. [Google Scholar] [CrossRef]
- Duren, W.; Weymouth, T.; Hull, T.; Omenn, G.S.; Athey, B.; Burant, C.; Karnovsky, A. MetDisease—Connecting metabolites to diseases via literature. Bioinformatics 2014, 30, 2239–2241. [Google Scholar] [CrossRef]
- Thongboonkerd, V.; McLeish, K.R.; Arthur, J.M.; Klein, J.B. Proteomic analysis of normal human urinary proteins isolated by acetone precipitation or ultracentrifugation. Kidney Int. 2002, 62, 1461–1469. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Function | Location | Reference |
|---|---|---|---|
| Urine Metabolome database (UMDB) | Metabolites of human urine | http://www.urinemetabolome.ca | Bouatra S et al., PLoS One. 2013 [83] |
| Human Urinary Proteomic Fingerprint Database (UPdb) | Share and exchange primary data derived from SELDI-, MALDI-, material-enhanced laser desorption/ionization (MELDI)-, CE-, LC-, and other TOF-MS analyses in urinary research | http://www.padb.org/ | Husi H et al., Int J Proteomics. 2013 [84] |
| ProteinProphetTM | Protein identification based on the peptides assigned to the MS/MS spectra | http://proteinprophet.sourceforge.net/ | Nesvizhskii AI et al., Anal Chem. 2003 [85] |
| PeptideProphetTM | Validates peptide assignments to the MS/MS spectra | http://peptideprophet.sourceforge.net/ | Keller A et al., Anal Chem. 2002 [86] |
| Urine proteomics for profiling of human disease | Resource of urinary proteins associated with common and rare human diseases | http://alexkentsis.net/urineproteomics/ | Kentsis A et al., Proteomics Clin Appl. 2009 [87] |
| Urinary Exosome Protein Database | Urinary exosomes from healthy human volunteers | https://hpcwebapps.cit.nih.gov/ESBL/Database/Exosome/ | Pisitkun T et al., Proc Natl Acad Sci USA. 2004 [9] |
| Max-Planck Unified (MAPU) proteome database | Body fluid (plasma, urine and cerebrospinal fluid) proteomes | http://www.mapuproteome.com/ | Zhang Y et al., Nucleic Acids Res. 2007 [52] |
| Human Protein Reference Database (HPRD) | Repository of proteomic information of human proteins | https://www.hprd.org | Marimuthu A et al., J. Proteome. 2011 [88] |
| The Kidney and Urinary Pathway Knowledge Base (KUPKB) | Knowledge related to the kidney and urinary pathways (KUP) | http://www.kupkb.org | Jupp S et al., J Biomed Semantics. 2011 [89] |
| Sys-BodyFluid | Reference database for body fluid proteomics and disease proteomics research | http://www.biosino.org/bodyfluid/ | Li SJ et al., Nucleic Acids Res. 2009 [90] |
| Gene Expression Omnibus (GEO) | Gene expression dataset | https://www.ncbi.nlm.nih.gov/geo/ | Barrett T, et al., Nucleic Acids Res. 2013 [54] |
| Human Kidney and Urine Proteome Project (HKUPP) | Proteomes of the kidney and urine | http://www.hkupp.org/ | Eric W et al., J Proteome Res. 2015 [91] |
| Visualization tool for risk factor analysis (VRIFA) | Computer-aided diagnosis and risk factor analysis | http://dm.postech.ac.kr/vrifa | Cho BH et al., Artif Intell Med. 2008 [92] |
| MosaiquesVisu software | Peak detection, mass deconvolution, 3D data visualizationand generating polypeptide lists | https://www.mhj-tools.com/sps-visu-micro/ | Neuhoff et al., Rapid Commun Mass Spectrom. 2004 [93] |
| The metabolomics service experts (MSOmics) | Service provider of metabolomics and for data analysis | http://msomics.com/index.html | Schrimpe A.C. et al., J Am Soc Mass Spectrom. 2016 [94] |
| Metabolite set enrichment analysis (MSEA) | Interprets human metabolite concentration changes in a biologically meaningful context | http://www.msea.ca | Xia J and Wishart D.S. Nucleic Acids Res. 2010 [95] |
| Podocyte mRNA Expression Database | mRNA expression data from FACS-sorted podocytes as analyzed by RNA-sequencing | http://helixweb.nih.gov/ESBL/Database/Podocyte_Transcriptome/index.htm | Kann M et al., J Am Soc Nephrol. 2015 [96] |
| MetScape 3.1 | Visualization and interpretation of metabolomic data using Cytoscape | http://metscape.med.umich.edu/ | Karnovsky A et al. [97] |
| CorrelationCalculator v1.0.1 | Large scale metabolic profiling | http://metscape.med.umich.edu/calculator.html | Basu S et al., Bioinformatics. 2017 [98] |
| MetDisease | MetDisease uses Medical Subject Headings (MeSH) disease terms mapped to PubChem compounds through literature to annotate compound networks. | http://metdisease.ncibi.org/ | Duren W et al., Bioinformatics. 2014 [99] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, P.; Antonydhason, V.; Gopal, J.; Haga, S.W.; Hasan, N.; Oh, J.-W. Bioinformatics for Renal and Urinary Proteomics: Call for Aggrandization. Int. J. Mol. Sci. 2020, 21, 961. https://doi.org/10.3390/ijms21030961
Paul P, Antonydhason V, Gopal J, Haga SW, Hasan N, Oh J-W. Bioinformatics for Renal and Urinary Proteomics: Call for Aggrandization. International Journal of Molecular Sciences. 2020; 21(3):961. https://doi.org/10.3390/ijms21030961
Chicago/Turabian StylePaul, Piby, Vimala Antonydhason, Judy Gopal, Steve W. Haga, Nazim Hasan, and Jae-Wook Oh. 2020. "Bioinformatics for Renal and Urinary Proteomics: Call for Aggrandization" International Journal of Molecular Sciences 21, no. 3: 961. https://doi.org/10.3390/ijms21030961
APA StylePaul, P., Antonydhason, V., Gopal, J., Haga, S. W., Hasan, N., & Oh, J.-W. (2020). Bioinformatics for Renal and Urinary Proteomics: Call for Aggrandization. International Journal of Molecular Sciences, 21(3), 961. https://doi.org/10.3390/ijms21030961