The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia


{kind=link}
{kind=link}
Abstract
1. Introduction
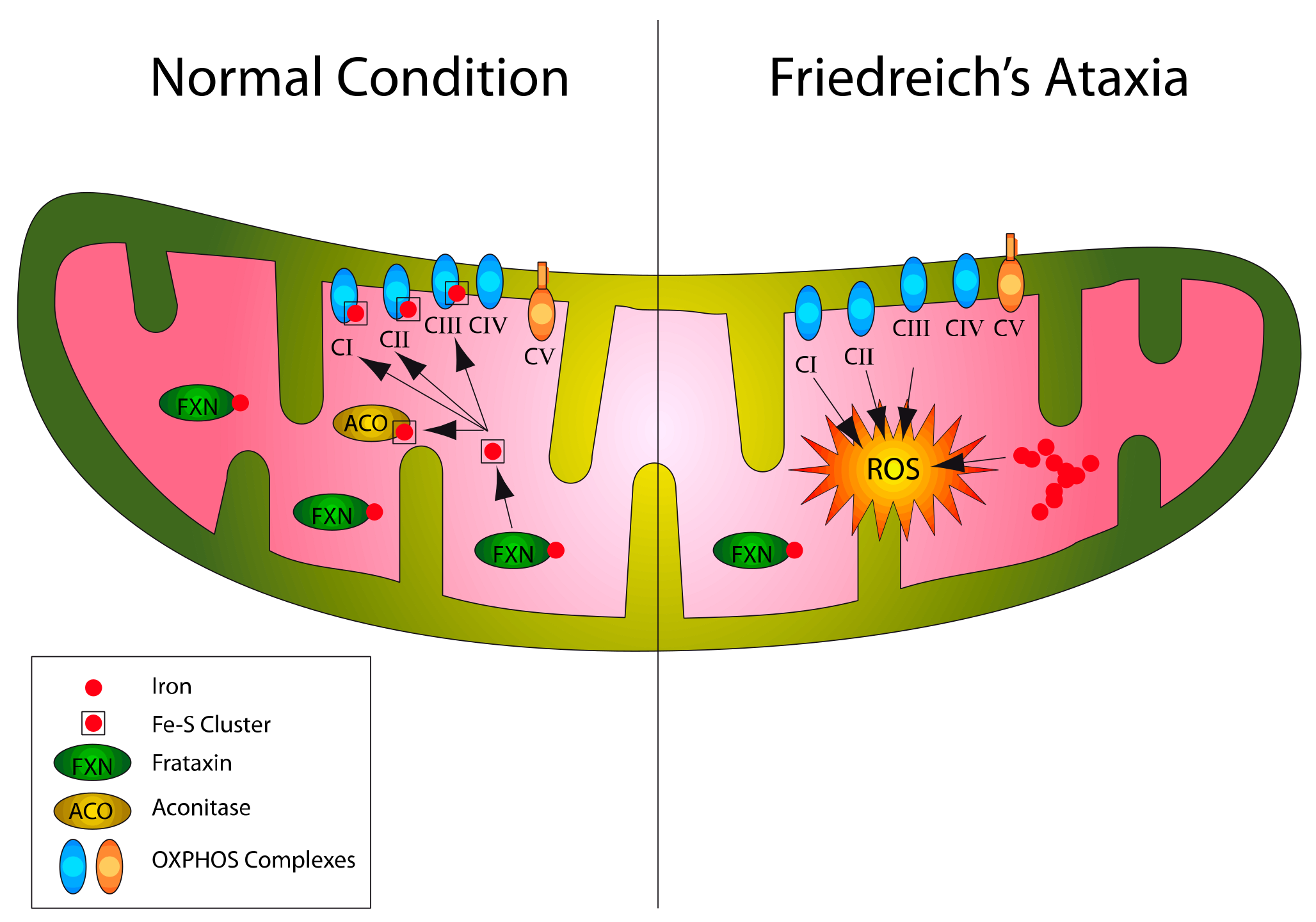
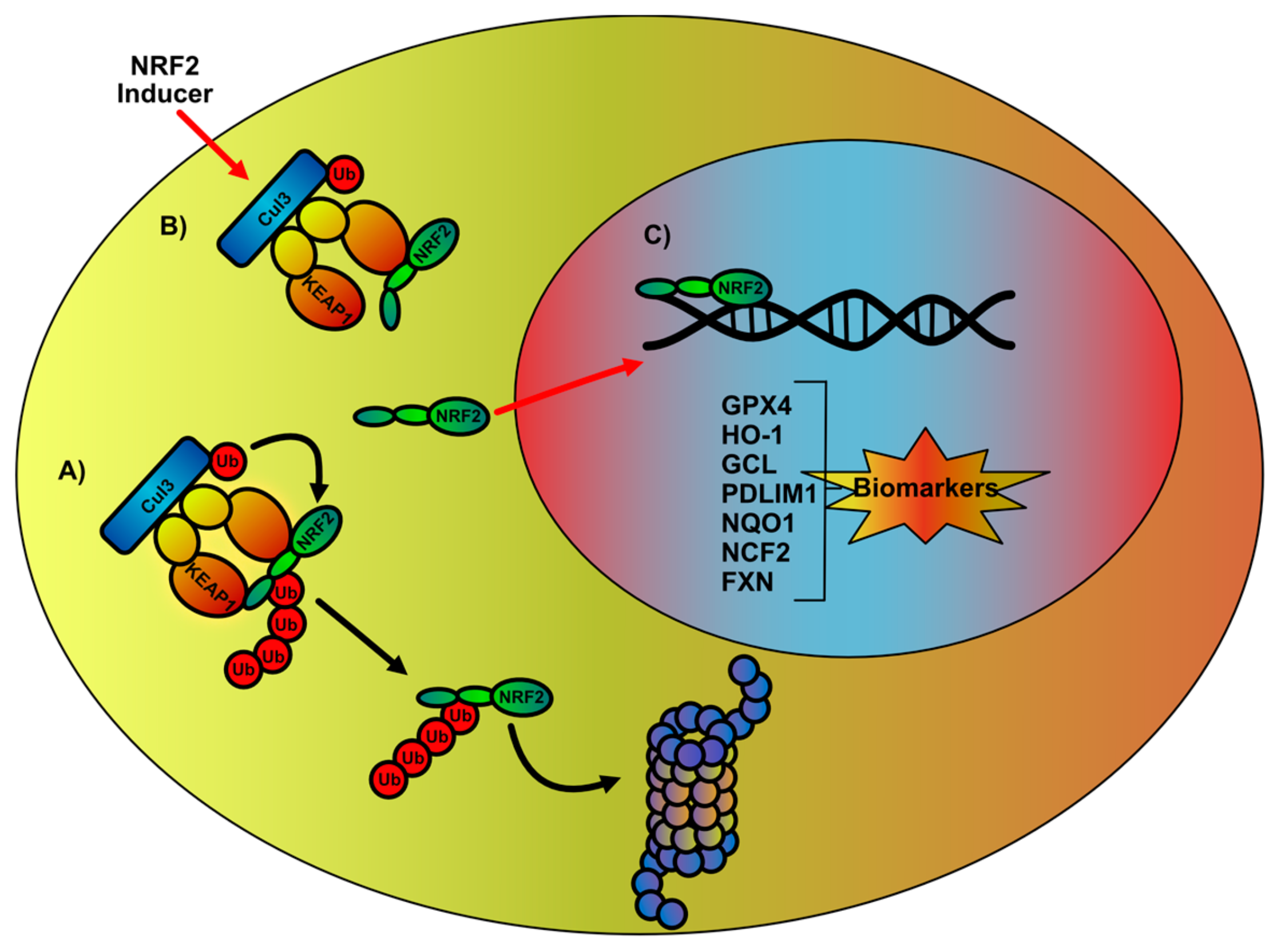
2. NRF2 Signaling Network Overview and Impairments in FA
3. Therapeutic Intervention in the Regulation of NRF2-Mediated Signaling Pathway
4. NRF2 Signaling Network as a Biomarker in the Evaluation of FA Therapy Outcome
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. Biomed. Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Chattopadhyay, A. Nrf2-ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Corenblum, M.J.; Ray, S.; Remley, Q.W.; Long, M.; Harder, B.; Zhang, D.D.; Barnes, C.A.; Madhavan, L. Reduced Nrf2 expression mediates the decline in neural stem cell function during a critical middle-age period. Aging Cell 2016, 15, 725–736. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front. Cell Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef]
- Robledinos-Anton, N.; Rojo, A.I.; Ferreiro, E.; Nunez, A.; Krause, K.H.; Jaquet, V.; Cuadrado, A. Transcription factor NRF2 controls the fate of neural stem cells in the subgranular zone of the hippocampus. Redox Biol. 2017, 13, 393–401. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef]
- Benarroch, E.E. Nrf2, cellular redox regulation, and neurologic implications. Neurology 2017, 88, 1942–1950. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef]
- Burk, K. Friedreich Ataxia: Current status and future prospects. Cerebellum Ataxias 2017, 4, 4. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Williamson, R.; Forrest, S.M. Friedreich ataxia: An overview. J. Med. Genet. 2000, 37, 1–8. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Cossee, M.; Durr, A.; Schmitt, M.; Dahl, N.; Trouillas, P.; Allinson, P.; Kostrzewa, M.; Nivelon-Chevallier, A.; Gustavson, K.H.; Kohlschutter, A.; et al. Friedreich’s ataxia: Point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999, 45, 200–206. [Google Scholar] [CrossRef]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; La Rosa, P.; Sette, C. Faulty RNA splicing: Consequences and therapeutic opportunities in brain and muscle disorders. Hum. Genet. 2017, 136, 1215–1235. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Bidichandani, S.I.; Ashizawa, T.; Patel, P.I. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am. J. Hum. Genet. 1998, 62, 111–121. [Google Scholar] [CrossRef]
- Sakamoto, N.; Chastain, P.D.; Parniewski, P.; Ohshima, K.; Pandolfo, M.; Griffith, J.D.; Wells, R.D. Sticky DNA: Self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol. Cell 1999, 3, 465–475. [Google Scholar] [CrossRef]
- Wells, R.D. DNA triplexes and Friedreich ataxia. FASEB J. 2008, 22, 1625–1634. [Google Scholar] [CrossRef]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef]
- Chutake, Y.K.; Costello, W.N.; Lam, C.; Bidichandani, S.I. Altered nucleosome positioning at the transcription start site and deficient transcriptional initiation in Friedreich ataxia. J. Biol. Chem. 2014, 289, 15194–15202. [Google Scholar] [CrossRef]
- Kumari, D.; Biacsi, R.E.; Usdin, K. Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J. Biol. Chem. 2011, 286, 4209–4215. [Google Scholar] [CrossRef]
- Burnett, R.; Melander, C.; Puckett, J.W.; Son, L.S.; Wells, R.D.; Dervan, P.B.; Gottesfeld, J.M. DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA.TTC repeats in Friedreich’s ataxia. Proc. Natl. Acad. Sci. USA 2006, 103, 11497–11502. [Google Scholar] [CrossRef]
- Soragni, E.; Miao, W.; Iudicello, M.; Jacoby, D.; De Mercanti, S.; Clerico, M.; Longo, F.; Piga, A.; Ku, S.; Campau, E.; et al. Epigenetic therapy for Friedreich ataxia. Ann. Neurol. 2014, 76, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Llorens, J.V.; Soriano, S.; Calap-Quintana, P.; Gonzalez-Cabo, P.; Molto, M.D. The Role of Iron in Friedreich’s Ataxia: Insights From Studies in Human Tissues and Cellular and Animal Models. Front. Neurosci. 2019, 13, 75. [Google Scholar] [CrossRef] [PubMed]
- Jasoliya, M.J.; McMackin, M.Z.; Henderson, C.K.; Perlman, S.L.; Cortopassi, G.A. Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans. Hum. Mol. Genet. 2017, 26, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999, 8, 425–430. [Google Scholar] [CrossRef]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51. [Google Scholar] [CrossRef]
- Haugen, A.C.; Di Prospero, N.A.; Parker, J.S.; Fannin, R.D.; Chou, J.; Meyer, J.N.; Halweg, C.; Collins, J.B.; Durr, A.; Fischbeck, K.; et al. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: Cellular model of pathology. PLoS Genet. 2010, 6, e1000812. [Google Scholar] [CrossRef]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef]
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef]
- Foury, F.; Cazzalini, O. Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett. 1997, 411, 373–377. [Google Scholar] [CrossRef]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Camakaris, J.; Brooks, H.; Evans-Whipp, T.; Thorburn, D.R.; Williamson, R.; Forrest, S.M. Direct evidence that mitochondrial iron accumulation occurs in Friedreich ataxia. Ann. Neurol. 1999, 45, 673–675. [Google Scholar] [CrossRef]
- Bradley, J.L.; Homayoun, S.; Hart, P.E.; Schapira, A.H.; Cooper, J.M. Role of oxidative damage in Friedreich’s ataxia. Neurochem Res. 2004, 29, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Seznec, H.; Simon, D.; Bouton, C.; Reutenauer, L.; Hertzog, A.; Golik, P.; Procaccio, V.; Patel, M.; Drapier, J.C.; Koenig, M.; et al. Friedreich ataxia: The oxidative stress paradox. Hum. Mol. Genet. 2005, 14, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Myers, L.M.; Lynch, D.R.; Farmer, J.M.; Friedman, L.S.; Lawson, J.A.; Wilson, R.B. Urinary isoprostanes in Friedreich ataxia: Lack of correlation with disease features. Mov. Disord. 2008, 23, 1920–1922. [Google Scholar] [CrossRef]
- Bulteau, A.L.; Dancis, A.; Gareil, M.; Montagne, J.J.; Camadro, J.M.; Lesuisse, E. Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic. Biol. Med. 2007, 42, 1561–1570. [Google Scholar] [CrossRef]
- Irazusta, V.; Moreno-Cermeno, A.; Cabiscol, E.; Ros, J.; Tamarit, J. Major targets of iron-induced protein oxidative damage in frataxin-deficient yeasts are magnesium-binding proteins. Free Radic. Biol. Med. 2008, 44, 1712–1723. [Google Scholar] [CrossRef]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich’s ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef]
- Llorens, J.V.; Navarro, J.A.; Martinez-Sebastian, M.J.; Baylies, M.K.; Schneuwly, S.; Botella, J.A.; Molto, M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 2007, 21, 333–344. [Google Scholar] [CrossRef]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S.; Webster, Z.; Blake, J.; Cooper, J.M.; King, R.; et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar] [CrossRef]
- Poburski, D.; Boerner, J.B.; Koenig, M.; Ristow, M.; Thierbach, R. Time-resolved functional analysis of acute impairment of frataxin expression in an inducible cell model of Friedreich ataxia. Biol. Open 2016, 5, 654–661. [Google Scholar] [CrossRef]
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; Pook, M.A.; Giunti, P.; Abramov, A.Y. ‘Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia’. Cell Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Dehmer, T.; Schols, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Burk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef]
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar] [CrossRef]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Investig. 2001, 31, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef] [PubMed]
- Anzovino, A.; Chiang, S.; Brown, B.E.; Hawkins, C.L.; Richardson, D.R.; Huang, M.L. Molecular Alterations in a Mouse Cardiac Model of Friedreich Ataxia: An Impaired Nrf2 Response Mediated via Upregulation of Keap1 and Activation of the Gsk3beta Axis. Am. J. Pathol. 2017, 187, 2858–2875. [Google Scholar] [CrossRef]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchere, F.; Lonn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): History, structure and function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Amoutzias, G.D.; Robertson, D.L.; Van de Peer, Y.; Oliver, S.G. Choose your partners: Dimerization in eukaryotic transcription factors. Trends Biochem. Sci. 2008, 33, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Nuclear import and export signals in control of Nrf2. J. Biol. Chem. 2005, 280, 29158–29168. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.X.; Zhang, Y.; Wang, Y.F.; Li, X.C.; Xiang, M.X.; Bian, C.; Chen, P. Upregulation of heme oxygenase-1 expression by hydroxysafflor yellow A conferring protection from anoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes. Int J. Cardiol. 2012, 160, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Wang, Y.; Zhu, X.; Jiang, M.; Song, E.; Song, Y. New application of the commercial sweetener rebaudioside a as a hepatoprotective candidate: Induction of the Nrf2 signaling pathway. Eur. J. Pharmacol. 2018, 822, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef]
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the Treatment of Friedreich’s Ataxia: A Comparison among Drugs. Int. J. Mol. Sci. 2019, 20, 5211. [Google Scholar] [CrossRef]
- Chantrel-Groussard, K.; Geromel, V.; Puccio, H.; Koenig, M.; Munnich, A.; Rotig, A.; Rustin, P. Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum. Mol. Genet. 2001, 10, 2061–2067. [Google Scholar] [CrossRef]
- Shoichet, S.A.; Baumer, A.T.; Stamenkovic, D.; Sauer, H.; Pfeiffer, A.F.; Kahn, C.R.; Muller-Wieland, D.; Richter, C.; Ristow, M. Frataxin promotes antioxidant defense in a thiol-dependent manner resulting in diminished malignant transformation in vitro. Hum. Mol. Genet. 2002, 11, 815–821. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef]
- He, X.; Ma, Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol. Pharmacol. 2009, 76, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [PubMed]
- Magesh, S.; Chen, Y.; Hu, L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Singh, K.; Connors, S.L.; Macklin, E.A.; Smith, K.D.; Fahey, J.W.; Talalay, P.; Zimmerman, A.W. Sulforaphane treatment of autism spectrum disorder (ASD). Proc. Natl. Acad. Sci. USA 2014, 111, 15550–15555. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int. J. Mol. Sci. 2017, 18, 2173. [Google Scholar] [CrossRef] [PubMed]
- Clulow, J.A.; Storck, E.M.; Lanyon-Hogg, T.; Kalesh, K.A.; Jones, L.H.; Tate, E.W. Competition-based, quantitative chemical proteomics in breast cancer cells identifies new target profiles for sulforaphane. Chem. Commun. (Camb.) 2017, 53, 5182–5185. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Hsu, A.; Williams, D.E.; Dashwood, R.H.; Stevens, J.F.; Yamamoto, M.; Ho, E. Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharm. Res. 2011, 28, 3171–3179. [Google Scholar] [CrossRef]
- Indelicato, E.; Bösch, S. Emerging therapeutics for the treatment of Friedreich’s ataxia. Expert Opin. Orphan Drugs 2018, 6, 57–67. [Google Scholar] [CrossRef]
- Reilly, J.F.; Yanez, G.; Bista, P.; Picarella, D.; Lee, D.; Vu, C.; Nichols, A.; Magrane, J. CAT-4001 improves mitochondrial function in a Friedreich’s ataxia model. In Proceedings of the 2nd International Ataxia Research Conference, Pisa, Italy, 27–30 September 2017. [Google Scholar]
- Esposti, M.D.; Ngo, A.; Ghelli, A.; Benelli, B.; Carelli, V.; McLennan, H.; Linnane, A.W. The interaction of Q analogs, particularly hydroxydecyl benzoquinone (idebenone), with the respiratory complexes of heart mitochondria. Arch. Biochem. Biophys. 1996, 330, 395–400. [Google Scholar] [CrossRef]
- James, A.M.; Sharpley, M.S.; Manas, A.R.; Frerman, F.E.; Hirst, J.; Smith, R.A.; Murphy, M.P. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef]
- Orsucci, D.; Mancuso, M.; Ienco, E.C.; LoGerfo, A.; Siciliano, G. Targeting mitochondrial dysfunction and neurodegeneration by means of coenzyme Q10 and its analogues. Curr. Med. Chem. 2011, 18, 4053–4064. [Google Scholar] [CrossRef]
- Kahn-Kirby, A.H.; Amagata, A.; Maeder, C.I.; Mei, J.J.; Sideris, S.; Kosaka, Y.; Hinman, A.; Malone, S.A.; Bruegger, J.J.; Wang, L.; et al. Targeting ferroptosis: A novel therapeutic strategy for the treatment of mitochondrial disease-related epilepsy. PLoS ONE 2019, 14, e0214250. [Google Scholar] [CrossRef]
- Suno, M.; Nagaoka, A. Inhibition of lipid peroxidation by a novel compound (CV-2619) in brain mitochondria and mode of action of the inhibition. Biochem. Biophys. Res. Commun. 1984, 125, 1046–1052. [Google Scholar] [CrossRef]
- Azim, S.A.; Darwish, H.A.; Rizk, M.Z.; Ali, S.A.; Kadry, M.O. Amelioration of titanium dioxide nanoparticles-induced liver injury in mice: Possible role of some antioxidants. Exp. Toxicol. Pathol. 2015, 67, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Pala, R.; Orhan, C.; Tuzcu, M.; Sahin, N.; Ali, S.; Cinar, V.; Atalay, M.; Sahin, K. Coenzyme Q10 Supplementation Modulates NFkappaB and Nrf2 Pathways in Exercise Training. J. Sports Sci. Med. 2016, 15, 196–203. [Google Scholar] [PubMed]
- Rao, V.A.; Klein, S.R.; Bonar, S.J.; Zielonka, J.; Mizuno, N.; Dickey, J.S.; Keller, P.W.; Joseph, J.; Kalyanaraman, B.; Shacter, E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J. Biol. Chem. 2010, 285, 34447–34459. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.G.; Seto, S.; Ganne, P.; Votruba, M. A randomized, placebo-controlled trial of the benzoquinone idebenone in a mouse model of OPA1-related dominant optic atrophy reveals a limited therapeutic effect on retinal ganglion cell dendropathy and visual function. Neuroscience 2016, 319, 92–106. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar]
- Prochaska, H.J.; Santamaria, A.B. Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: A screening assay for anticarcinogenic enzyme inducers. Anal. Biochem. 1988, 169, 328–336. [Google Scholar] [CrossRef]
- Li, W.W.; Heinze, J.; Haehnel, W. Site-specific binding of quinones to proteins through thiol addition and addition-elimination reactions. J. Am. Chem. Soc. 2005, 127, 6140–6141. [Google Scholar] [CrossRef]
- Mazzei, L.; Cianci, M.; Musiani, F.; Ciurli, S. Inactivation of urease by 1,4-benzoquinone: Chemistry at the protein surface. Dalton Trans. 2016, 45, 5455–5459. [Google Scholar] [CrossRef]
- Mishra, P.; Paital, B.; Jena, S.; Swain, S.S.; Kumar, S.; Yadav, M.K.; Chainy, G.B.N.; Samanta, L. Possible activation of NRF2 by Vitamin E/Curcumin against altered thyroid hormone induced oxidative stress via NFkB/AKT/mTOR/KEAP1 signalling in rat heart. Sci. Rep. 2019, 9, 7408. [Google Scholar] [CrossRef]
- Geromel, V.; Darin, N.; Chretien, D.; Benit, P.; DeLonlay, P.; Rotig, A.; Munnich, A.; Rustin, P. Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: Rationale and comparative benefits. Mol. Genet. Metab. 2002, 77, 21–30. [Google Scholar] [CrossRef]
- Glorieux, C.; Buc Calderon, P. Cancer Cell Sensitivity to Redox-Cycling Quinones is Influenced by NAD(P)H: Quinone Oxidoreductase 1 Polymorphism. Antioxidants (Basel) 2019, 8, 369. [Google Scholar] [CrossRef]
- Jaber, S.; Polster, B.M. Idebenone and neuroprotection: Antioxidant, pro-oxidant, or electron carrier? J. Bioenerg. Biomembr. 2015, 47, 111–118. [Google Scholar] [CrossRef]
- Rietjens, I.M.; Boersma, M.G.; Haan, L.; Spenkelink, B.; Awad, H.M.; Cnubben, N.H.; van Zanden, J.J.; Woude, H.; Alink, G.M.; Koeman, J.H. The pro-oxidant chemistry of the natural antioxidants vitamin C, vitamin E, carotenoids and flavonoids. Environ. Toxicol. Pharmacol. 2002, 11, 321–333. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Calsolaro, V.; Choub, A.; Siciliano, G. Coenzyme Q10 and Neurological Diseases. Pharmaceuticals (Basel) 2009, 2, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Spector, R.; Johanson, C.E. Vitamin transport and homeostasis in mammalian brain: Focus on Vitamins B and E. J. Neurochem. 2007, 103, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Turnaturi, R.; Parenti, C.; Pasquinucci, L. Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy. Nanomaterials (Basel) 2018, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Korlipara, L.V.; Hart, P.E.; Bradley, J.L.; Schapira, A.H. Coenzyme Q10 and vitamin E deficiency in Friedreich’s ataxia: Predictor of efficacy of vitamin E and coenzyme Q10 therapy. Eur. J. Neurol. 2008, 15, 1371–1379. [Google Scholar] [CrossRef]
- Hart, P.E.; Lodi, R.; Rajagopalan, B.; Bradley, J.L.; Crilley, J.G.; Turner, C.; Blamire, A.M.; Manners, D.; Styles, P.; Schapira, A.H.; et al. Antioxidant treatment of patients with Friedreich ataxia: Four-year follow-up. Arch. Neurol. 2005, 62, 621–626. [Google Scholar] [CrossRef]
- Lodi, R.; Hart, P.E.; Rajagopalan, B.; Taylor, D.J.; Crilley, J.G.; Bradley, J.L.; Blamire, A.M.; Manners, D.; Styles, P.; Schapira, A.H.; et al. Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich’s ataxia. Ann. Neurol. 2001, 49, 590–596. [Google Scholar] [CrossRef]
- Artuch, R.; Aracil, A.; Mas, A.; Colome, C.; Rissech, M.; Monros, E.; Pineda, M. Friedreich’s ataxia: Idebenone treatment in early stage patients. Neuropediatrics 2002, 33, 190–193. [Google Scholar] [CrossRef]
- Brandsema, J.F.; Stephens, D.; Hartley, J.; Yoon, G. Intermediate-dose idebenone and quality of life in Friedreich ataxia. Pediatr. Neurol. 2010, 42, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Hausse, A.O.; Aggoun, Y.; Bonnet, D.; Sidi, D.; Munnich, A.; Rotig, A.; Rustin, P. Idebenone and reduced cardiac hypertrophy in Friedreich’s ataxia. Heart 2002, 87, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Arpa, J.; Montero, R.; Aracil, A.; Dominguez, F.; Galvan, M.; Mas, A.; Martorell, L.; Sierra, C.; Brandi, N.; et al. Idebenone treatment in paediatric and adult patients with Friedreich ataxia: Long-term follow-up. Eur. J. Paediatr. Neurol. 2008, 12, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Rustin, P.; Rotig, A.; Munnich, A.; Sidi, D. Heart hypertrophy and function are improved by idebenone in Friedreich’s ataxia. Free Radic. Res. 2002, 36, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Rustin, P.; von Kleist-Retzow, J.C.; Chantrel-Groussard, K.; Sidi, D.; Munnich, A.; Rotig, A. Effect of idebenone on cardiomyopathy in Friedreich’s ataxia: A preliminary study. Lancet 1999, 354, 477–479. [Google Scholar] [CrossRef]
- Di Prospero, N.A.; Baker, A.; Jeffries, N.; Fischbeck, K.H. Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: A randomised, placebo-controlled trial. Lancet Neurol. 2007, 6, 878–886. [Google Scholar] [CrossRef]
- Lagedrost, S.J.; Sutton, M.S.; Cohen, M.S.; Satou, G.M.; Kaufman, B.D.; Perlman, S.L.; Rummey, C.; Meier, T.; Lynch, D.R. Idebenone in Friedreich ataxia cardiomyopathy-results from a 6-month phase III study (IONIA). Am. Heart J. 2011, 161, 639–645. [Google Scholar] [CrossRef]
- Lynch, D.R.; Perlman, S.L.; Meier, T. A phase 3, double-blind, placebo-controlled trial of idebenone in friedreich ataxia. Arch. Neurol. 2010, 67, 941–947. [Google Scholar] [CrossRef]
- Meier, T.; Perlman, S.L.; Rummey, C.; Coppard, N.J.; Lynch, D.R. Assessment of neurological efficacy of idebenone in pediatric patients with Friedreich’s ataxia: Data from a 6-month controlled study followed by a 12-month open-label extension study. J. Neurol. 2012, 259, 284–291. [Google Scholar] [CrossRef]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V. Drug Development for the Therapy of Mitochondrial Diseases. Trends Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.; Freeman, M.; Shaw, J.; Gooch, C.; Huang, Y.X.; Klein, M.; Miller, G.; Zesiewicz, T. EPI-743 for Friedreichs Ataxia Patients with Point Mutations. Neurology 2016, 86, 5.388. [Google Scholar]
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Gulcin, I. Antioxidant properties of resveratrol: A structure-activity insight. Innov. Food Sci. Emerg. 2010, 11, 210–218. [Google Scholar] [CrossRef]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef]
- Tanigawa, S.; Fujii, M.; Hou, D.X. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic. Biol. Med. 2007, 42, 1690–1703. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef]
- Li, L.; Voullaire, L.; Sandi, C.; Pook, M.A.; Ioannou, P.A.; Delatycki, M.B.; Sarsero, J.P. Pharmacological screening using an FXN-EGFP cellular genomic reporter assay for the therapy of Friedreich ataxia. PLoS ONE 2013, 8, e55940. [Google Scholar] [CrossRef]
- Yiu, E.M.; Tai, G.; Peverill, R.E.; Lee, K.J.; Croft, K.D.; Mori, T.A.; Scheiber-Mojdehkar, B.; Sturm, B.; Praschberger, M.; Vogel, A.P.; et al. An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on frataxin levels. J. Neurol. 2015, 262, 1344–1353. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef]
- Walsh, J.; Jenkins, R.E.; Wong, M.; Olayanju, A.; Powell, H.; Copple, I.; O’Neill, P.M.; Goldring, C.E.; Kitteringham, N.R.; Park, B.K. Identification and quantification of the basal and inducible Nrf2-dependent proteomes in mouse liver: Biochemical, pharmacological and toxicological implications. J. Proteomics 2014, 108, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Shekh-Ahmad, T.; Eckel, R.; Dayalan Naidu, S.; Higgins, M.; Yamamoto, M.; Dinkova-Kostova, A.T.; Kovac, S.; Abramov, A.Y.; Walker, M.C. KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain 2018, 141, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Clay, A.; Hearle, P.; Schadt, K.; Lynch, D.R. New developments in pharmacotherapy for Friedreich ataxia. Expert Opin. Pharmacother. 2019, 20, 1855–1867. [Google Scholar] [CrossRef] [PubMed]
- Strawser, C.; Schadt, K.; Hauser, L.; McCormick, A.; Wells, M.; Larkindale, J.; Lin, H.; Lynch, D.R. Pharmacological therapeutics in Friedreich ataxia: The present state. Expert Rev. Neurother. 2017, 17, 895–907. [Google Scholar] [CrossRef]
- Blair, I.A.; Farmer, J.; Hersch, S.; Larkindale, J.; Lynch, D.R.; Napierala, J.; Napierala, M.; Payne, R.M.; Subramony, S.H. The current state of biomarker research for Friedreich’s ataxia: A report from the 2018 FARA biomarker meeting. Future Sci. OA 2019, 5, FSO398. [Google Scholar] [CrossRef]
- Friedman, L.S.; Farmer, J.M.; Perlman, S.; Wilmot, G.; Gomez, C.M.; Bushara, K.O.; Mathews, K.D.; Subramony, S.H.; Ashizawa, T.; Balcer, L.J.; et al. Measuring the rate of progression in Friedreich ataxia: Implications for clinical trial design. Mov. Disord. 2010, 25, 426–432. [Google Scholar] [CrossRef]
- Patel, M.; Isaacs, C.J.; Seyer, L.; Brigatti, K.; Gelbard, S.; Strawser, C.; Foerster, D.; Shinnick, J.; Schadt, K.; Yiu, E.M.; et al. Progression of Friedreich ataxia: Quantitative characterization over 5 years. Ann. Clin. Transl. Neurol. 2016, 3, 684–694. [Google Scholar] [CrossRef]
- Reetz, K.; Dogan, I.; Hohenfeld, C.; Didszun, C.; Giunti, P.; Mariotti, C.; Durr, A.; Boesch, S.; Klopstock, T.; Rodriguez de Rivera Garrido, F.J.; et al. Nonataxia symptoms in Friedreich Ataxia: Report from the Registry of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS). Neurology 2018, 91, e917–e930. [Google Scholar] [CrossRef]
- Tai, G.; Yiu, E.M.; Delatycki, M.B.; Corben, L.A. How does performance of the Friedreich Ataxia Functional Composite compare to rating scales? J. Neurol. 2017, 264, 1768–1776. [Google Scholar] [CrossRef]
- Hayashi, G.; Cortopassi, G. Lymphoblast Oxidative Stress Genes as Potential Biomarkers of Disease Severity and Drug Effect in Friedreich’s Ataxia. PLoS ONE 2016, 11, e0153574. [Google Scholar] [CrossRef]
- Yuzawa, S.; Miyano, K.; Honbou, K.; Inagaki, F.; Sumimoto, H. The domain organization of p67 phox, a protein required for activation of the superoxide-producing NADPH oxidase in phagocytes. J. Innate. Immun. 2009, 1, 543–555. [Google Scholar] [CrossRef]
- Ono, R.; Kaisho, T.; Tanaka, T. PDLIM1 inhibits NF-kappaB-mediated inflammatory signaling by sequestering the p65 subunit of NF-kappaB in the cytoplasm. Sci. Rep. 2015, 5, 18327. [Google Scholar] [CrossRef] [PubMed]
- Copple, I.M.; den Hollander, W.; Callegaro, G.; Mutter, F.E.; Maggs, J.L.; Schofield, A.L.; Rainbow, L.; Fang, Y.; Sutherland, J.J.; Ellis, E.C.; et al. Characterisation of the NRF2 transcriptional network and its response to chemical insult in primary human hepatocytes: Implications for prediction of drug-induced liver injury. Arch. Toxicol. 2019, 93, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Atochina-Vasserman, E.N.; Tomer, Y.; Kadire, H.; Beers, M.F. Surfactant protein D protects against acute hyperoxic lung injury. Am. J. Respir. Crit. Care Med. 2008, 178, 805–813. [Google Scholar] [CrossRef]
- Angeloni, C.; Turroni, S.; Bianchi, L.; Fabbri, D.; Motori, E.; Malaguti, M.; Leoncini, E.; Maraldi, T.; Bini, L.; Brigidi, P.; et al. Novel targets of sulforaphane in primary cardiomyocytes identified by proteomic analysis. PLoS ONE 2013, 8, e83283. [Google Scholar] [CrossRef]
- Hasegawa, T.; Ohno, K.; Funahashi, S.; Miyazaki, K.; Nagano, A.; Sato, K. CLP36 interacts with palladin in dorsal root ganglion neurons. Neurosci. Lett. 2010, 476, 53–57. [Google Scholar] [CrossRef]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Petrillo, S.; Schirinzi, T.; Di Lazzaro, G.; D’Amico, J.; Colona, V.L.; Bertini, E.; Pierantozzi, M.; Mari, L.; Mercuri, N.B.; Piemonte, F.; et al. Systemic activation of Nrf2 pathway in Parkinson’s disease. Mov. Disord 2019. [Google Scholar] [CrossRef] [PubMed]
- Sahdeo, S.; Scott, B.D.; McMackin, M.Z.; Jasoliya, M.; Brown, B.; Wulff, H.; Perlman, S.L.; Pook, M.A.; Cortopassi, G.A. Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum. Mol. Genet. 2014, 23, 6848–6862. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. Int. J. Mol. Sci. 2020, 21, 916. https://doi.org/10.3390/ijms21030916
La Rosa P, Bertini ES, Piemonte F. The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. International Journal of Molecular Sciences. 2020; 21(3):916. https://doi.org/10.3390/ijms21030916
Chicago/Turabian StyleLa Rosa, Piergiorgio, Enrico Silvio Bertini, and Fiorella Piemonte. 2020. "The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia" International Journal of Molecular Sciences 21, no. 3: 916. https://doi.org/10.3390/ijms21030916
APA StyleLa Rosa, P., Bertini, E. S., & Piemonte, F. (2020). The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. International Journal of Molecular Sciences, 21(3), 916. https://doi.org/10.3390/ijms21030916