The Roles of Lipoprotein in Psoriasis

{kind=link}
Abstract
1. Introduction
2. Psoriasis
2.1. The Etiology of Psoriasis
2.2. The Molecular Mechanism of Psoriasis
3. The Relationships among Psoriasis, Cardiovascular Diseases, and Dyslipidemia
3.1. Normal Function of Fat and Cholesterol in Skin Cells
3.2. The Correlation Between Psoriasis and Atherosclerosis
3.3. The Relationship Between Psoriasis and Blood Lipids
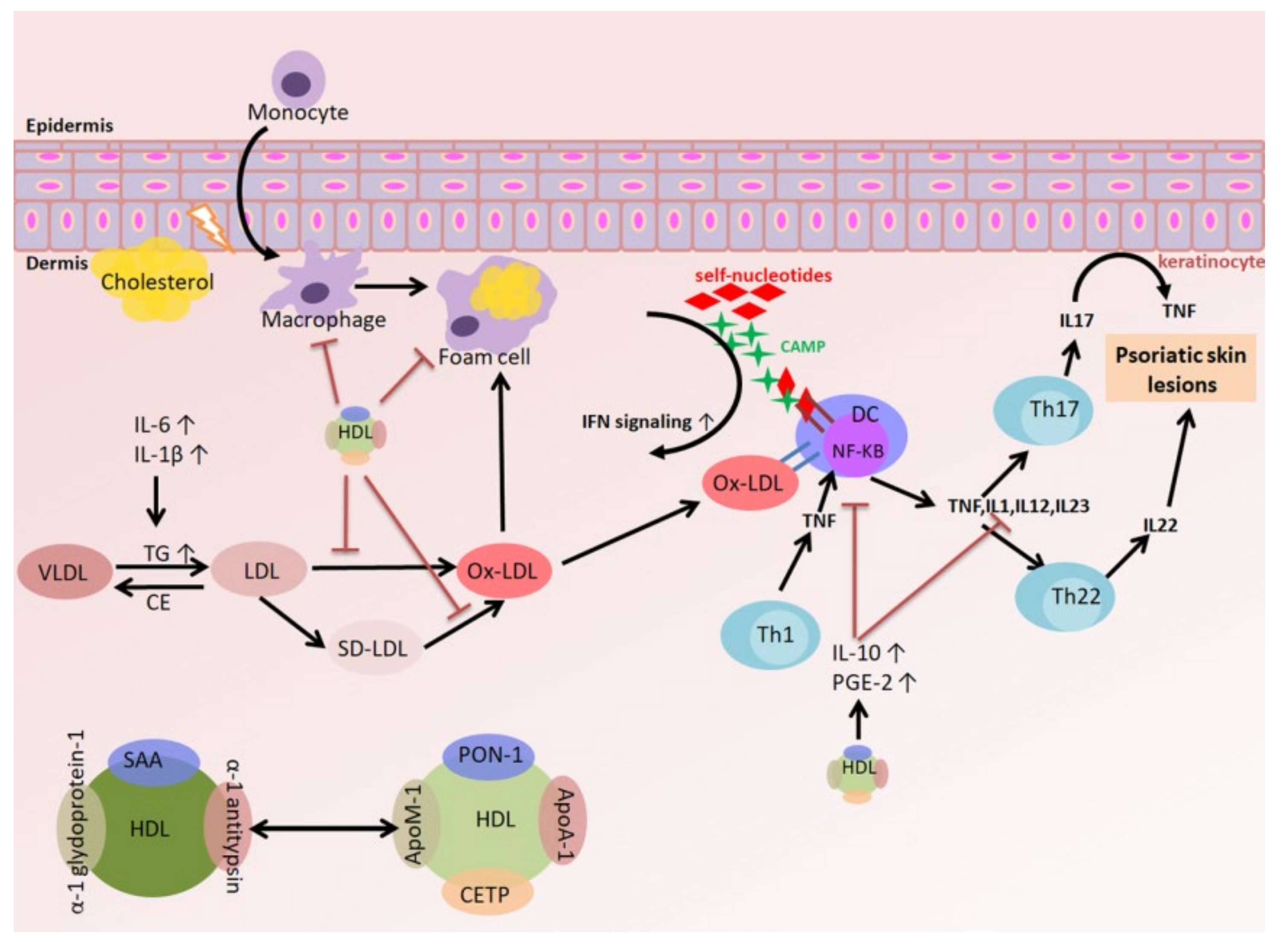
3.4. The Relationships among Oxidized LDL (oxLDL), the Inflammatory Response, and Psoriasis
3.5. The Relationships among HDL, the Inflammatory Response, and Psoriasis
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| apoA-I | Apolipoprotein A1 |
| ABCA1 | ATP-binding cassette A1 |
| aHDL | autoantibodies against HDL |
| AMP | Antimicrobial peptide |
| apo | apolipoprotein |
| BMI | Body mass index |
| C3 | Complement 3 |
| CAMP | Cathelicidin antimicrobial peptide |
| CCL20 | C-C motif chemokine ligand 20 |
| CD | Cluster of differentiation |
| CE | Cholesteryl ester |
| CER | Ceramide |
| CETP | Cholesteryl ester transfer protein |
| DC | Dendritic cell |
| ER | Endoplasmic reticulum |
| FFA | Free fatty acid |
| GM-CSF | Granulocyte macrophage colony stimulating factor |
| HDL | High density lipoprotein |
| HDL-C | High-density lipoprotein cholesterol |
| HDL-CE | High density lipoprotein cholesteryl ester |
| HFD | High fat diet |
| HSL | Hormone-sensitive lipase |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| IFN | Interferon |
| IL | Interleukin |
| IMQ | Imiquimod |
| JAK | Janus kinase |
| LCAT | Lecithin-cholesterol acyltransferase |
| LD | Lipid droplet |
| LDL | Low density lipoprotein |
| LDL-R | Low density lipoprotein receptor |
| Lp(a) | Lipoprotein(a) |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MHC | Major histocompatibility complex |
| NF-κB | Nuclear factor-κB |
| NK | Natural killer |
| ox-LDL | Oxidized LDL |
| pDC | Plasmacytoid dendritic cell |
| PI3K | Phosphoinositide 3 kinase |
| PON | Paraoxonase |
| PsA | Psoriatic arthritis |
| PV | Psoriasis vulgaris |
| RCT | Reverse cholesterol transport |
| SAA | Serum amyloid A |
| SFA | Saturated fatty acid |
| SR-B1 | Scavenger receptor class B type 1 |
| STAT | Signal transducer and activator of transcription |
| Th | T helper cell |
| Tfh | T follicular helper cells |
| TG | Triglyceride |
| TGF-β | Transforming growth factor beta |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| Treg | Regulatory T cell |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VLDL | Very-low-density lipoprotein |
References
- Ghazizadeh, R.; Tosa, M.; Ghazizadeh, M. Clinical improvement in psoriasis with treatment of associated hyperlipidemia. Am. J. Med. Sci. 2011, 341, 394–398. [Google Scholar] [CrossRef]
- Harrington, C.L.; Dey, A.K.; Yunus, R.; Joshi, A.A.; Mehta, N.N. Psoriasis as a human model of disease to study inflammatory atherogenesis. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H867–H873. [Google Scholar] [CrossRef]
- Kimball, A.B.; Szapary, P.; Mrowietz, U.; Reich, K.; Langley, R.G.; You, Y.; Hsu, M.C.; Yeilding, N.; Rader, D.J.; Mehta, N.N. Underdiagnosis and undertreatment of cardiovascular risk factors in patients with moderate to severe psoriasis. J. Am. Acad Derm. 2012, 67, 76–85. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Sorci-Thomas, M.G.; Thomas, M.J. Microdomains, Inflammation, and Atherosclerosis. Circ. Res. 2016, 118, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Amezaga Urruela, M.; Suarez-Almazor, M.E. Lipid paradox in rheumatoid arthritis: Changes with rheumatoid arthritis therapies. Curr. Rheumatol. Rep. 2012, 14, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Thomas, M.J.; Sorci-Thomas, M.G. High-Density Lipoprotein Functionality as a New Pharmacological Target on Cardiovascular Disease: Unifying Mechanism That Explains High-Density Lipoprotein Protection Toward the Progression of Atherosclerosis. J. Cardiovasc. Pharm. 2018, 71, 325–331. [Google Scholar] [CrossRef]
- Wang, S.H.; Yuan, S.G.; Peng, D.Q.; Zhao, S.P. HDL and ApoA-I inhibit antigen presentation-mediated T cell activation by disrupting lipid rafts in antigen presenting cells. Atherosclerosis 2012, 225, 105–114. [Google Scholar] [CrossRef]
- Whetzel, A.M.; Sturek, J.M.; Nagelin, M.H.; Bolick, D.T.; Gebre, A.K.; Parks, J.S.; Bruce, A.C.; Skaflen, M.D.; Hedrick, C.C. ABCG1 deficiency in mice promotes endothelial activation and monocyte-endothelial interactions. Arter. Thromb Vasc. Biol. 2010, 30, 809–817. [Google Scholar] [CrossRef]
- Batuca, J.R.; Ames, P.R.; Amaral, M.; Favas, C.; Isenberg, D.A.; Delgado Alves, J. Anti-atherogenic and anti-inflammatory properties of high-density lipoprotein are affected by specific antibodies in systemic lupus erythematosus. Rheumatol. (Oxf.) 2009, 48, 26–31. [Google Scholar] [CrossRef]
- Spah, F. Inflammation in atherosclerosis and psoriasis: Common pathogenic mechanisms and the potential for an integrated treatment approach. Br. J. Derm. 2008, 159, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Feldman, S.R.; Stern, R.S.; Thomas, J.; Rolstad, T.; Margolis, D.J. Determinants of quality of life in patients with psoriasis: A study from the US population. J. Am. Acad Derm. 2004, 51, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Thrash, B.; Menter, A. Comorbidities in psoriasis patients. Semin Cutan Med. Surg. 2010, 29, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Langan, S.M.; Seminara, N.M.; Shin, D.B.; Troxel, A.B.; Kimmel, S.E.; Mehta, N.N.; Margolis, D.J.; Gelfand, J.M. Prevalence of metabolic syndrome in patients with psoriasis: A population-based study in the United Kingdom. J. Invest. Derm. 2012, 132, 556–562. [Google Scholar] [CrossRef]
- Yeung, H.; Takeshita, J.; Mehta, N.N.; Kimmel, S.E.; Ogdie, A.; Margolis, D.J.; Shin, D.B.; Attor, R.; Troxel, A.B.; Gelfand, J.M. Psoriasis severity and the prevalence of major medical comorbidity: A population-based study. Jama Derm. 2013, 149, 1173–1179. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B. Risk of myocardial infarction in patients with psoriasis. JAMA 2006, 296, 1735–1741. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Parisi, R.; Symmons, D.P.; Griffiths, C.E.; Ashcroft, D.M.; Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team. Global epidemiology of psoriasis: A systematic review of incidence and prevalence. J. Invest. Derm. 2013, 133, 377–385. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Yang, J.; Sundrud, M.S.; Skepner, J.; Yamagata, T. Targeting Th17 cells in autoimmune diseases. Trends Pharm. Sci. 2014, 35, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Tsuji, G.; Chiba, T.; Kadono, T. Cardiovascular and Metabolic Diseases Comorbid with Psoriasis: Beyond the Skin. Intern. Med. 2017, 56, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, J.; Dobrowolska, A.; Rychlewska-Hanczewska, A.; Adamski, Z. New insights into the role of T cells in pathogenesis of psoriasis and psoriatic arthritis. Autoimmunity 2016, 49, 435–450. [Google Scholar] [CrossRef]
- Greb, J.E.; Goldminz, A.M.; Elder, J.T.; Lebwohl, M.G.; Gladman, D.D.; Wu, J.J.; Mehta, N.N.; Finlay, A.Y.; Gottlieb, A.B. Psoriasis. Nat. Rev. Dis. Primers 2016, 2, 16082. [Google Scholar] [CrossRef]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, A.; Nograles, K.E.; Johnson-Huang, L.M.; Fuentes-Duculan, J.; Cardinale, I.; Bonifacio, K.M.; Gulati, N.; Mitsui, H.; Guttman-Yassky, E.; Suarez-Farinas, M.; et al. IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS ONE 2014, 9, e90284. [Google Scholar] [CrossRef]
- Lavocat, F.; Ndongo-Thiam, N.; Miossec, P. Interleukin-25 Produced by Synoviocytes Has Anti-inflammatory Effects by Acting As a Receptor Antagonist for Interleukin-17A Function. Front. Immunol. 2017, 8, 647. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suarez-Farinas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J. Invest. Derm. 2011, 131, 677–687. [Google Scholar] [CrossRef]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef]
- Takahashi, H.; Numasaki, M.; Lotze, M.T.; Sasaki, H. Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol. Lett. 2005, 98, 189–193. [Google Scholar] [CrossRef]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Keijsers, R.R.; Joosten, I.; van Erp, P.E.; Koenen, H.J.; van de Kerkhof, P.C. Cellular sources of IL-17 in psoriasis: A paradigm shift? Exp. Derm. 2014, 23, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, Z.; Deng, L.; Zhang, G.; Yuan, X.; Huang, L.; Xu, W.; Shen, L. Modulation of STAT3 and STAT5 activity rectifies the imbalance of Th17 and Treg cells in patients with acute coronary syndrome. Clin. Immunol. 2015, 157, 65–77. [Google Scholar] [CrossRef]
- Del Rosso, J.Q.; Levin, J. Clinical relevance of maintaining the structural and functional integrity of the stratum corneum: Why is it important to you? J. Drugs Derm. 2011, 10, s5–s12. [Google Scholar]
- Harding, C.R. The stratum corneum: Structure and function in health and disease. Dermatol. Ther. 2004, 17, 6–15. [Google Scholar] [CrossRef]
- Proksch, E.; Brandner, J.M.; Jensen, J.M. The skin: An indispensable barrier. Exp. Derm. 2008, 17, 1063–1072. [Google Scholar] [CrossRef]
- van Smeden, J.; Hoppel, L.; van der Heijden, R.; Hankemeier, T.; Vreeken, R.J.; Bouwstra, J.A. LC/MS analysis of stratum corneum lipids: Ceramide profiling and discovery. J. Lipid Res. 2011, 52, 1211–1221. [Google Scholar] [CrossRef]
- Komatsu, M.; Takahashi, T.; Abe, T.; Takahashi, I.; Ida, H.; Takada, G. Evidence for the association of ultraviolet-C and H(2)O(2)-induced apoptosis with acid sphingomyelinase activation. Biochim. Biophys. Acta 2001, 1533, 47–54. [Google Scholar] [CrossRef]
- Charles, A.G.; Han, T.Y.; Liu, Y.Y.; Hansen, N.; Giuliano, A.E.; Cabot, M.C. Taxol-induced ceramide generation and apoptosis in human breast cancer cells. Cancer Chemother Pharm. 2001, 47, 444–450. [Google Scholar] [CrossRef]
- Motta, S.; Monti, M.; Sesana, S.; Mellesi, L.; Ghidoni, R.; Caputo, R. Abnormality of water barrier function in psoriasis. Role of ceramide fractions. Arch. Derm. 1994, 130, 452–456. [Google Scholar] [CrossRef]
- Motta, S.; Monti, M.; Sesana, S.; Caputo, R.; Carelli, S.; Ghidoni, R. Ceramide composition of the psoriatic scale. Biochim. Biophys. Acta 1993, 1182, 147–151. [Google Scholar] [CrossRef]
- Alessandrini, F.; Stachowitz, S.; Ring, J.; Behrendt, H. The level of prosaposin is decreased in the skin of patients with psoriasis vulgaris. J. Invest. Derm. 2001, 116, 394–400. [Google Scholar] [CrossRef]
- Nemes, Z.; Marekov, L.N.; Fesus, L.; Steinert, P.M. A novel function for transglutaminase 1: Attachment of long-chain omega-hydroxyceramides to involucrin by ester bond formation. Proc. Natl. Acad. Sci. USA 1999, 96, 8402–8407. [Google Scholar] [CrossRef]
- Incardona, J.P.; Eaton, S. Cholesterol in signal transduction. Curr. Opin. Cell Biol. 2000, 12, 193–203. [Google Scholar] [CrossRef]
- Kraemer, F.B.; Shen, W.J.; Harada, K.; Patel, S.; Osuga, J.; Ishibashi, S.; Azhar, S. Hormone-sensitive lipase is required for high-density lipoprotein cholesteryl ester-supported adrenal steroidogenesis. Mol. Endocrinol. 2004, 18, 549–557. [Google Scholar] [CrossRef]
- Shen, W.J.; Azhar, S.; Kraemer, F.B. Lipid droplets and steroidogenic cells. Exp. Cell Res. 2016, 340, 209–214. [Google Scholar] [CrossRef]
- Shen, W.J.; Hu, J.; Hu, Z.; Kraemer, F.B.; Azhar, S. Scavenger receptor class B type I (SR-BI): A versatile receptor with multiple functions and actions. Metabolism 2014, 63, 875–886. [Google Scholar] [CrossRef]
- Ueda, Y.; Royer, L.; Gong, E.; Zhang, J.; Cooper, P.N.; Francone, O.; Rubin, E.M. Lower plasma levels and accelerated clearance of high density lipoprotein (HDL) and non-HDL cholesterol in scavenger receptor class B type I transgenic mice. J. Biol. Chem. 1999, 274, 7165–7171. [Google Scholar] [CrossRef]
- Lim, H.Y.; Thiam, C.H.; Yeo, K.P.; Bisoendial, R.; Hii, C.S.; McGrath, K.C.; Tan, K.W.; Heather, A.; Alexander, J.S.; Angeli, V. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 2013, 17, 671–684. [Google Scholar] [CrossRef]
- Kaye, J.A.; Li, L.; Jick, S.S. Incidence of risk factors for myocardial infarction and other vascular diseases in patients with psoriasis. Br. J. Derm. 2008, 159, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Dixit, V.D. Immunological complications of obesity. Nat. Immunol. 2012, 13, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Herbert, D.; Franz, S.; Popkova, Y.; Anderegg, U.; Schiller, J.; Schwede, K.; Lorz, A.; Simon, J.C.; Saalbach, A. High-Fat Diet Exacerbates Early Psoriatic Skin Inflammation Independent of Obesity: Saturated Fatty Acids as Key Players. J. Invest. Derm. 2018, 138, 1999–2009. [Google Scholar] [CrossRef]
- Farshchian, M.; Zamanian, A.; Farshchian, M.; Monsef, A.R.; Mahjub, H. Serum lipid level in Iranian patients with psoriasis. J. Eur. Acad. Derm. Venereol. 2007, 21, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B.; Gelfand, J.M. Prevalence of cardiovascular risk factors in patients with psoriasis. J. Am. Acad. Derm. 2006, 55, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Pirillo, A.; Norata, G.D. Vascular inflammation and low-density lipoproteins: Is cholesterol the link? A lesson from the clinical trials. Br. J. Pharm. 2017, 174, 3973–3985. [Google Scholar] [CrossRef] [PubMed]
- Paiva-Lopes, M.J.; Delgado Alves, J. Psoriasis-associated vascular disease: The role of HDL. J. Biomed. Sci. 2017, 24, 73. [Google Scholar] [CrossRef]
- Goren, A.; Ozsolak, F.; Shoresh, N.; Ku, M.; Adli, M.; Hart, C.; Gymrek, M.; Zuk, O.; Regev, A.; Milos, P.M.; et al. Chromatin profiling by directly sequencing small quantities of immunoprecipitated DNA. Nat. Methods 2010, 7, 47–49. [Google Scholar] [CrossRef][Green Version]
- Ono, K. Current concept of reverse cholesterol transport and novel strategy for atheroprotection. J. Cardiol. 2012, 60, 339–343. [Google Scholar] [CrossRef]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Translation of high-density lipoprotein function into clinical practice: Current prospects and future challenges. Circulation 2013, 128, 1256–1267. [Google Scholar] [CrossRef]
- Barter, P.J.; Nicholls, S.; Rye, K.A.; Anantharamaiah, G.M.; Navab, M.; Fogelman, A.M. Antiinflammatory properties of HDL. Circ. Res. 2004, 95, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D.; Lim, H.Y.; Lee, H.G.; Yoon, D.Y.; Choe, Y.K.; Choi, I.; Paik, S.G.; Kim, Y.S.; Yang, Y.; Lim, J.S. Apolipoprotein A-I induces IL-10 and PGE2 production in human monocytes and inhibits dendritic cell differentiation and maturation. Biochem. Biophys. Res. Commun. 2005, 338, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Marsche, G.; Saemann, M.D.; Heinemann, A.; Holzer, M. Inflammation alters HDL composition and function: Implications for HDL-raising therapies. Pharmacol. Ther. 2013, 137, 341–351. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, C.-M.; Chen, C.-C.; Chu, C.-K.; Wang, K.-H.; Huang, C.-Y.; Lee, A.-W. The Roles of Lipoprotein in Psoriasis. Int. J. Mol. Sci. 2020, 21, 859. https://doi.org/10.3390/ijms21030859
Shih C-M, Chen C-C, Chu C-K, Wang K-H, Huang C-Y, Lee A-W. The Roles of Lipoprotein in Psoriasis. International Journal of Molecular Sciences. 2020; 21(3):859. https://doi.org/10.3390/ijms21030859
Chicago/Turabian StyleShih, Chun-Ming, Chang-Cyuan Chen, Chen-Kuo Chu, Kuo-Hsien Wang, Chun-Yao Huang, and Ai-Wei Lee. 2020. "The Roles of Lipoprotein in Psoriasis" International Journal of Molecular Sciences 21, no. 3: 859. https://doi.org/10.3390/ijms21030859
APA StyleShih, C.-M., Chen, C.-C., Chu, C.-K., Wang, K.-H., Huang, C.-Y., & Lee, A.-W. (2020). The Roles of Lipoprotein in Psoriasis. International Journal of Molecular Sciences, 21(3), 859. https://doi.org/10.3390/ijms21030859