FGF21 Protects against Aggravated Blood-Brain Barrier Disruption after Ischemic Focal Stroke in Diabetic db/db Male Mice via Cerebrovascular PPARγ Activation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
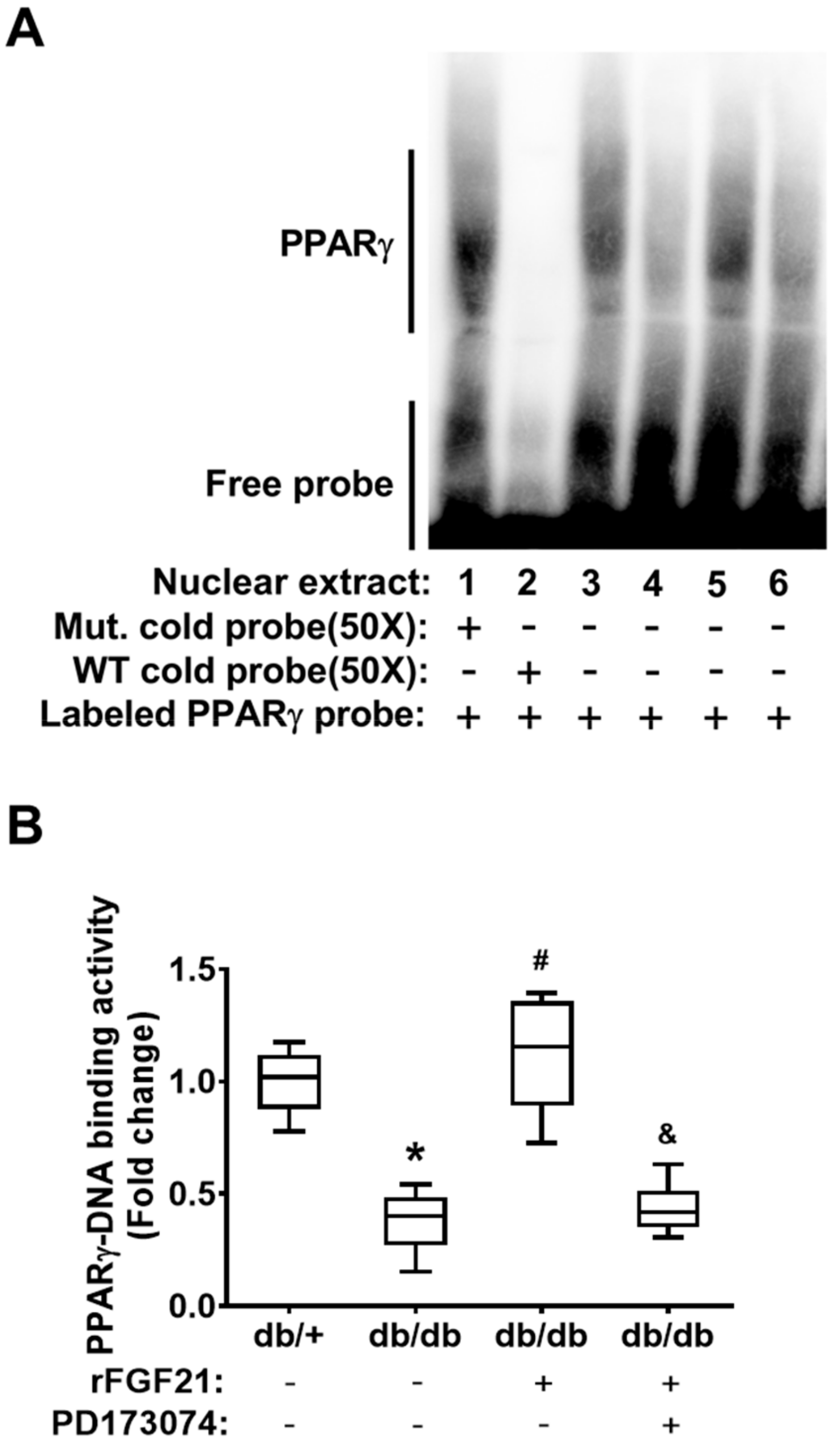
2.1. rFGF21 Increases PPARγ DNA-Binding Activity via FGFR1 at a Peri-infarct Area after Distal Middle Cerebral Occlusion (dMCAO) in db/db Mice
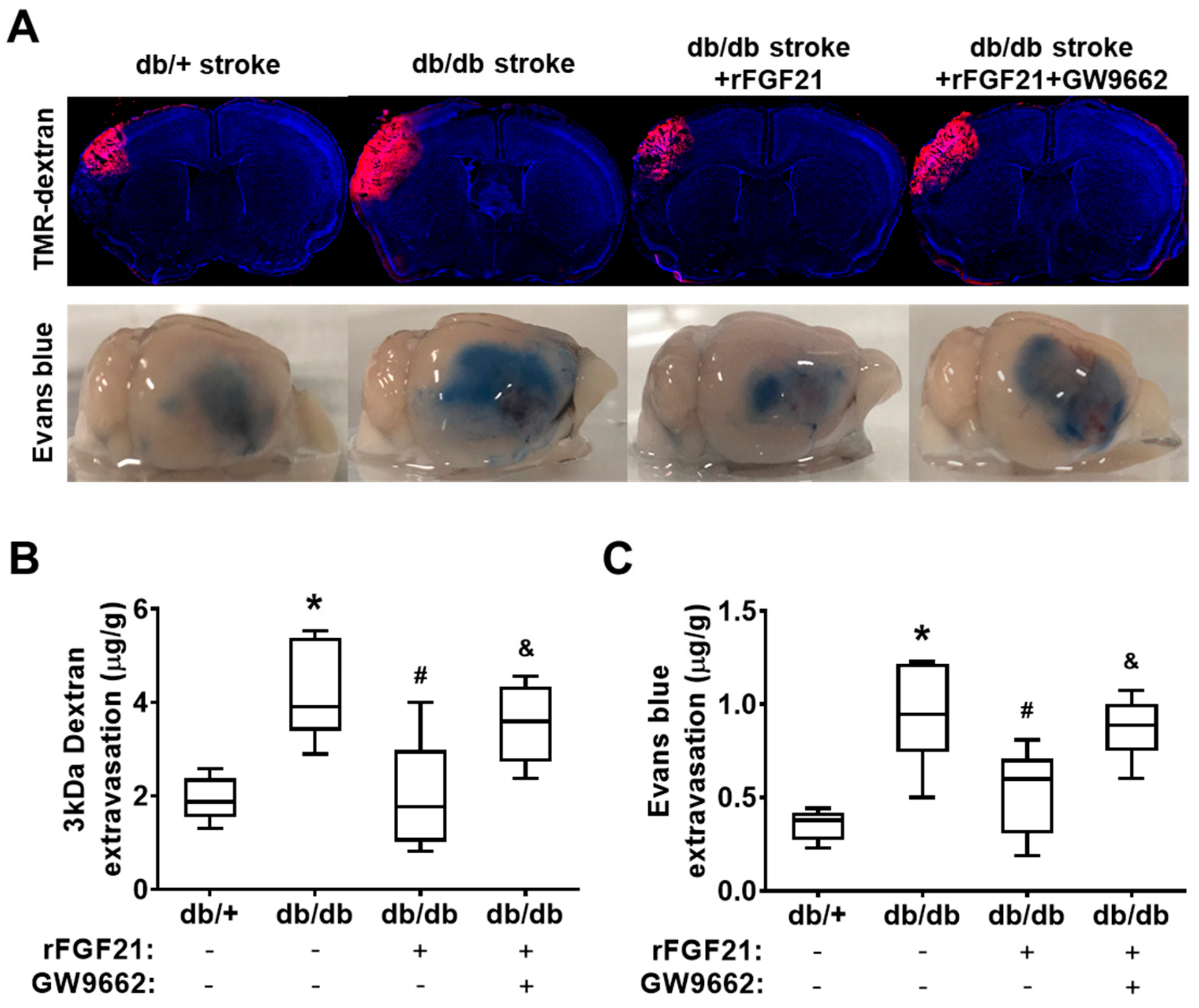
2.2. rFGF21 Reduces BBB Extravasation via PPARγ Activation at a Peri-Infarct Area after dMCAO in db/db Mice
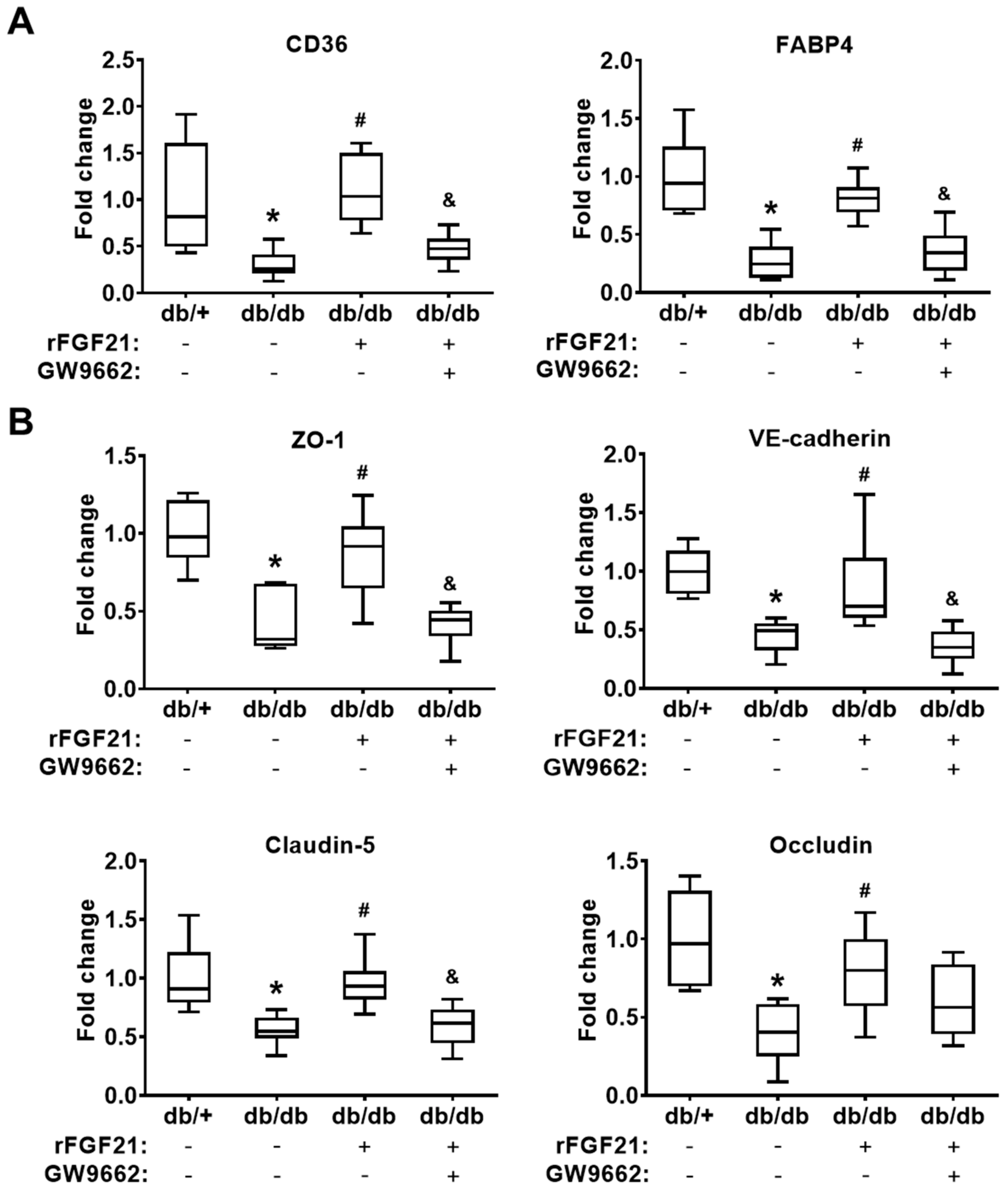
2.3. rFGF21 Inhibits Reduction of Junction Protein mRNA Expression via Cerebrovascular PPARγ Activation after dMCAO in db/db Mice
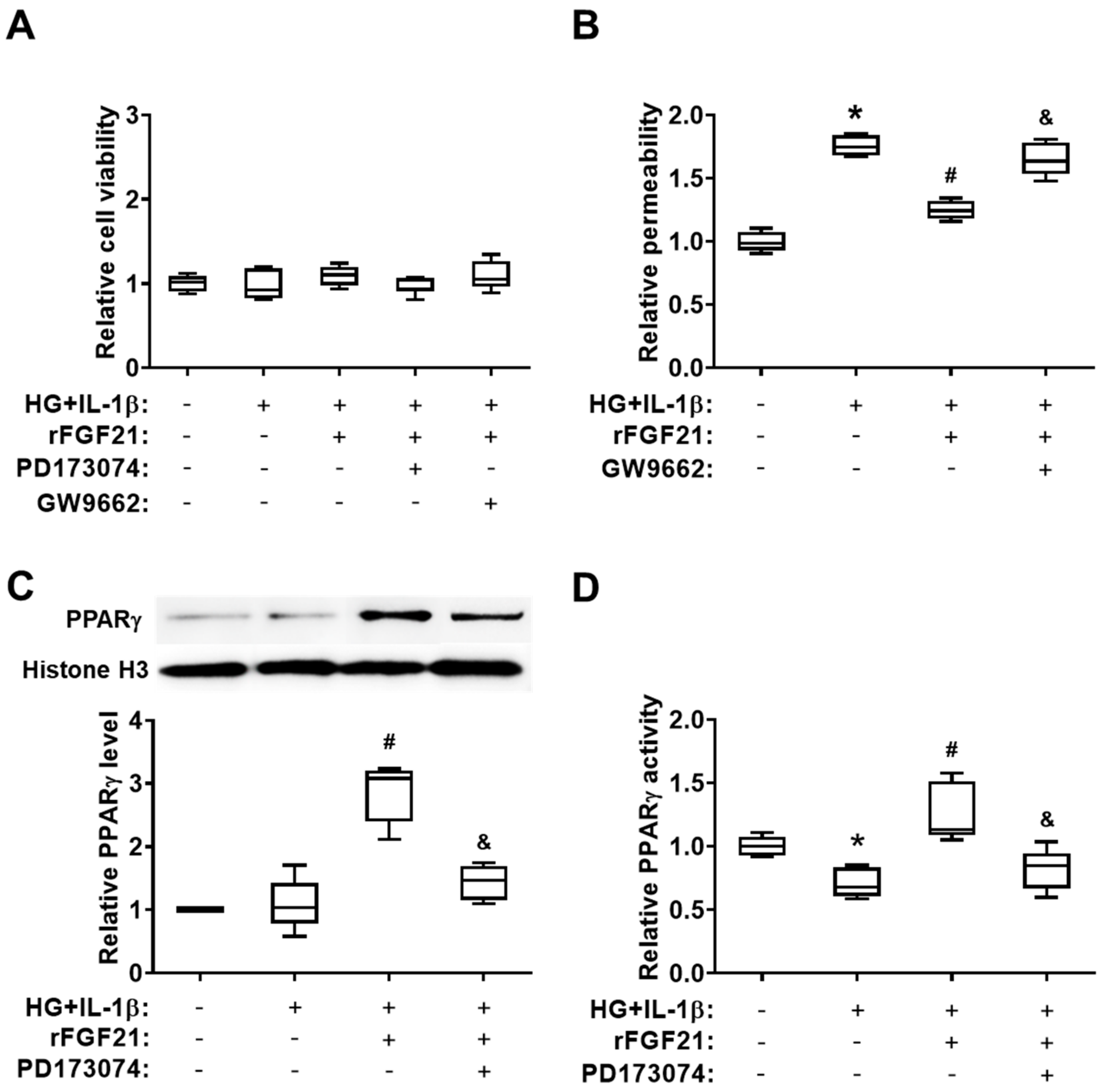
2.4. rFGF21 Ameliorates Transendothelial Permeability via Promoting FGFR1-mediated PPARγ Activity in Cultured HBMECs
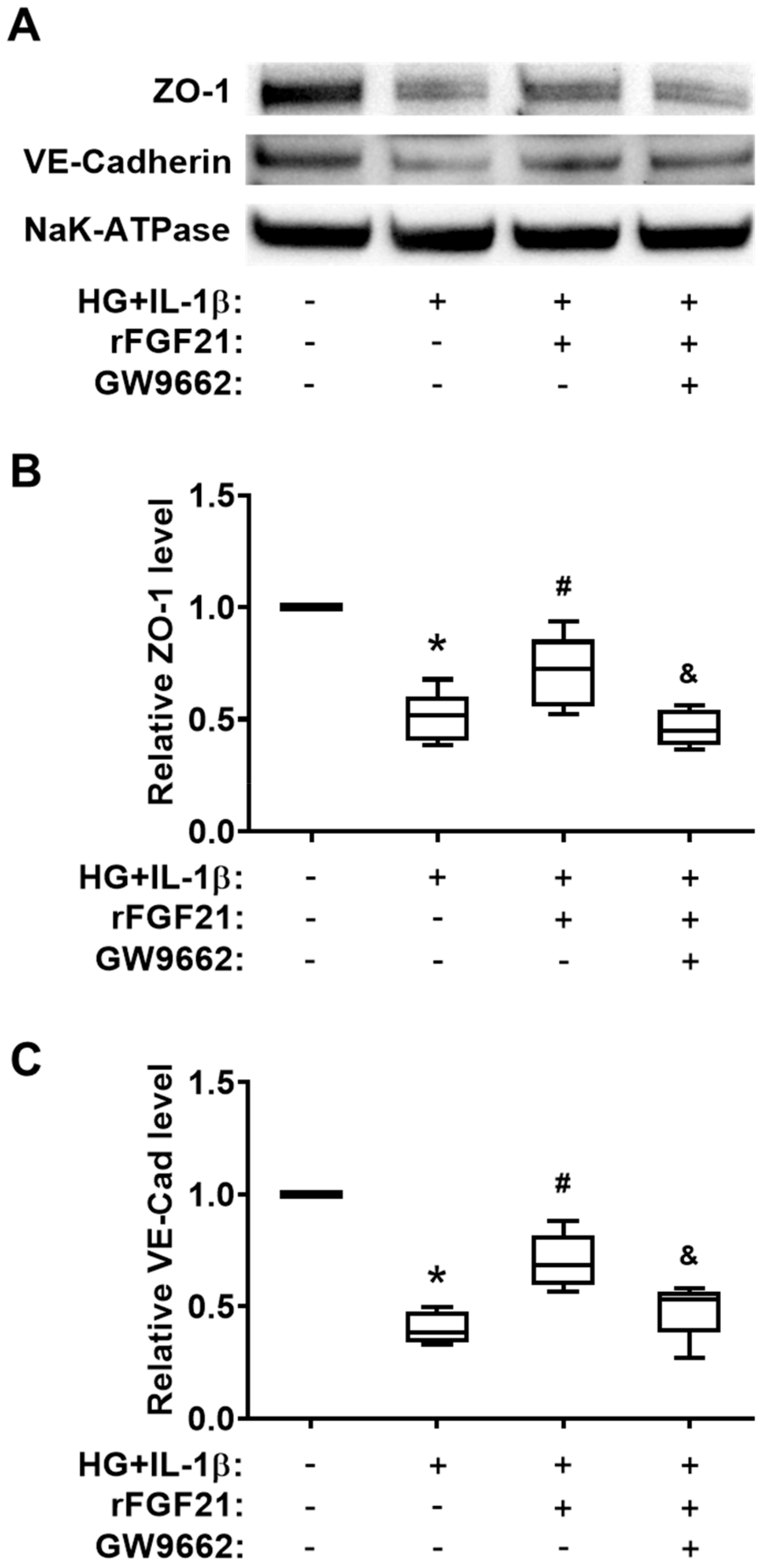
2.5. rFGF21 Increases Junction Protein Expression via Promoting PPARγ in Cultured HBMECs
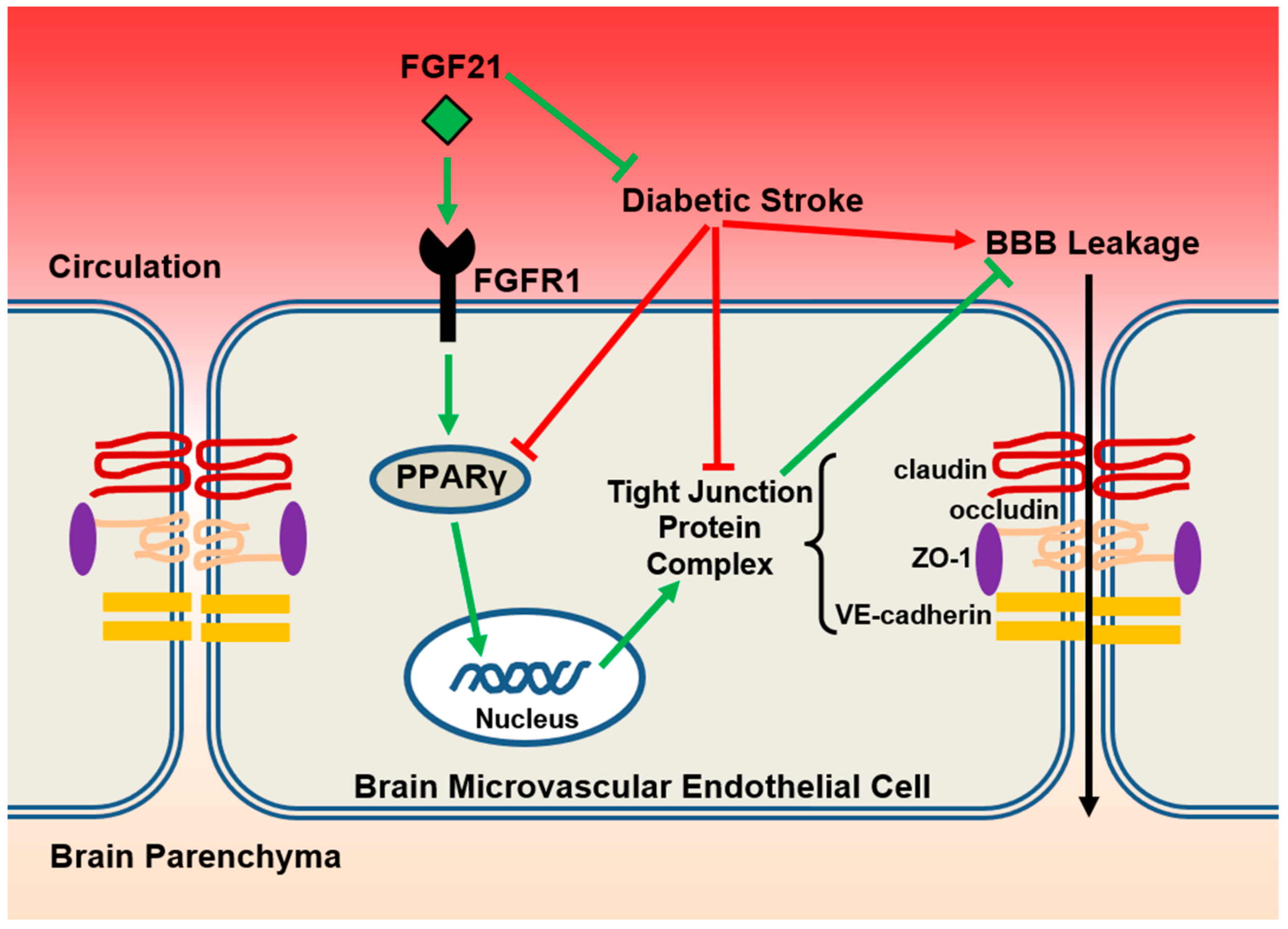
3. Discussion
4. Materials and Methods
4.1. Mouse Model of Focal Cerebral Ischemia
4.2. Cultured HBMECs and Injury Model
4.3. Experimental Groups
4.4. Mouse BBB Permeability Assay
4.5. Endothelial Monolayer Permeability Assay
4.6. Nuclear Extraction and EMSA
4.7. Mouse Brain Microvascular Endothelial Cell Isolation
4.8. RT-PCR Assay
4.9. PPARγ Activity in Vitro Assay
4.10. Western Blot
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| T2DM | type 2 diabetes mellitus |
| BBB | blood—brain barrier |
| FGF21 | fibroblast growth factor 21 |
| rFGF21 | recombinant fibroblast growth factor 21 |
| FGFR1 | fibroblast growth factor receptor 1 |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| dMCAO | distal middle cerebral artery occlusion |
| HBMEC | human brain microvascular endothelial cell |
| IL-1β | interleukin-1β |
| FITC | fluorescein isothiocyanate |
| EMSA | electrophoretic mobility shift assay |
References
- Beckman, J.A.; Paneni, F.; Cosentino, F.; Creager, M.A. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part II. Eur. Heart J. 2013, 34, 2444–2452. [Google Scholar] [CrossRef] [PubMed]
- Air, E.L.; Kissela, B.M. Diabetes, the metabolic syndrome, and ischemic stroke: Epidemiology and possible mechanisms. Diabetes Care 2007, 30, 3131–3140. [Google Scholar] [CrossRef] [PubMed]
- Tureyen, K.; Bowen, K.; Liang, J.; Dempsey, R.J.; Vemuganti, R. Exacerbated brain damage, edema and inflammation in type-2 diabetic mice subjected to focal ischemia. J. Neurochem. 2011, 116, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Kernan, W.N.; Launer, L.J.; Goldstein, L.B. What is the future of stroke prevention?: Debate: Polypill versus personalized risk factor modification. Stroke 2010, 41, S35–S38. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar] [CrossRef]
- Starr, J.M.; Wardlaw, J.; Ferguson, K.; MacLullich, A.; Deary, I.J.; Marshall, I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J. Neurol. Neurosurg. Psychiatry 2003, 74, 70–76. [Google Scholar] [CrossRef]
- Huber, J.D.; VanGilder, R.L.; Houser, K.A. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2660–H2668. [Google Scholar] [CrossRef]
- Ding, G.; Yan, T.; Chen, J.; Chopp, M.; Li, L.; Li, Q.; Cui, C.; Ning, R.; Jiang, Q. Persistent cerebrovascular damage after stroke in type two diabetic rats measured by magnetic resonance imaging. Stroke 2015, 46, 507–512. [Google Scholar] [CrossRef]
- Reeson, P.; Jeffery, A.; Brown, C.E. Illuminating the Effects of Stroke on the Diabetic Brain: Insights From Imaging Neural and Vascular Networks in Experimental Animal Models. Diabetes 2016, 65, 1779–1788. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, N.; Wang, Q.; Yu, Z.; Lin, L.; Yuan, J.; Guo, S.; Ahn, B.J.; Wang, X.J.; Li, X.; et al. Endocrine Regulator rFGF21 (Recombinant Human Fibroblast Growth Factor 21) Improves Neurological Outcomes Following Focal Ischemic Stroke of Type 2 Diabetes Mellitus Male Mice. Stroke 2018, 49, 3039–3049. [Google Scholar] [CrossRef]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Yu, R.T.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Yang, T.; Liu, H.; Han, L.; Zhang, K.; Hu, X.; Zhang, X.; Yin, K.J.; Gao, Y.; Bennett, M.V.L.; et al. Peroxisome proliferator-activated receptor gamma (PPARgamma): A master gatekeeper in CNS injury and repair. Prog. Neurobiol. 2018, 163–164, 27–58. [Google Scholar] [CrossRef] [PubMed]
- Min, L.J.; Mogi, M.; Shudou, M.; Jing, F.; Tsukuda, K.; Ohshima, K.; Iwanami, J.; Horiuchi, M. Peroxisome proliferator-activated receptor-gamma activation with angiotensin II type 1 receptor blockade is pivotal for the prevention of blood-brain barrier impairment and cognitive decline in type 2 diabetic mice. Hypertension 2012, 59, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Aronowski, J.; Zhao, X. Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 2011, 42, 1781–1786. [Google Scholar] [CrossRef]
- Yu, Z.; Lin, L.; Jiang, Y.; Chin, I.; Wang, X.; Li, X.; Lo, E.H.; Wang, X. Recombinant FGF21 Protects Against Blood-Brain Barrier Leakage Through Nrf2 Upregulation in Type 2 Diabetes Mice. Mol. Neurobiol. 2019, 56, 2314–2327. [Google Scholar] [CrossRef]
- Marechal, L.; Laviolette, M.; Rodrigue-Way, A.; Sow, B.; Brochu, M.; Caron, V.; Tremblay, A. The CD36-PPARgamma Pathway in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Briot, A.; Decaunes, P.; Volat, F.; Belles, C.; Coupaye, M.; Ledoux, S.; Bouloumie, A. Senescence Alters PPARgamma (Peroxisome Proliferator-Activated Receptor Gamma)-Dependent Fatty Acid Handling in Human Adipose Tissue Microvascular Endothelial Cells and Favors Inflammation. Arter. Thromb. Vasc. Biol. 2018, 38, 1134–1146. [Google Scholar] [CrossRef]
- Woo, Y.C.; Xu, A.; Wang, Y.; Lam, K.S. Fibroblast growth factor 21 as an emerging metabolic regulator: Clinical perspectives. Clin. Endocrinol. 2013, 78, 489–496. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. FGF21 as a mediator of adaptive responses to stress and metabolic benefits of anti-diabetic drugs. J. Endocrinol. 2015, 226, R1–R16. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; DiMarchi, R. FGF21 Revolutions: Recent Advances Illuminating FGF21 Biology and Medicinal Properties. Trends Endocrinol. Metab. 2015, 26, 608–617. [Google Scholar] [CrossRef]
- Planavila, A.; Redondo-Angulo, I.; Villarroya, F. FGF21 and Cardiac Physiopathology. Front. Endocrinol. (Lausanne) 2015, 6, 133. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A.; Estrada, E.Y.; Dencoff, J.E. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke 1998, 29, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Belayev, L.; Busto, R.; Zhao, W.; Ginsberg, M.D. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996, 739, 88–96. [Google Scholar] [CrossRef]
- Haelewyn, B.; David, H.N.; Colloc’h, N.; Colomb, D.G., Jr.; Risso, J.J.; Abraini, J.H. Interactions between nitrous oxide and tissue plasminogen activator in a rat model of thromboembolic stroke. Anesthesiology 2011, 115, 1044–1053. [Google Scholar] [CrossRef]
- Wang, Z.; Leng, Y.; Tsai, L.K.; Leeds, P.; Chuang, D.M. Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: The roles of HDAC and MMP-9 inhibition. J. Cereb. Blood Flow Metab. 2011, 31, 52–57. [Google Scholar] [CrossRef]
- Ketsawatsomkron, P.; Pelham, C.J.; Groh, S.; Keen, H.L.; Faraci, F.M.; Sigmund, C.D. Does peroxisome proliferator-activated receptor-gamma (PPAR gamma) protect from hypertension directly through effects in the vasculature? J. Biol. Chem. 2010, 285, 9311–9316. [Google Scholar] [CrossRef]
- Wang, Q.; Yuan, J.; Yu, Z.; Lin, L.; Jiang, Y.; Cao, Z.; Zhuang, P.; Whalen, M.J.; Song, B.; Wang, X.J.; et al. FGF21 Attenuates High-Fat Diet-Induced Cognitive Impairment via Metabolic Regulation and Anti-inflammation of Obese Mice. Mol. Neurobiol. 2018, 55, 4702–4717. [Google Scholar] [CrossRef]
- Hsuchou, H.; Pan, W.; Kastin, A.J. The fasting polypeptide FGF21 can enter brain from blood. Peptides 2007, 28, 2382–2386. [Google Scholar] [CrossRef]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef]
- Prakash, R.; Carmichael, S.T. Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr. Opin. Neurol. 2015, 28, 556–564. [Google Scholar] [CrossRef]
- Serlin, Y.; Levy, J.; Shalev, H. Vascular pathology and blood-brain barrier disruption in cognitive and psychiatric complications of type 2 diabetes mellitus. Cardiovasc. Psychiatry Neurol. 2011, 2011, 609202. [Google Scholar] [CrossRef]
- Kamada, H.; Yu, F.; Nito, C.; Chan, P.H. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: Relation to blood-brain barrier dysfunction. Stroke 2007, 38, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Reeson, P.; Tennant, K.A.; Gerrow, K.; Wang, J.; Weiser Novak, S.; Thompson, K.; Lockhart, K.L.; Holmes, A.; Nahirney, P.C.; Brown, C.E. Delayed inhibition of VEGF signaling after stroke attenuates blood-brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J. Neurosci. 2015, 35, 5128–5143. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pr. 2010, 87, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Mankovsky, B.N.; Ziegler, D. Stroke in patients with diabetes mellitus. Diabetes Metab. Res. Rev. 2004, 20, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Poppe, A.Y.; Majumdar, S.R.; Jeerakathil, T.; Ghali, W.; Buchan, A.M.; Hill, M.D. Canadian Alteplase for Stroke Effectiveness Study, I. Admission hyperglycemia predicts a worse outcome in stroke patients treated with intravenous thrombolysis. Diabetes Care 2009, 32, 617–622. [Google Scholar] [CrossRef]
- Williams, L.S.; Rotich, J.; Qi, R.; Fineberg, N.; Espay, A.; Bruno, A.; Fineberg, S.E.; Tierney, W.R. Effects of admission hyperglycemia on mortality and costs in acute ischemic stroke. Neurology 2002, 59, 67–71. [Google Scholar] [CrossRef]
- McMurray, F.; Cox, R.D. Mouse models and type 2 diabetes: Translational opportunities. Mamm Genome 2011, 22, 390–400. [Google Scholar] [CrossRef]
- Cipolletta, E.; Gambardella, J.; Fiordelisi, A.; Del Giudice, C.; Di Vaia, E.; Ciccarelli, M.; Sala, M.; Campiglia, P.; Coscioni, E.; Trimarco, B.; et al. Antidiabetic and Cardioprotective Effects of Pharmacological Inhibition of GRK2 in db/db Mice. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- King, A.J. The use of animal models in diabetes research. Br. J. Pharm. 2012, 166, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Shakya, A.K.; Perez-Pinzon, M.A.; Dave, K.R. Cerebral ischemic damage in diabetes: An inflammatory perspective. J. Neuroinflammation 2017, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Foo, J.P.; Liu, S.; Lim, S.C. The role of fibroblast growth factor 21 in diabetes and its complications: A review from clinical perspective. Diabetes Res. Clin. Pr. 2015, 108, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Sa-Nguanmoo, P.; Chattipakorn, N.; Chattipakorn, S.C. Potential roles of fibroblast growth factor 21 in the brain. Metab Brain Dis 2016, 31, 239–248. [Google Scholar] [CrossRef]
- Li, G.; Simon, M.J.; Cancel, L.M.; Shi, Z.D.; Ji, X.; Tarbell, J.M.; Morrison, B., 3rd; Fu, B.M. Permeability of endothelial and astrocyte cocultures: In vitro blood-brain barrier models for drug delivery studies. Ann. Biomed. Eng. 2010, 38, 2499–2511. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Pearson, A.; Campbell, J.; Shnyder, S.D.; Knowles, M.A.; Ashworth, A.; Turner, N.C. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3-mutant cancer. Cancer Discov. 2013, 3, 1058–1071. [Google Scholar] [CrossRef]
- Li, P.; Song, Y.; Zan, W.; Qin, L.; Han, S.; Jiang, B.; Dou, H.; Shao, C.; Gong, Y. Lack of CUL4B in Adipocytes Promotes PPARgamma-Mediated Adipose Tissue Expansion and Insulin Sensitivity. Diabetes 2017, 66, 300–313. [Google Scholar] [CrossRef]
- Hind, W.H.; England, T.J.; O’Sullivan, S.E. Cannabidiol protects an in vitro model of the blood-brain barrier from oxygen-glucose deprivation via PPARgamma and 5-HT1A receptors. Br. J. Pharm. 2016, 173, 815–825. [Google Scholar] [CrossRef]
- Yanagida, K.; Liu, C.H.; Faraco, G.; Galvani, S.; Smith, H.K.; Burg, N.; Anrather, J.; Sanchez, T.; Iadecola, C.; Hla, T. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc. Natl. Acad. Sci. USA 2017, 114, 4531–4536. [Google Scholar] [CrossRef]
- Liu, N.; Jiang, Y.; Chung, J.Y.; Li, Y.; Yu, Z.; Kim, J.W.; Lok, J.M.; Whalen, M.J.; Wang, X. Annexin A2 Deficiency Exacerbates Neuroinflammation and Long-Term Neurological Deficits after Traumatic Brain Injury in Mice. Int J. Mol. Sci 2019, 20. [Google Scholar] [CrossRef]
- Boulay, A.C.; Saubamea, B.; Decleves, X.; Cohen-Salmon, M. Purification of Mouse Brain Vessels. J. Vis. Exp. 2015, 105, e53208. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Lin, L.; Liu, N.; Wang, Q.; Yuan, J.; Li, Y.; Chung, K.K.; Guo, S.; Yu, Z.; Wang, X. FGF21 Protects against Aggravated Blood-Brain Barrier Disruption after Ischemic Focal Stroke in Diabetic db/db Male Mice via Cerebrovascular PPARγ Activation. Int. J. Mol. Sci. 2020, 21, 824. https://doi.org/10.3390/ijms21030824
Jiang Y, Lin L, Liu N, Wang Q, Yuan J, Li Y, Chung KK, Guo S, Yu Z, Wang X. FGF21 Protects against Aggravated Blood-Brain Barrier Disruption after Ischemic Focal Stroke in Diabetic db/db Male Mice via Cerebrovascular PPARγ Activation. International Journal of Molecular Sciences. 2020; 21(3):824. https://doi.org/10.3390/ijms21030824
Chicago/Turabian StyleJiang, Yinghua, Li Lin, Ning Liu, Qingzhi Wang, Jing Yuan, Yadan Li, Kelly K. Chung, Shuzhen Guo, Zhanyang Yu, and Xiaoying Wang. 2020. "FGF21 Protects against Aggravated Blood-Brain Barrier Disruption after Ischemic Focal Stroke in Diabetic db/db Male Mice via Cerebrovascular PPARγ Activation" International Journal of Molecular Sciences 21, no. 3: 824. https://doi.org/10.3390/ijms21030824
APA StyleJiang, Y., Lin, L., Liu, N., Wang, Q., Yuan, J., Li, Y., Chung, K. K., Guo, S., Yu, Z., & Wang, X. (2020). FGF21 Protects against Aggravated Blood-Brain Barrier Disruption after Ischemic Focal Stroke in Diabetic db/db Male Mice via Cerebrovascular PPARγ Activation. International Journal of Molecular Sciences, 21(3), 824. https://doi.org/10.3390/ijms21030824