Abstract
The ventral tegmental area (VTA) plays an important role in the reward and motivational processes that facilitate the development of drug addiction. Presynaptic α1-AR activation modulates glutamate and Gamma-aminobutyric acid (GABA) release. This work elucidates the role of VTA presynaptic α1-ARs and their modulation on glutamatergic and GABAergic neurotransmission during cocaine sensitization. Excitatory and inhibitory currents (EPSCs and IPSCs) measured by a whole cell voltage clamp show that α1-ARs activation increases EPSCs amplitude after 1 day of cocaine treatment but not after 5 days of cocaine injections. The absence of a pharmacological response to an α1-ARs agonist highlights the desensitization of the receptor after repeated cocaine administration. The desensitization of α1-ARs persists after a 7-day withdrawal period. In contrast, the modulation of α1-ARs on GABA neurotransmission, shown by decreases in IPSCs’ amplitude, is not affected by acute or chronic cocaine injections. Taken together, these data suggest that α1-ARs may enhance DA neuronal excitability after repeated cocaine administration through the reduction of GABA inhibition onto VTA dopamine (DA) neurons even in the absence of α1-ARs’ function on glutamate release and protein kinase C (PKC) activation. α1-AR modulatory changes in cocaine sensitization increase our knowledge of the role of the noradrenergic system in cocaine addiction and may provide possible avenues for therapeutics.
1. Introduction
The mesocorticolimbic system is composed of dopamine (DA) neurons projecting mainly from the ventral tegmental area (VTA) to cortical and ventral forebrain structures [1,2,3]. The activation of VTA DA neurons has been implicated in motivated behaviors as well as in mediating the reinforcing actions of drug abuse [4,5,6]. VTA DA neurons receive noradrenergic (NE) inputs from the locus coeruleus and other pontine structures [7,8], and tracing studies have shown that NE afferents have extrasynaptic and synaptic connections on VTA DA neurons [9]. Moreover, the VTA contains alpha-1 adrenoreceptors (α1-ARs) [10], which are located primarily in presynaptic elements [11,12,13]. NE inputs have been shown to facilitate VTA DA neuronal transmission and induce changes in burst firing via α1-ARs [14,15,16]. Also, α1-ARs participate in the development of stress and anxiety responses, and in addiction-related behaviors as well [17,18,19,20]. Furthermore, activation of α1-ARs at presynaptic terminals increase glutamate (Glu) and decreased GABA release onto VTA DA neurons, changing their excitability [12,13].
Diverse brain nuclei send Glu and GABAergic inputs to the VTA. The Glu innervation to the VTA includes afferents from the prefrontal cortex, the lateral hypothalamus, medial habenula, bed nucleus of the stria terminalis, dorsal raphe, laterodorsal, and pedunculopontine tegmental nuclei [21,22,23,24,25,26,27]. Also, the presence of local Glu neurons has been demonstrated [28,29]. On the other hand, the mesopontine tegmentum, the lateral habenula via the rostromedial tegmentum, nucleus accumbens, and the periaqueductal gray provide GABAergic innervation to the VTA [30,31,32].
Chronic cocaine exposure leads to a progressive increase in locomotor activity in rats termed, behavioral sensitization [33,34]. As a result of chronic drug exposure, neuroadaptations in mesocorticolimbic synapses, including DA, Glu, and GABA neurotransmission, occur [35,36,37]. In the VTA, DA cell activation plays an important role in cocaine sensitization [38,39]. For example, increases in the AMPA/NMDA ratio [39,40,41] and VTA DA firing [42] develop during cocaine sensitization. On the other hand, daily exposure to cocaine reduces the GABAA receptor-mediated inhibition of DA neurons facilitating neuronal excitation [43]. Cocaine-induced inhibition of monoamine reuptake may further enhance the availability of noradrenaline at synapses and cause stronger stimulation of α1-ARs [44]. Also, postsynaptic α1-ARs activation on DA neurons contributes to increase DA levels, motor activity, and enhance the pacemaker and burst firing frequency of VTA cells [45,46]. α1b-AR knockout animals and α1-AR blockade demonstrate the critical role of this receptor for the development of addictive processes related to cocaine sensitization and self-administration [19,47,48,49]. Furthermore, prazosin (α1-AR antagonist) attenuates a cocaine-induced reinstatement of drug-seeking behavior in rats [46,50]. Prazosin can also reduce relapse in cocaine addicts [51]. Moreover, α1-ARs’ density changes in different rat brain regions after cocaine sensitization [52]. Investigations have shown that decreased α1-AR regulation of an mGlu-R-mediated current might be a cellular mechanism associated with the effects of cocaine withdrawal in a self-administration protocol [53]. In sum, these studies suggest that α1-ARs contribute to the behavioral and neurochemical effects of cocaine addiction.
Electrophysiological studies of VTA α1-AR direct modulation on Glu and GABA terminals at different stages of cocaine sensitization have not been reported. Here, we tested the contribution of presynaptic α1-ARs activation in VTA DA neuronal excitability: glutamatergic and GABAergic modulation after acute and chronic cocaine treatment and after a withdrawal period.
2. Results
2.1. Presynaptic α1-AR Modulation of Glutamate Release on VTA DA Neurons after Acute Cocaine Injection
We first tested the effect of α1-ARs activation on glutamate release in animals treated with one cocaine injection. Animals were injected with either saline (0.9%; n = 10) or cocaine (15 mg/kg, i.p.; n = 10), and motor activity was recorded at 10 min intervals for 60 min. For the total activity (Figure 1A), an unpaired t-test (df = 18, t = 4.104, p = 0.0007) showed differences between saline (n = 10; 1003 ± 118.6 photocell counts) and cocaine (n = 10; 2907 ± 448.4 photocell counts) treated animals. These results indicate that acute cocaine-treated animals present the characteristic of an increased locomotion response to cocaine.

Figure 1.
α1-ARs-mediated effect on AMPA EPSCs is preserved in ventral tegmental area (VTA) dopamine (DA) neurons after one cocaine/saline injection. (A) The bar graph shows total locomotor activity recorded from saline- (0.9%, white bar; n = 10) and cocaine-treated (15 mg/kg i.p., black bar; n = 10) animals. The locomotion of cocaine-treated animals was significantly increased when compared to saline-treated controls (Unpaired t-test p < 0.001). (B,C) Representative recordings from saline- and cocaine-treated animals, respectively, showing that phenylephrine superfusion (10 μM, 10 min) induces a significant increase in AMPA EPSCs amplitude in VTA DA neurons in both groups. (D) A time course summary of phenylephrine’s effect on AMPA EPSCs recorded from VTA DA neurons from saline- (n = 10) and cocaine- (n = 10) treated animals at 8 min from control recordings (2 min intervals), 5 and 10 min phenylephrine (10 μM), and 5 and 10 min washout (One-way ANOVA, Newman-Keuls post-hoc). (E) The bar graph shows that 10 min of phenylephrine application resulted in a ~28% increase in AMPA EPSCs amplitude in VTA DA neurons from saline- (n = 10/10) and cocaine- (n = 10/10) treated animals when compared to control recordings (One-way ANOVA, Newman-Keuls post-hoc.). One-way ANOVA, Newman-Keuls post-hoc * p < 0.05. Unpaired t-test *** p < 0.001. Two-way repeated-measures ANOVA, Bonferroni post-hoc # p < 0.05. n = the number of cells recorded/number of animals.
Twenty-four hours after the injection, brain slices were prepared and AMPA EPSCs were recorded on VTA DA neurons from saline- and cocaine-treated animals. Ten minutes of phenylephrine superfusion (10 μM) increased AMPA EPSCs amplitude in both animal groups: saline group: 127.76 ± 7.22% of control (n = 10; One-way ANOVA F2,29 = 4.81, p < 0.05, Figure 1B,C); cocaine group: 127.99 ± 9.13% of control (n = 10; One-way ANOVA F2,29 = 7.14, p < 0.05, Figure 1C–E). No differences were found between the DA-cells of saline- and cocaine-treated animals after 10 min of phenylephrine superfusion (Two-way repeated-measures ANOVA, treatment factor F1,126 = 2.092, p = 0.16, time factor F7,126 = 8.922, p < 0.0001, interaction F7, 17 = 2.96. p < 0.01). Although, Bonferroni post hoc test detected differences between treatments (saline vs cocaine) after 5 and 10 min of washout (p < 0.05 and p < 0.01, respectively). These results suggest that α1-ARs are functional after one cocaine injection.
2.2. α1-ARs-Mediated AMPA EPSCs Effect is Absent in VTA DA Neurons from Cocaine Sensitized Animals
To determine if presynaptic α1-AR modulation on glutamate release was altered in VTA DA neurons after behavioral sensitization, animals were injected with either saline (0.9%; n = 5) or cocaine (15 mg/kg, i.p.; n = 8) for five consecutive days. For total activity (Figure 2A), Two-way repeated measures ANOVA showed differences in treatment: saline vs. cocaine (F1,44 = 14.80; p = 0.0027). Also, total activity between the days (F4,44 = 4.09; p = 0.0066) and the interaction between treatment and days showed differences (F4,44 = 7.41; p = 0.0001). A Bonferroni post hoc analysis revealed that cocaine-treated animals had a higher total activity than saline-treated animals on days 4 (p = 0.001) and 5 (p = 0.001). The time course of locomotor responses (Figure 2B) compares day 1 and day 5 after saline or cocaine injection. Two-way repeated measures ANOVA showed differences in treatment (F3,110 = 23.22; p < 0.0001) and time (F5,110 = 13.661; p < 0.0001). The interaction between time and treatment showed significant differences (F15,110 = 2.21, p < 0.01). The Bonferroni post hoc analysis demonstrated an increase in locomotor activity on day 5 of cocaine treatment during the 60 min recording after cocaine injection compared to one day of cocaine treatment (p < 0.001). Together, these results indicate that the animals used in this study were successfully sensitized to cocaine.

Figure 2.
α1-ARs-mediated effect on AMPA EPSCs is absent in the VTA DA neurons of cocaine sensitized animals. (A) The bar graph shows the total locomotor activity recorded from saline- (0.9%, white bars; n = 5) and cocaine- (15 mg/kg i.p., black bars; n = 8) treated animals. Cocaine-injected animals show a progressive increase in locomotor activity over 5 days. On days 4 and 5, the total activity was significantly increased when compared to day 1 in cocaine-treated animals (Two-way repeated measures ANOVA, Bonferroni post hoc test p < 0.001). (B) The time course of locomotor activity on days 1 (grey) and 5 (black) of cocaine- (15 mg/kg, i.p.) (filled) or saline- (unfilled) treated animals. Cocaine sensitization was manifested as an increase in locomotor activity during the first 10–50 min after cocaine injection in comparison with day 1. The data (pcc/60 min; Mean ± SEM) were analyzed by Two-way repeated measures ANOVA, Bonferroni post hoc test (p < 0.001). (C,D) Representative recordings from saline- and cocaine-treated animals, respectively, illustrating that phenylephrine superfusion (10 μM, 10 min) induces a significant increment in AMPA EPSCs amplitude only in VTA DA neurons from saline-treated animals. (E) A time course summary of the effects of phenylephrine bath application on AMPA EPSCs amplitude recorded from VTA DA neurons from saline-treated animals (n = 8/5) and VTA DA neurons from cocaine-treated animals (n = 9/8) at 8 min after control recording (2 min intervals), 5 and 10 min phenylephrine (10 μM), and 5 and 10 min washout. A 10 min phenylephrine application increased the AMPA EPSCs amplitude only in neurons from saline-treated animals compared to control recordings (One-way ANOVA, Newman-Keuls post-hoc, p < 0.05). A washout of phenylephrine’s response was observed. (F). A bar graph showing that, in neurons from saline-treated animals (n = 8), phenylephrine application resulted in an ~36% increase in AMPA EPSCs amplitude compared to control recordings (One-way ANOVA, Newman-Keuls post-hoc, p < 0.05). No significant differences were observed on VTA DA neurons in cocaine-treated animals (n = 9) after 10 min of phenylephrine application (104.6 ± 5.3%). Two-way repeated measures ANOVA showed in factor time, at 10 min of phenylephrine superfusion (F7,105 = 2.831, Bonferroni post hoc, p < 0.05) * p < 0.05; ** p < 0.01, *** p < 0.001. n = the number of cells recorded/number of animals.
Recordings of VTA DA neurons from saline-treated animals show that a bath application of the selective α1-AR agonist, phenylephrine (10 μM), for 10 min, but not for 5 min, increased AMPA EPSCs peak amplitude to 136.9 ± 16.2% of control (n = 8; Figure 2C–F). However, on VTA DA neurons from 5-days cocaine-treated animals, phenylephrine superfusion was unable to exert its excitatory action on AMPA EPSCs after a 5- or 10-min application (control 99.9 ± 1.04%; phenylephrine 10 min 104.6 ± 5.33%; n = 9; Two-way repeated measures ANOVA treatment factor F1.105 = 0.644, p = 0.43; time factor F7,105 = 2.831, p < 0.01; interaction F7,105 = 1.64, p = 0.13; Bonferroni post hoc, p < 0.05, Figure 2E,F). These results strongly suggest that α1-ARs have been altered by 5 days of cocaine administration.
2.3. Presynaptic PKC Activity is Decreased in VTA Slices of Cocaine Sensitized Animals
To determine the mechanism of α1-AR physiological alteration by the cocaine sensitization process, we recorded spontaneous EPSCs (sEPSCs) from VTA DA neurons of saline- and cocaine-treated animals in the presence of a PKC activator. For this, rats were injected with either saline (0.9%; n = 6) or cocaine (15 mg/kg, i.p.; n = 6) for five consecutive days. Animals treated with 5 days of cocaine showed an increase the in total locomotor activity in comparison to saline-injected animals (two-way repeated measures ANOVA, treatment F1,10 = 78.72, p < 0.0001; time F4,10 = 3.69, p < 0,05, Figure 3A). Bonferroni post hoc analysis revealed that cocaine treated animals had higher total activity on day 5 than day 1 (p < 0.001). The time course of locomotor responses (Figure 3B) compares day 1 and day 5 after saline or cocaine injection. The Two-way repeated measures ANOVA showed differences in treatment (F3,100 = 28.63; p < 0.0001) and time (F5,100 = 28.63; p < 0.0001). Interactions between time and treatment were significantly different (F15,100 = 4.71, p < 0.0001). Bonferroni post-hoc showed significant differences between saline and cocaine treatment on day 1 until 40 min after injection (p < 0.01). Also, Bonferroni post-hoc showed significant differences between day 1 and 5 of cocaine injection after 40 min of the treatment (p < 0.001). Together, these results indicate that animals were successfully sensitized to cocaine.

Figure 3.
Phorbol 12-myristate 13-acetate (PMA) pre-treatment on VTA DA neurons from cocaine-sensitized animals failed to increase sEPSCs frequency or amplitude. (A) The bar graph shows the total locomotor activity recorded from saline- (0.9%, white bars; n = 6) and cocaine- (15 mg/kg i.p., black bars; n = 6) treated animals. Cocaine-injected animals showed a progressive increase in locomotor activity over days. On day 5, total activity was significantly increased when compared to day 1 in cocaine-treated animals (Two-way repeated measures ANOVA, time F4,40 = 3.69, p < 0.05; treatment F1,40 = 78.72, p < 0.001). (B) The time course of locomotor activity on days 1 (grey) and 5 (black) of cocaine- (15 mg/kg, i.p.; filled; n = 6) or saline- (unfilled; n = 6) treated animals. Cocaine sensitization was manifested as an increase in locomotor activity during the first 40 min after cocaine injection in comparison with day 1. The data (pcc/60 min; Mean ± SEM) were analyzed by Two-way repeated measures ANOVA, time F8, 160 = 33.99, treatment F3,160 = 25.97, p < 0.001, Bonferroni post hoc test, p < 0.01. (C). Representative recordings from the neurons of saline-treated animals, illustrating that PMA pretreatment (0.5 μM; 30 min) increases sEPSC frequency but not the amplitude. (D). Representative recordings from neurons of cocaine-sensitized animals illustrating that PMA pretreatment failed to increase the frequency or amplitude of sEPSCs. (E) and (G) The cumulative probability of the inter-event interval and amplitude cumulative distribution, respectively, from control- and PMA-treated neurons of saline-injected animals. Plots were constructed from the cells used in (C,F,H). The cumulative probability of inter-event interval and amplitude cumulative distribution (respectively) from control- and PMA-treated neurons of cocaine-sensitized animals. The plots were constructed from the cells used in (D,I). A summary graph showing that PMA treatment increased the mean sEPSC frequency in VTA DA cells from saline- (n = 13/6) but not cocaine- (n = 9/6) injected animals. (J) Summary graph shows that PMA treatment failed to increase the mean sEPSC amplitude in VTA DA cells from saline- (n = 8/6) and cocaine- (n = 9/6) injected animals. An unpaired t-test. * p < 0.05, ** p < 0.01, *** p < 0.001. n = the number of cells recorded/number of animals.
In order to determine PKC’s role in Glu presynaptic terminals, sEPSCs were recorded from VTA DA neurons in brain slices of saline- and cocaine-treated animals preincubated for 30 min with phorbol 12-myristate 13-acetate (PMA; 0.5 µM; PKC activator). In addition, intracellular GÖ6976 application (PKC antagonist; 1 µM) via the pipette was used to exclude PKC postsynaptic actions. The brain slices of both treatment groups (saline and cocaine) were incubated in artificial cerebrospinal fluid (ACSF) without PMA as control. PMA increased sEPSCs frequency when compared to saline brain slices without PMA pretreatment (Figure 3C); Figure 3E,G show that PMA pretreatment shifts to the left the cumulative probability in the inter-event interval without changing the amplitude distribution, respectively. In contrast, sEPSCs sample recordings of PMA pretreated slices from cocaine-injected animals showed no differences in frequency or amplitude (Figure 3D,F,H). PMA pretreatment significantly increased the sEPSC frequency of VTA DA neurons from saline-treated animals (control 3.253 ± 0.9110 Hz, n = 12; PMA 13.05 ± 3.498 Hz, n = 13; unpaired t-test, p < 0.05; Figure 3I) but not the amplitude (control 14.54 ± 0.37 pA, n = 12; PMA 14.51 ± 0.49, n = 13; unpaired t-test, p = 0.95; Figure 3J). In slices from cocaine-injected animals, PMA pretreatment failed to increase the frequency (control 4.06 ± 0.88, n = 8; PMA 8.22 ± 3.16, n = 9; unpaired t-test, p = 0.24; Figure 3I) or amplitude (control 15.01 ± 0.43, n = 8; PMA 14.11 ± 0.40, n = 9; unpaired t-test, p = 0.15; Figure 3J). Altogether, these results demonstrate that presynaptic PKC activation may play a role in glutamate release decline after repeated cocaine injections.
2.4. Activation of α1-ARs Failed to Increase AMPA EPSCs Amplitude after a 7-Day Withdrawal Period
To evaluate if the cocaine induced alteration in α1-AR’s effect on AMPA EPSCs was still present after a withdrawal period, we recorded AMPA EPSCs on VTA DA neurons of saline- and cocaine-injected animals that went through a 7-day withdrawal period. Male rats were injected with either saline (0.9%; n = 12) or cocaine (15 mg/kg, i.p.; n = 8) for five consecutive days to induce behavioral sensitization [54]. Two-way repeated measures ANOVA showed significant differences in day factor (F5,72 = 3.28; p < 0.05) and treatment factor (F1,72 = 75.42, p < 0.0001) on the total locomotor activity of cocaine-treated animals in comparison to saline-treated animals (Figure 4A). The interaction between treatment and day showed significant differences (F4,72 = 5.30, p < 0.001). The Bonferroni post hoc test presented differences on all treatment days between saline and cocaine injected animals (Figure 4A).

Figure 4.
α1-ARs-mediated effect on AMPA EPSCs is still absent in VTA DA neurons after a 7-day withdrawal period. (A) A bar graph showing the total locomotor activity recorded from saline- (0.9%, white bars; n = 12) and cocaine- (15 mg/kg i.p., black bars; n = 8) treated animals. Cocaine-injected animals showed a progressive increase in locomotor activity over 5 days when compared to saline-injected animals (Two-way repeated measures ANOVA, day factor F5,72 = 3.28; p < 0.05; treatment factor F1,72 = 75.42, p < 0.0001). On day 5, the total activity was significantly increased when compared to day 1 in cocaine-treated animals (Bonferroni post hoc test p < 0.0001). (B) The time course of locomotor activity on days 1 (grey) and 5 (black) of cocaine- (15 mg/kg, i.p.; n = 8) (filled) or saline- (unfilled; n = 12) treated animals. Cocaine sensitization was manifested as an increase in locomotor activity during the first 40 min after cocaine injection in comparison with day 1. The data (pcc/60 min; Mean ± SEM) were analyzed by Two-way repeated measures ANOVA (treatment factor F3,180 = 72.73; p < 0.0001; time factor F5,180 = 110.2; p < 0.0001), Bonferroni post hoc test (p < 0.001). (C,D) Representative recordings from saline- and cocaine-treated animals after 7 days of withdrawal, respectively, illustrating that phenylephrine superfusion (10 μM, 10 min) induces a significant increment in AMPA EPSCs amplitude only in VTA DA neurons from saline-treated animals. (E) The time course summary of the effects of phenylephrine bath application on AMPA EPSCs amplitude recorded from 12 VTA DA neurons from saline-treated animals and 14 VTA DA neurons from cocaine-treated animals at 8 min of control (2 min intervals), 5 and 10 min phenylephrine (10 μM), and 5 and 10 min washout. A 10 min phenylephrine application increases the AMPA EPSCs amplitude only in neurons from saline-treated animals (Two-way repeated measures ANOVA, time factor F7,168 = 11.68, p < 0.0001; treatment factor F1,168 = 21.20, p < 0.001). A Bonferroni post hoc test demonstrated significant differences between saline and cocaine EPSCs recordings after 5 and 10 min of phenylephrine superfusion (p < 0.001). (F) A bar graph showing that, in neurons from saline-treated animals (n = 12/12), phenylephrine application resulted in a ~40% increase in AMPA EPSCs amplitude. No significant differences were observed on VTA DA neurons from cocaine-treated animals after 10 min of phenylephrine application when compared to control EPSCs recordings (99.6 ± 3.3%, n = 14/8, One-way ANOVA, Newman-Keuls post-hoc, p < 0.001). *** p < 0.001, **** p < 0.0001. n = the number of cells recorded/number of animals.
The time course of locomotor responses (Figure 4B) compares day 1 and day 5 after saline or cocaine injection. Two-way repeated measures ANOVA showed differences in treatment (F3,180 = 72.73; p < 0.0001) and time (F5,180 = 110.2; p < 0.0001). The interaction between treatment and time showed differences (F15,180 = 17.54; p < 0.0001). The Bonferroni post hoc analysis demonstrated an increase in locomotor activity after 5 days of cocaine treatment during the first 40 min after cocaine injection compared to their first cocaine treatment (p < 0.0001). These results indicate that the animals used in this study were successfully sensitized to cocaine.
After a 7-day withdrawal period, electrophysiological recordings of the VTA DA neurons of saline-injected animals treated with the α1-AR agonist (phenylephrine, 10 μM; 10 min) showed an increase in AMPA EPSCs peak amplitude to 140.5 ± 5.4% of control (n = 12; One-way ANOVA F2,38 = 26.57, p < 0.001, Figure 4C,E,F). However, in the slices from animals treated with cocaine for 5 days, which were submitted to a 7-day withdrawal period, phenylephrine superfusion was unable to exert its excitatory action on AMPA EPSCs after 5 or 10 min of application (control 100.2 ± 1.6%; phenylephrine 10 min 99.6 ± 3.3%; n = 14; One-way ANOVA F2,42 = 0.97, p = 0.38, Figure 4 D–F). Two-way repeated measures ANOVA showed significant differences in time (F7,168 = 11.68, p < 0.001 and treatment factor (F1,168 = 21.20, p < 0.001). The interaction between time and treatment showed significant differences (F7,168 = 12.42, p < 0.0001). The Bonferroni post hoc test demonstrated significant differences between saline and cocaine EPSCs recordings on 5 and 10 min of phenylephrine superfusion (p < 0.0001). These results strongly suggest that α1-ARs are still desensitized after 7 days of cocaine withdrawal.
2.5. α1-ARs Activation Decrease GABAA IPSCs Amplitude in VTA DA Neurons from Acute Saline and Cocaine-Treated Animals
To study the presynaptic α1-AR modulation of GABA release onto VTA DA neurons after an acute cocaine injection, male rats were injected with either saline (0.9%; n = 7) or cocaine (15 mg/kg, i.p.; n = 7) for one day. The total activity (Figure 5A) showed differences between saline- (n = 7; 1166 ± 280.7 photocell counts) vs. cocaine- (n = 7; 4158 ± 439.5 photocell counts, Unpaired t-test, df = 12, t = 5.73, p < 0.0001) treated animals. These results indicate that acute cocaine-treated animals presented the characteristic increased locomotion response to the drug.
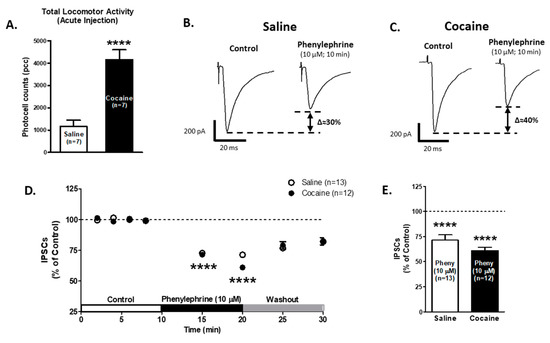
Figure 5.
α1-ARs-mediated effect on GABAA IPSCs amplitude is not affected in VTA DA neurons after one saline or cocaine injection. (A) A bar graph showing the total locomotor activity recorded from saline- (0.9%, white bar; n = 7) and cocaine- (15 mg/kg i.p., black bar; n = 7) treated animals. The locomotion of cocaine treated animals was significantly increased compared to saline-treated controls (Unpaired t-test, p < 0.0001). (B,C) Representative recordings from saline- and cocaine-treated animals, respectively, show that phenylephrine superfusion (10 μM, 10 min) induces a significant decrease in GABAA IPSCs amplitude in VTA DA neurons from both groups. (D) The time course summary of phenylephrine effects on GABA IPSCs recorded from VTA DA neurons from saline- (n = 13) and cocaine- (n = 12) treated animals at 8 min of control (2 min intervals), 5 and 10 min phenylephrine (10 μM), and 5 and 10 min washout. A 10 min phenylephrine application decreases the GABAA IPSCs amplitude in neurons from saline- and cocaine-treated animals (One-way ANOVA, Newman-Keuls post hoc test, p < 0.0001). (E) A bar graph showing that phenylephrine application results in a ~30% decrease in GABAA IPSCs amplitude in VTA DA neurons from saline- (n = 13/7) and cocaine- (n = 12/7) treated animals (One-way ANOVA, Newman-Keuls post-hoc, p < 0.0001). **** p < 0.0001. n = the number of cells recorded/number of animals.
Phenylephrine application (10 μM) on VTA DA neurons decreased GABAA IPSCs amplitude after 10 min, saline group: 71.3 ± 5.2% of control (n = 13; One-way ANOVA F2,36 = 17.75, Newman-Keuls post hoc test p < 0.0001, Figure 5B,D,E); cocaine group: 61.0 ± 3.1% of control (n = 12; One-way ANOVA F2,34 = 32.74, Newman-Keuls post hoc test p < 0.0001, Figure 5C–E). The two-way repeated measures ANOVA showed significant differences in time factor (F7,161 = 30.23, p < 0.0001) and no significant differences in treatment factor (F1,16 = 0.49, p = 0.48). The interaction between the treatment and time showed no differences (F7,161 = 0.53, p = 0.80). The Bonferroni post hoc test did not present differences between saline and cocaine IPSCs recordings (p > 0.05). These results suggest that α1-ARs modulation on VTA GABA neurotransmission is functional after one cocaine injection.
2.6. Modulation of α1-ARs on GABA Release is Present in the VTA DA Neurons of Sensitized Animals
For total activity (Figure 6A), Two-way repeated measures ANOVA showed differences in treatment: saline (n = 10) vs. cocaine (n = 10) (F1,58 = 35.01; p < 0.0001). Additionally, the total activity between the days showed differences (F4,68 = 5.33; p = 0.0009). The interaction between treatment and days showed differences (F4,68 = 8.01; p <0.0001). The Bonferroni post hoc analysis revealed that cocaine-treated animals had a higher total activity than saline-treated animals on days 3 (p = 0.0001), 4, and 5 (p = 0.0001). The time course of locomotor responses (Figure 6B) compares day 1 and day 5 after saline or cocaine injection. The Two-way repeated measures ANOVA showed differences in treatment (F3,150 = 13.21; p < 0.0001) and time (F5,150 = 39.44; p < 0.0001). The interactions between time and treatment showed significant differences (F15,150 = 2.75, p = 0.0009). The Bonferroni post hoc analysis demonstrated an increase in locomotor activity after 5 days of cocaine treatment for 40 min after cocaine injection compared to their first cocaine treatment (p < 0.05). Together, these results indicate that the animals used in this study were successfully sensitized to cocaine.

Figure 6.
α1-ARs-mediated GABAA IPSCs effect is present in VTA DA neurons from cocaine-sensitized animals. (A) A bar graph showing total locomotor activity recorded from saline- (0.9%, white bars; n = 10) and cocaine- (15 mg/kg i.p., black bars; n = 9) treated animals. Cocaine-injected animals showed a progressive increase in locomotor activity over 5 days. On days 3, 4, and 5, the total activity was significantly increased when compared to day 1 in cocaine-treated animals (Two-way repeated measures ANOVA, Bonferroni post hoc test, p < 0.001). (B) The time course of locomotor activity on days 1 (grey) and 5 (black) of cocaine- (15 mg/kg, i.p.) (filled) or saline- (unfilled) treated animals. Cocaine sensitization was manifested as an increase in locomotor activity during the first 10–40 min after cocaine injection in comparison with day 1. The data (pcc/60 min; Mean ± SEM) were analyzed by Two-way repeated measures ANOVA, Bonferroni post hoc test (p < 0.05). (C,D) Representative recordings from saline- and cocaine-treated animals, respectively, illustrate that phenylephrine superfusion (10 μM, 10 min) induces a significant reduction on GABAA IPSCs amplitude in VTA DA neurons from saline- and cocaine-treated animals. (E) The time course summary of the effects of phenylephrine bath application on GABAA IPSCs amplitude, recorded from 8 VTA DA neurons from saline-treated animals and 11 VTA DA neurons from cocaine-treated animals at 8 min of control (2 min intervals), 5 and 10 min phenylephrine (10 μM), and 5 and 10 min washout. A 10 min phenylephrine application increases the GABAA IPSCs amplitude in neurons from saline- and cocaine-treated animals (One-way ANOVA, Newman-Keuls post hoc test, p < 0.001). (F) A Bar graph showing that, in neurons from saline- (n = 8/10) and cocaine- (n = 11/9) treated animals, phenylephrine application resulted in a ~50% reduction on GABAA IPSCs amplitude (One-way ANOVA, Newman-Keuls post-hoc, p < 0.001). * p < 0.05; *** p < 0.0001. n = the number of cells recorded/number of animals.
In slices from saline-treated animals, bath application of the selective α1-AR agonist, phenylephrine (10 μM) after 10 min, decreased GABAA IPSCs amplitude to 50.0 ± 5.4% of control (n = 8; One-way ANOVA F2,24 = 62.61, Newman-Keuls post hoc test, p < 0.0001, Figure 6C,E,F). Similarly, 10 min of phenylephrine superfusion decreased GABAA IPSCs amplitude to 50.3 ± 6.9% of control (n = 11; One-way ANOVA F2,30 = 27.05, Newman-Keuls post hoc test, p < 0.0001, Figure 6D–F) in slices from animals treated with cocaine for 5 days. No significant differences were found by Two-way repeated measures ANOVA in the treatment factor (F1,119 = 0.32, p = 0.57), nonetheless, the time factor was significantly different (F7,119 = 56.12, p < 0.0001). The interaction between time and treatment showed no significant differences (F7,119 = 0.48, p = 0.84). Additionally, the Bonferroni post hoc test showed no differences between saline and cocaine IPSCs recordings (p > 0.05). These results demonstrate that α1-ARs modulation on GABA neurotransmission is present after 5 days of cocaine administration.
3. Discussion
The cocaine sensitization paradigm modifies the α1-ARs activation of glutamatergic, but not GABAergic, transmission. The present results showed that α1-AR-mediated activation on glutamatergic transmission is no longer present after 5 days of cocaine sensitization protocol, even after withdrawal period. In addition, presynaptic the PKC activity related to glutamatergic transmission decreased in VTA neurons of cocaine-sensitized animals. On the other hand, α1-AR- regulation of GABAergic transmission is unaffected by acute and repeated cocaine treatment. As a possible limitation of this study future investigations should address sex differences in the observed responses.
Cocaine rapidly increases monoamines (dopamine, serotonin, and norepinephrine) concentration in the synaptic cleft by blocking their reuptake [55,56]. A single injection or repeated cocaine administration increases glutamatergic and decreases GABAergic synaptic transmission onto VTA DA neurons [40,57]. Changes on glutamatergic and GABAergic neurotransmission after cocaine injection could be due, in part, to norepinephrine action on α1-ARs. Therefore, α1-ARs activation, after cocaine injection, could modify VTA DA neuronal excitability and increase DA release onto the nucleus accumbens (NAcc) [45]. In accordance with this, the present results show that α1-ARs activation increases glutamatergic transmission onto VTA DA neurons 24 h after an acute cocaine injection. This activation of α1-ARs could generate positive feedback onto VTA DA cells that might be important for the effects induced by drug abuse [35,40,45,58]. We have shown that α1-ARs increase presynaptic glutamate release onto VTA DA neurons [12]. Thus, after an acute cocaine injection, presynaptic α1-ARs activation could help promote long-term potentiation (LTP) at VTA DA neurons synapses via increases in extracellular glutamate. Cocaine-induced synaptic plasticity in the VTA has been associated with maladaptive behaviors and the development of addiction [59,60,61,62]. Long-term changes in the VTA are induced by a single and multiple cocaine injections [40,41]. In addition, LTP, as the persistent increase in AMPA/NMDA ratio on VTA DA cells, is critical for the maintenance of cocaine sensitization [63]. These changes in synaptic plasticity in the VTA have been implicated in the development of drug addiction and cocaine sensitization [5,54,64,65].
Different studies have shown that α1-ARs have a critical role in drug addiction. Pre-exposure to prazosin (α1-ARs antagonist) decreases behavioral sensitization and psychostimulants’ self-administration [19,49,66]. Similar results were obtained with α1b-AR knockout animals [47,48]. An alteration in α1-ARs activation inhibits DA release on NAcc after D-amphetamine systemic administration [66,67]. These findings indicate that α1-ARs activation is an important substrate in the development of drug addiction to psychostimulants. Our results show that α1-ARs activation failed to increase glutamatergic transmission onto VTA DA neurons in sensitized animals. Nalepa et al., [52] indicated that cocaine sensitization was associated with changes in α1-ARs density in certain regions of rats’ brains. Although this study does not show results on α1-ARs at the VTA, it suggests that these receptors are susceptible to changes and therefore their effect could be absent after cocaine sensitization. We postulate that after the initiation of cocaine sensitization, α1-ARs located on glutamatergic afferents to VTA DA neurons are desensitized. Receptor desensitization is described as a decrease in the response to an agonist after persistent stimulation and constitutes a regulation process of many G-protein coupled receptors [68,69,70,71,72]. The desensitization process involves the stimulation of G-protein coupled receptor kinases, β-arrestins, and PKC activation that induces inactivation and receptor internalization [68,70]. α1-ARs desensitization has been extensively described [69,71,73,74]. This process could be a homeostatic response to prevent prolonged receptor stimulation and the detrimental effects associated with this protracted stimulation.
The blockade of α1-ARs or 5-HT2A receptors only attenuates the behavioral response to cocaine, D-amphetamine, and morphine administration [19,45,47,48]. In addition, the simultaneous blockade of α1-ARs and 5-HT2A receptors entirely abolish the locomotor response and development of behavioral sensitization to psychostimulants and opiates [48]. Therefore, α1-ARs and 5-HT2A receptors appear to be physiologically interrelated and could mediate the locomotor response and development of behavioral sensitization to psychostimulants and opiates. On the other hand, studies have shown that repeated cocaine injections produce a persistent increase in the capacity of cocaine to elevate extracellular glutamate levels via an increase in D1 receptor stimulation of glutamate release at the VTA [75]. The α1-ARs desensitization after repeated cocaine administration could induce a shift towards neural pathways controlled by other receptors. Thus, the increase in glutamatergic transmission that was mediated by presynaptic α1-ARs could be compensated by 5-HT2A or D1 receptors’ activation.
Here, we demonstrate that presynaptic PKC activation, in the presence of phorbol ester (PMA), did not increase glutamate release onto VTA DA neurons after chronic cocaine exposure. In agreement with our results, it has been shown that PKC increases its activity after an acute cocaine injection but not after chronic (7 days) cocaine administration [76,77]. Studies have also demonstrated that cocaine temporarily increases PKC activity in the VTA. Repeated cocaine exposure increases PKC activity at 2 but not 6 or 24 h after the last injection [76,77]. No significant cocaine-induced changes in PKC activity in the VTA were detected 24 h after a cocaine challenge injection [76]. In VTA DA neurons from naïve animals, PKC activation with a phorbol ester causes an increase in both mEPSC frequency and amplitude [78]. Altogether, these data suggest that repeated cocaine injection induces a transitory increase (2 h but not 6 or 24 h) in PKC activity in the VTA during the initiation of cocaine sensitization. Also, the modification of PKC activity could be part of the transient processes that occur at the VTA during the initiation of cocaine sensitization. Therefore, our results suggest that changes in PKC activity in the VTA after repeated cocaine injections may be involved in the initiation of behavioral sensitization.
The desensitization of α1-AR has been shown to be associated with decreases in the number of receptor sites due to prolonged exposure to NE or agonists (phenylephrine), which results in decreased responsiveness [74]. Our results show that after 7 days of withdrawal, α1-AR-mediated increased glutamatergic transmission onto VTA DA neurons is still absent. Therefore, the increased NE in the synaptic cleft induced by cocaine administration could overstimulate and cause a long-lasting desensitization of α1-ARs.
The desensitization of α1-AR showed here could be part of several neuroadaptations induced at the mesocorticolimbic system by chronic cocaine treatment. Some of these neuroadaptations include: (A) an increase in GluR1 subunit with increases in AMPA/NMDA ratio on VTA DA cells (5–7 days after cessation of drug administration) [41,43,79,80]; (B) an increased glutamate transmission [75]; (C) a reduced D1-stimulated GABA transmission in the VTA [81]; (D) enhanced basal levels of extracellular DA [54]; (E) increased tyrosine hydroxylase synthesis [82]; and (F) a decrease in Ih current and DA neurons capacitance [83,84]. In addition, a decrease in nerve terminal glutamate immunoreactivity within the VTA in cocaine sensitization, after a withdrawal period, has been reported [85,86,87]. Most of these modifications are temporary but necessary to trigger other alterations for the maintenance and expression of addiction-related behaviors [34,88,89].
In contrast to our results of α1-AR modulation on glutamatergic neurotransmission after cocaine treatment, a decrease in GABAergic transmission onto VTA DA neurons by α1-AR activation is still observed following both the acute injection and 24 h after the initiation of cocaine sensitization. Cocaine administration decreases evoked GABAA currents on VTA neurons [43,90]. Other investigators have shown that different addictive drugs, including cocaine, attenuated long-term potentiation of GABAergic synapses onto VTA dopamine neurons [91]. In addition, it has been demonstrated that increased GABA neurotransmission attenuates cocaine behavioral sensitization [92]. Therefore, after drug administration, DA neurons could be subjected to less inhibition and consequently have increased excitability in response to a given stimulus.
Our results on naïve rats demonstrate that α1-ARs modulation on GABAergic release is dependent on voltage-sensitive sodium channels, since there are no changes in the frequency or amplitude of mIPSCs (in the presence of TTX) [13]. Steffesen et al. [93], demonstrated that cocaine reduced GABA release onto VTA DA neurons by blocking voltage-sensitive sodium channels present on presynaptic GABAergic terminals. Thus, cocaine-induced NE increase can activate α1-ARs and diminish GABA release onto VTA DA neurons.
The stimulation of GABA neurons at the rostromedial tegmentum (RMTg) increases IPSCs’ amplitude in VTA DA cells that project to the NAcc lateral shell [94]. The VTA DA–NAcc lateral shell connection is highly associated with reward behavior [3,95]. The activation of VTA GABA neurons in VTA DA cells has been linked with the disruption of reward behaviors [96]. Taken together, these data suggest that the activation of GABA-RMTg inputs and local VTA GABA neurons can induce an inhibition on VTA DA cells and disturb reward-related behaviors. Consequently, a decrease in these two inhibitory sources onto VTA DA cells can facilitate the development of reward behaviors. After acute and repeated cocaine injections, α1-ARs activation induces a decrease in GABA neurotransmission onto VTA DA neurons. Therefore, we can hypothesize that the decrease in GABA release via α1-AR activation can reverse neurotransmitter actions related to aversive behaviors, allowing neurons to increase their excitation. This increased excitability will enhance DA release in the NAcc and facilitate the development of rewarding behaviors.
In conclusion, the present data show that, after the initiation of cocaine sensitization, the α1-ARs present on glutamatergic terminals onto VTA DA neurons are desensitized and their PKC-mediated response is absent. Additionally, this receptor desensitization is still present after a withdrawal period. On the other hand, the α1-ARs modulation of GABA neurotransmission remains intact after chronic cocaine treatment (Figure 7). Therefore, the cocaine-induced enhanced DA cell excitability could be increased through a reduction of GABA inhibition on VTA DA neurons even if the α1-ARs function on glutamate release and PKC activation is not present. Altogether, these changes in α1-ARs neuromodulation could be part of potential homeostatic alterations occurring at the VTA after the initiation of cocaine sensitization. The understanding of the neuropathological mechanisms and the neurochemical imbalances that are behind cocaine addiction could help in the development of effective pharmacological treatments.

Figure 7.
Proposed model for α1-ARs action on glutamatergic and GABAergic terminals during the initiation and expression of cocaine sensitization. After cocaine sensitization and a withdrawal period, the α1-ARs present on glutamate terminals on VTA DA neurons are desensitized. In addition, a PKC-mediated increase in glutamate release on VTA DA cells is absent after 5 days of cocaine injections. In contrast, the α1-AR-mediated decrease on GABA release on VTA DA neurons remains unaltered.
4. Materials and Methods
4.1. Animals
All experimental procedures were performed according to the US Public Health Service publication “Guide for the Care and Use of Laboratory Animals” and were approved by the Animal Care and Use Committee at the University of Puerto Rico Medical Sciences Campus (IACUC# 4050108; 9/4/2014)). Sprague-Dawley male rats weighing between 250–300 g were housed two per cage and maintained at a constant temperature and humidity with a 12-h hour light/dark cycle. Water and food were provided ad libitum throughout the course of the experiment.
4.2. Sensitization Protocol and Behavior
The animals were divided randomly into two groups (saline and cocaine). Two days before the beginning of the experiment, each group was habituated (for 1 h) to the infrared photocell box. On experimental day one, the animals were placed for 15 min in the photocell box. After the 15-min habituation, the animals were treated with either 15 mg/kg i.p. of cocaine (Sigma, St. Louis, MO, USA) or isovolumetric saline injections. Immediately after the injections, locomotor activity was recorded at 10 min intervals for 60 min. This procedure was repeated for five (5) consecutive days. A significant statistical difference of p < 0.05 between day 1 and day 5, (Two-way ANOVA) was considered a successful sensitization protocol [33]. Twenty-four hours after the last cocaine injection or following the 7-day withdrawal period, animals were anesthetized with a 90 mg/kg of chloral-hydrate i.p. (Sigma, St. Louis, MO, USA) and their brains rapidly removed.
4.3. Electrophysiological Studies
EPSCs and IPSCs from the VTA DA neurons of rats previously treated with 1 or 5 injections of cocaine or saline were measured using whole cell voltage clamp techniques. Phenylephrine superfusion was used to determine whether the cocaine-induced potentiation of AMPA or GABAA mediated currents are altered by α1-AR [12,13,16]. To determine the effect of cocaine sensitization on presynaptic PKC activation, slices were preincubated for 30 min with phorbol 12-myristate 13-acetate (PMA; 0.5 µM). Also, an internal solution containing GO6976 (PKC antagonist; 1 µM) via the recording pipette was used to exclude PKC postsynaptic actions.
4.4. Slice Preparation
Horizontal slices (220 µM) containing the VTA were cut using a vibratome (VT1000S, Leica, Wetzlar, Germany). The rat midbrain was placed on an ice-cold oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM) 127 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, and 25 D(+)-glucose and was saturated with a 95%O2–5%CO2 gas mixture to a pH = 7.4. Slices were transferred to an intermediate chamber and incubated at 32 °C in the same solution for 45 min before the initiation of electrophysiological recordings. MK-801 (10 μM, Tocris, Ellisville, MO, USA) was added to the incubation solutions to block N-methyl-D-aspartate (NMDA)-mediated excitotoxicity.
4.5. Electrophysiological Recordings
VTA slices were totally submerged in a recording chamber (500 µL) with ACSF superfusion at 1–2 mL/min at 32°C. Whole cell voltage clamp recordings were obtained from visually identified neurons in the VTA using an infrared microscope with differential interference contrast (DIC) optics (BX51WI Olympus, Japan). The recordings were acquired through data acquisition software (pClamp 10, Molecular Devices, Sunnyvale, CA, USA). All recordings were performed in putative DA neurons identified by the presence of a large hyperpolarization-activated cation current (Ih > 200 pA), evoked by 1-s hyperpolarizing steps from −60 to −130 mV. Ih is present in about 84% VTA DA neurons and VTA GABA cells rarely express this conductance [97,98]. Therefore, the contribution of non-dopaminergic neurons to the experimental recordings performed in this study is not likely to be significant. Whole-cell voltage-clamp recordings were made at a holding potential of −70 mV unless otherwise indicated.
AMPA Excitatory Postsynaptic Currents (EPSCs) Recordings: Picrotoxin (100 μM, Sigma, St. Louis, MO, USA) was added to the ACSF during the recording procedures to block GABAA receptor-mediated inhibitory postsynaptic currents (IPSCs). Borosilicate glass patch pipettes (O.D.1.5 mm, I.D:1,0 mm WPI, Sarasota, FL, USA) were pulled to a final resistance of 3–6 MΩ and filled with (in mM): 115 CH3SO4K (Methyl potassium sulfate); 20 KCl; 1.5 MgCl2; 5 HEPES; 1 EGTA; 2 ATP; 0.2 GTP; 10 Creatine Phosphate (CP); pH 7.25, 290 mOsm. (Na) GTP, (Mg) ATP, and (Na) CP were added fresh daily.
GABAA Inhibitory Postsynaptic Currents (IPSCs) Recordings: The superfusion medium contained 2-amino-5-phosphonopentanoic acid (AP5; 100 μM) and either 6-cyano-2,3-dihydroxy-7 nitroquinox saline (CNQX; 10 μM) or 6,7-Dinitroquinoxaline-2,3-dione (DNQX; 10 μM) to block fast NMDA- or AMPA-mediated synaptic potentials, respectively. In all experiments, eticlopride (100 μM) was included in the superfusion solution to block any possible effects mediated by the dopamine D2 receptor. Whole-cell voltage-clamp recordings were made using microelectrodes filled with a solution containing (in mM): 70 K+ gluconate, 80 KCl, 1 EGTA, 5 HEPES, 2 MgATP, and 0.3 GTP. At the end of each experiment, picrotoxin (100 μM) was added to completely abolish all evoked or spontaneous GABAA mediated postsynaptic currents.
The data were collected through a Multiclamp 700B amplifier (Axon Instruments, Foster City, CA, USA), filtered at 1 kHz, digitized at 5 kHz using Digidata 1440A (Axon Instruments, Foster City, CA, USA), and stored in a PC computer and analyzed off-line using GraphPad Prism 5 (GraphPad Software, Inc) software. The pipette’s liquid junction potential was offset and compensated using standard Multiclamp 700B circuitry. The seal’s quality used was 4–6 GΩ. The series resistances were not compensated and were monitored during the entire experiment. The data were discarded if changes (in seal) of more than 15% occurred.
4.6. Recording of Synaptic Currents
A bipolar stainless-steel stimulating electrode (FHC Inc, Bowdoin, ME, USA) was placed approximately 100 μm rostral to the recording electrode and was used to stimulate afferents at 0.1Hz by applying a brief (400 μs; low pass filter 1 kHz, digitized 5 KHz) electrical pulse (100–300 μA). AMPA EPSCs and GABAA IPSCs were recorded at −70 mV. All EPSCs and IPSCs shown in the figures are averages of 5 current traces. AMPA EPSCs’ and GABAA IPSCs’ amplitudes were calculated by taking a 1 ms window around the peak of the EPSC or IPSC and comparing this to a 5 ms window immediately before the stimulation artifact. Peak EPSCs’ or IPSCs’ amplitudes were averaged during the control recordings. This value was used to normalize control and treatment recordings. This procedure allowed data recording as percentages of the control condition for appropriate statistical comparisons. Spontaneous AMPA EPSC’s (sEPSCs) signals were recorded at −70 mV, filtered at 1 kHz and digitized at 5 kHz using pCLAMP 10 software (Molecular Devices, Sunnyvale, CA, USA). For a given cell, sEPSCs were collected (1 sweep for each condition, 3min/sweep) for a control and phenylephrine’s period. The recorded sEPSCs were analyzed afterward using Mini Analysis program 6.0.7 (Synaptosoft Inc. Decatur, GA, USA). Detection criteria were set at >6 pA, <1.3 ms rise time, and <0.1 ms decay time. The choice of this cutoff amplitude for the acceptance of sEPSCs was made to obtain a high signal-to-noise ratio. Each event was also visually inspected to prevent noise disturbance during the analysis.
4.7. Drugs
Cocaine hydrochloride, phenylephrine hydrochloride ([R]-[−]-1-[3-Hydroxyphenyl]-2-methylaminoethanol hydrochloride), phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma–Aldrich (St. Louis, MO, USA), GO6976 was purchased from Tocris (Ellisville, MO, USA).
4.8. Data Analysis
All data were presented as mean ± SEM. The statistical significance was assessed using an Unpaired t-test, One-Way ANOVA with Newman-Keuls as a post hoc analysis or Two-Way repeated measures ANOVA with time and treatment as factors, and Bonferroni as a post hoc analysis, except for the significance of horizontal shifts to the cumulative probability distribution plots obtained from single cell recordings. For the latter case, the Kolmogorov–Smirnov test was used. p values were reported throughout the text and the significance was set at p < 0.05.
Author Contributions
Conceptualization, M.C.V.-M. and C.A.J.-R.; methodology, M.C.V.-M.; validation, M.C.V.-M., B.S.-V., M.E.V.-H., R.V.-T. and C.A.J.-R.; formal analysis, M.C.V.-M.; investigation, M.C.V.-M. and C.A.J.-R.; resources, R.V.-T. and C.A.J.-R..; data curation, M.C.V.-M.; writing—original draft preparation, M.C.V.-M.; writing—review and editing, M.C.V.-M., B.S.-V., M.E.V.-H., R.V.-T. and C.A.J.-R.; visualization, M.C.V.-M.; supervision, C.A.J.-R.; project administration, R.V.-T., C.A.J.-R.; funding acquisition, C.A.J.-R. All authors have read and agreed to the published version of the manuscript
Funding
This research was funded by MBRS-SCORE, grant number GM-08224, SC1GM0848-05A1 and GM 084854; the National Science Foundation Partnerships for International Research and Education (OISE-1545803) to CAJR and the Research Initiative for Scientific Enhancement (RISE) Program (R25GM061838). Infrastructure support was provided in part by the National Institute on Minority Health and Health Disparities RCMI Grant: 2U54MD007600.
Acknowledgments
M.C.V.-M. received economic support from Universidad Industrial de Santander, Colombia.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| α1-AR | Alpha 1 Adrenergic Receptor |
| µM | Micromolar |
| 5-HT | 5-hydroxytryptamine |
| ACSF | Artificial cerebrospinal fluid |
| AMPA | 2-Amino-3-(5-Methyl-3-Oxo-1, 2-Oxazol-4-yl) Propanoic Acid |
| DA | Dopamine |
| EPSC | Inhibitory postsynaptic current |
| GABA | Gamma-aminobutyric acid |
| Glu | Glutamatergic |
| i.p. | Intraperitoneal |
| Ih | H current |
| IPSC | Inhibitory postsynaptic current |
| kg | Kilogram |
| LTP | Long-term potentiation |
| mg | Milligram |
| min | Minutes |
| ms | Milliseconds |
| NAcc | Nucleus Accumbens |
| NE | Norepinephrine |
| NMDA | N-Methyl d-Aspartate |
| pcc | Photocell counts |
| PKC | Protein kinase C |
| PMA | Phorbol 12-myristate 13-acetate |
| RMTg | Rostromedial tegmentum |
| sEPSC | Spontaneous excitatory postsynaptic current |
| VTA | Ventral Tegmental Area |
References
- Dahlstrom, A.; Fuxe, K. Localization of monoamines in the lower brain stem. Experientia 1964, 20, 398–399. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, U. Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol. Scand. 1971, 367, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Lammel, S.; Ion, D.I.; Roeper, J.; Malenka, R.C. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron 2011, 70, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Schultz, W. Getting formal with dopamine and reward. Neuron 2002, 36, 241–263. [Google Scholar] [CrossRef]
- Kauer, J.A. Learning mechanisms in addiction: Synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu. Rev. Physiol. 2004, 66, 447–475. [Google Scholar] [CrossRef]
- Grace, A.A.; Floresco, S.B.; Goto, Y.; Lodge, D.J. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007, 30, 220–227. [Google Scholar] [CrossRef]
- Jones, B.E.; Halaris, A.E.; McIlhany, M.; Moore, R.Y. Ascending projections of the locus coeruleus in the rat. I. Axonal transport in central noradrenaline neurons. Brain Res. 1977, 127, 1–21. [Google Scholar] [CrossRef]
- Mejias-Aponte, C.A.; Drouin, C.; Aston-Jones, G. Adrenergic and noradrenergic innervation of the midbrain ventral tegmental area and retrorubral field: Prominent inputs from medullary homeostatic centers. J. Neurosci. 2009, 29, 3613–3626. [Google Scholar] [CrossRef]
- Liprando, L.A.; Miner, L.H.; Blakely, R.D.; Lewis, D.A.; Sesack, S.R. Ultrastructural interactions between terminals expressing the norepinephrine transporter and dopamine neurons in the rat and monkey ventral tegmental area. Synapse 2004, 52, 233–244. [Google Scholar] [CrossRef]
- Greene, J.G.; Dingledine, R.; Greenamyre, J.T. Gene expression profiling of rat midbrain dopamine neurons: Implications for selective vulnerability in parkinsonism. Neurobiol. Dis. 2005, 18, 19–31. [Google Scholar] [CrossRef]
- Rommelfanger, K.S.; Mitrano, D.A.; Smith, Y.; Weinshenker, D. Light and electron microscopic localization of alpha-1 adrenergic receptor immunoreactivity in the rat striatum and ventral midbrain. Neuroscience 2009, 158, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Velasquez-Martinez, M.C.; Vazquez-Torres, R.; Jimenez-Rivera, C.A. Activation of alpha1-adrenoceptors enhances glutamate release onto ventral tegmental area dopamine cells. Neuroscience 2012, 216, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Velasquez-Martinez, M.C.; Vazquez-Torres, R.; Rojas, L.V.; Sanabria, P.; Jimenez-Rivera, C.A. Alpha-1 adrenoreceptors modulate GABA release onto ventral tegmental area dopamine neurons. Neuropharmacology 2015, 88, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Grenhoff, J.; Nisell, M.; Ferre, S.; Aston-Jones, G.; Svensson, T.H. Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J. Neural Transm./Gen. Sect. JNT 1993, 93, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Grenhoff, J.; Svensson, T.H. Prazosin modulates the firing pattern of dopamine neurons in rat ventral tegmental area. Eur. J. Pharmacol. 1993, 233, 79–84. [Google Scholar] [CrossRef]
- Paladini, C.A.; Williams, J.T. Noradrenergic inhibition of midbrain dopamine neurons. J. Neurosci. 2004, 24, 4568–4575. [Google Scholar] [CrossRef]
- Cecchi, M.; Khoshbouei, H.; Javors, M.; Morilak, D.A. Modulatory effects of norepinephrine in the lateral bed nucleus of the stria terminalis on behavioral and neuroendocrine responses to acute stress. Neuroscience 2002, 112, 13–21. [Google Scholar] [CrossRef]
- Hague, C.; Chen, Z.; Uberti, M.; Minneman, K.P. Alpha(1)-adrenergic receptor subtypes: Non-identical triplets with different dancing partners? Life Sci. 2003, 74, 411–418. [Google Scholar] [CrossRef]
- Jimenez-Rivera, C.A.; Feliu-Mojer, M.; Vazquez-Torres, R. Alpha-noradrenergic receptors modulate the development and expression of cocaine sensitization. Ann. N. Y. Acad. Sci. 2006, 1074, 390–402. [Google Scholar] [CrossRef]
- Greenwell, T.N.; Walker, B.M.; Cottone, P.; Zorrilla, E.P.; Koob, G.F. The alpha1 adrenergic receptor antagonist prazosin reduces heroin self-administration in rats with extended access to heroin administration. Pharmacol. Biochem. Behav. 2009, 91, 295–302. [Google Scholar] [CrossRef]
- Charara, A.; Smith, Y.; Parent, A. Glutamatergic inputs from the pedunculopontine nucleus to midbrain dopaminergic neurons in primates: Phaseolus vulgaris-leucoagglutinin anterograde labeling combined with postembedding glutamate and GABA immunohistochemistry. J. Comp. Neurol. 1996, 364, 254–266. [Google Scholar] [CrossRef]
- Georges, F.; Aston-Jones, G. Activation of ventral tegmental area cells by the bed nucleus of the stria terminalis: A novel excitatory amino acid input to midbrain dopamine neurons. J. Neurosci. 2002, 22, 5173–5187. [Google Scholar] [CrossRef] [PubMed]
- Lodge, D.J.; Grace, A.A. The laterodorsal tegmentum is essential for burst firing of ventral tegmental area dopamine neurons. Proc. Natl. Acad. Sci. USA 2006, 103, 5167–5172. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Liu, C.L.; Yang, S.; Jin, G.Z.; Bunney, B.S.; Shi, W.X. Functional coupling between the prefrontal cortex and dopamine neurons in the ventral tegmental area. J. Neurosci. 2007, 27, 5414–5421. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Derst, C.; Veh, R.W.; Zahm, D.S. Glutamatergic afferents of the ventral tegmental area in the rat. J. Neurosci. 2007, 27, 5730–5743. [Google Scholar] [CrossRef] [PubMed]
- Omelchenko, N.; Sesack, S.R. Glutamate synaptic inputs to ventral tegmental area neurons in the rat derive primarily from subcortical sources. Neuroscience 2007, 146, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhang, S.; Wang, H.L.; Wang, H.; de Jesus Aceves Buendia, J.; Hoffman, A.F.; Lupica, C.R.; Seal, R.P.; Morales, M. A glutamatergic reward input from the dorsal raphe to ventral tegmental area dopamine neurons. Nat. Commun. 2014, 5, 5390. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sheen, W.; Morales, M. Glutamatergic neurons are present in the rat ventral tegmental area. Eur. J. Neurosci. 2007, 25, 106–118. [Google Scholar] [CrossRef]
- Dobi, A.; Margolis, E.B.; Wang, H.L.; Harvey, B.K.; Morales, M. Glutamatergic and nonglutamatergic neurons of the ventral tegmental area establish local synaptic contacts with dopaminergic and nondopaminergic neurons. J. Neurosci. 2010, 30, 218–229. [Google Scholar] [CrossRef]
- Jhou, T.C.; Fields, H.L.; Baxter, M.G.; Saper, C.B.; Holland, P.C. The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron 2009, 61, 786–800. [Google Scholar] [CrossRef]
- Jhou, T.C.; Geisler, S.; Marinelli, M.; Degarmo, B.A.; Zahm, D.S. The mesopontine rostromedial tegmental nucleus: A structure targeted by the lateral habenula that projects to the ventral tegmental area of Tsai and substantia nigra compacta. J. Comp. Neurol. 2009, 513, 566–596. [Google Scholar] [CrossRef] [PubMed]
- Polter, A.M.; Barcomb, K.; Tsuda, A.C.; Kauer, J.A. Synaptic function and plasticity in identified inhibitory inputs onto VTA dopamine neurons. Eur. J. Neurosci. 2018, 47, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Kalivas, P.W.; Stewart, J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res. Brain Res. Rev. 1991, 16, 223–244. [Google Scholar] [CrossRef]
- Steketee, J.D.; Kalivas, P.W. Drug wanting: Behavioral sensitization and relapse to drug-seeking behavior. Pharmacol. Rev. 2011, 63, 348–365. [Google Scholar] [CrossRef]
- You, Z.B.; Wang, B.; Zitzman, D.; Azari, S.; Wise, R.A. A role for conditioned ventral tegmental glutamate release in cocaine seeking. J. Neurosci. 2007, 27, 10546–10555. [Google Scholar] [CrossRef]
- Wise, R.A. Ventral tegmental glutamate: A role in stress-, cue-, and cocaine-induced reinstatement of cocaine-seeking. Neuropharmacology 2009, 56 (Suppl. 1), 174–176. [Google Scholar] [CrossRef]
- Michaeli, A.; Matzner, H.; Poltyrev, T.; Yaka, R. Modifications of the input currents on VTA dopamine neurons following acute versus chronic cocaine exposure. Neuropharmacology 2012, 62, 1834–1840. [Google Scholar] [CrossRef]
- Wolf, M.E. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog. Neurobiol. 1998, 54, 679–720. [Google Scholar] [CrossRef]
- Dunn, J.M.; Inderwies, B.R.; Licata, S.C.; Pierce, R.C. Repeated administration of AMPA or a metabotropic glutamate receptor agonist into the rat ventral tegmental area augments the subsequent behavioral hyperactivity induced by cocaine. Psychopharmacology 2005, 179, 172–180. [Google Scholar] [CrossRef]
- Ungless, M.A.; Whistler, J.L.; Malenka, R.C.; Bonci, A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 2001, 411, 583–587. [Google Scholar] [CrossRef]
- Borgland, S.L.; Malenka, R.C.; Bonci, A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: Electrophysiological and behavioral correlates in individual rats. J. Neurosci. 2004, 24, 7482–7490. [Google Scholar] [CrossRef] [PubMed]
- White, F.J.; Hu, X.T.; Zhang, X.F.; Wolf, M.E. Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J. Pharmacol. Exp. Ther. 1995, 273, 445–454. [Google Scholar] [PubMed]
- Liu, Q.S.; Pu, L.; Poo, M.M. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature 2005, 437, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Wieczerzak, K.; Witarski, T.; Kowalska, M.; Nawrat, D.; Roman, A.; Bielawski, A.; Nalepa, I. Effect of cocaine on responsiveness of alpha(1)-adrenergic receptors in rat cerebral cortex: Modulation by GABA-mimetic drugs. Pharmacol. Rep. 2008, 60, 980–984. [Google Scholar] [PubMed]
- Goertz, R.B.; Wanat, M.J.; Gomez, J.A.; Brown, Z.J.; Phillips, P.E.; Paladini, C.A. Cocaine increases dopaminergic neuron and motor activity via midbrain alpha1 adrenergic signaling. Neuropsychopharmacology 2015, 40, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Solecki, W.B.; Szklarczyk, K.; Pradel, K.; Kwiatkowska, K.; Dobrzanski, G.; Przewlocki, R. Noradrenergic signaling in the VTA modulates cocaine craving. Addict. Biol. 2018, 23, 596–609. [Google Scholar] [CrossRef]
- Drouin, C.; Darracq, L.; Trovero, F.; Blanc, G.; Glowinski, J.; Cotecchia, S.; Tassin, J.P. Alpha1b-adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J. Neurosci. 2002, 22, 2873–2884. [Google Scholar] [CrossRef]
- Auclair, A.; Drouin, C.; Cotecchia, S.; Glowinski, J.; Tassin, J.P. 5-HT2A and alpha1b-adrenergic receptors entirely mediate dopamine release, locomotor response and behavioural sensitization to opiates and psychostimulants. Eur. J. Neurosci. 2004, 20, 3073–3084. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Kosten, T.A. Previous exposure to cocaine enhances cocaine self-administration in an alpha 1-adrenergic receptor dependent manner. Neuropsychopharmacology 2007, 32, 638–645. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Kosten, T.A. Prazosin, an alpha-1 adrenergic antagonist, reduces cocaine-induced reinstatement of drug-seeking. Biol. Psychiatry 2005, 57, 1202–1204. [Google Scholar] [CrossRef]
- Newton, T.F.; De La Garza, R., 2nd; Brown, G.; Kosten, T.R.; Mahoney, J.J., 3rd; Haile, C.N. Noradrenergic alpha(1) receptor antagonist treatment attenuates positive subjective effects of cocaine in humans: A randomized trial. PLoS ONE 2012, 7, e30854. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, I.; Witarski, T.; Kowalska, M.; Filip, M.; Vetulani, J. Effect of cocaine sensitization on alpha1-adrenoceptors in brain regions of the rat: An autoradiographic analysis. Pharmacol. Rep. 2006, 58, 827–835. [Google Scholar] [PubMed]
- Paladini, C.A.; Mitchell, J.M.; Williams, J.T.; Mark, G.P. Cocaine self-administration selectively decreases noradrenergic regulation of metabotropic glutamate receptor-mediated inhibition in dopamine neurons. J. Neurosci. 2004, 24, 5209–5215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kalivas, P.W.; Duffy, P. Time course of extracellular dopamine and behavioral sensitization to cocaine. I. Dopamine axon terminals. J. Neurosci. 1993, 13, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Ritz, M.C.; Lamb, R.J.; Goldberg, S.R.; Kuhar, M.J. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science 1987, 237, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Johanson, C.E.; Fischman, M.W. The pharmacology of cocaine related to its abuse. Pharmacol. Rev. 1989, 41, 3–52. [Google Scholar] [PubMed]
- Nugent, F.S.; Kauer, J.A. LTP of GABAergic synapses in the ventral tegmental area and beyond. J. Physiol. 2008, 586, 1487–1493. [Google Scholar] [CrossRef]
- Saal, D.; Dong, Y.; Bonci, A.; Malenka, R.C. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 2003, 37, 577–582. [Google Scholar] [CrossRef]
- Nestler, E.J. The neurobiology of cocaine addiction. Sci. Pract. Perspect. 2005, 3, 4–10. [Google Scholar] [CrossRef]
- Kauer, J.A.; Malenka, R.C. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007, 8, 844–858. [Google Scholar] [CrossRef]
- Luscher, C.; Malenka, R.C. Drug-evoked synaptic plasticity in addiction: From molecular changes to circuit remodeling. Neuron 2011, 69, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Luscher, C. Cocaine-evoked synaptic plasticity of excitatory transmission in the ventral tegmental area. Cold Spring Harb. Perspect. Med. 2013, 3, a012013. [Google Scholar] [CrossRef] [PubMed]
- Velez-Hernandez, M.E.; Vazquez-Torres, R.; Velasquez-Martinez, M.C.; Jimenez, L.; Baez, F.; Sacktor, T.C.; Jimenez-Rivera, C.A. Inhibition of Protein kinase Mzeta (PKMzeta) in the mesolimbic system alters cocaine sensitization in rats. J. Drug Alcohol. Res. 2013, 2, 235669. [Google Scholar] [CrossRef] [PubMed]
- Kalivas, P.W.; Churchill, L.; Klitenick, M.A. GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience 1993, 57, 1047–1060. [Google Scholar] [CrossRef]
- Ikemoto, S.; Wise, R.A. Mapping of chemical trigger zones for reward. Neuropharmacology 2004, 47 (Suppl. 1), 190–201. [Google Scholar] [CrossRef]
- Darracq, L.; Blanc, G.; Glowinski, J.; Tassin, J.P. Importance of the noradrenaline-dopamine coupling in the locomotor activating effects of D-amphetamine. J. Neurosci. 1998, 18, 2729–2739. [Google Scholar] [CrossRef]
- Auclair, A.; Cotecchia, S.; Glowinski, J.; Tassin, J.P. D-amphetamine fails to increase extracellular dopamine levels in mice lacking alpha 1b-adrenergic receptors: Relationship between functional and nonfunctional dopamine release. J. Neurosci. 2002, 22, 9150–9154. [Google Scholar] [CrossRef]
- Kohout, T.A.; Lefkowitz, R.J. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 2003, 63, 9–18. [Google Scholar] [CrossRef]
- Toews, M.L.; Prinster, S.C.; Schulte, N.A. Regulation of alpha-1B adrenergic receptor localization, trafficking, function, and stability. Life Sci. 2003, 74, 379–389. [Google Scholar] [CrossRef]
- Reiter, E.; Lefkowitz, R.J. GRKs and beta-arrestins: Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 2006, 17, 159–165. [Google Scholar] [CrossRef]
- Garcia-Sainz, J.A.; Romero-Avila, M.T.; Alcantara-Hernandez, R. Mechanisms involved in alpha1B-adrenoceptor desensitization. IUBMB Life 2011, 63, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Pennock, R.L.; Dicken, M.S.; Hentges, S.T. Multiple inhibitory G-protein-coupled receptors resist acute desensitization in the presynaptic but not postsynaptic compartments of neurons. J. Neurosci. 2012, 32, 10192–10200. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sainz, J.A.; Gottfried-Blackmore, A.; Vazquez-Prado, J.; Romero-Avila, M.T. Protein kinase C-mediated phosphorylation and desensitization of human alpha(1b)-adrenoceptors. Eur. J. Pharmacol. 1999, 385, 263–271. [Google Scholar] [CrossRef]
- Chalothorn, D.; McCune, D.F.; Edelmann, S.E.; Garcia-Cazarin, M.L.; Tsujimoto, G.; Piascik, M.T. Differences in the cellular localization and agonist-mediated internalization properties of the alpha(1)-adrenoceptor subtypes. Mol. Pharmacol. 2002, 61, 1008–1016. [Google Scholar] [CrossRef]
- Kalivas, P.W.; Duffy, P. Repeated cocaine administration alters extracellular glutamate in the ventral tegmental area. J. Neurochem. 1998, 70, 1497–1502. [Google Scholar] [CrossRef]
- Steketee, J.D.; Rowe, L.A.; Chandler, L.J. The effects of acute and repeated cocaine injections on protein kinase C activity and isoform levels in dopaminergic brain regions. Neuropharmacology 1998, 37, 339–347. [Google Scholar] [CrossRef]
- Steketee, J.D. Intra-ventral tegmental area administration of H7 delays, but does not prevent the development of cocaine-induced sensitization. Brain Res. Bull. 1997, 43, 565–571. [Google Scholar] [CrossRef]
- Luu, P.; Malenka, R.C. Spike timing-dependent long-term potentiation in ventral tegmental area dopamine cells requires PKC. J. Neurophysiol. 2008, 100, 533–538. [Google Scholar] [CrossRef][Green Version]
- Mameli, M.; Luscher, C. Synaptic plasticity and addiction: Learning mechanisms gone awry. Neuropharmacology 2011, 61, 1052–1059. [Google Scholar] [CrossRef]
- Mameli, M.; Bellone, C.; Brown, M.T.; Luscher, C. Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat. Neurosci. 2011, 14, 414–416. [Google Scholar] [CrossRef]
- Bonci, A.; Williams, J.T. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron 1996, 16, 631–639. [Google Scholar] [CrossRef]
- Sorg, B.A.; Chen, S.Y.; Kalivas, P.W. Time course of tyrosine hydroxylase expression after behavioral sensitization to cocaine. J. Pharmacol. Exp. Ther. 1993, 266, 424–430. [Google Scholar] [PubMed]
- Arencibia-Albite, F.; Vazquez, R.; Velasquez-Martinez, M.C.; Jimenez-Rivera, C.A. Cocaine sensitization inhibits the hyperpolarization-activated cation current Ih and reduces cell size in dopamine neurons of the ventral tegmental area. J. Neurophysiol. 2012, 107, 2271–2282. [Google Scholar] [CrossRef] [PubMed]
- Arencibia-Albite, F.; Vazquez-Torres, R.; Jimenez-Rivera, C.A. Cocaine sensitization increases subthreshold activity in dopamine neurons from the ventral tegmental area. J. Neurophysiol. 2017, 117, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Kozell, L.B.; Meshul, C.K. The effects of acute or repeated cocaine administration on nerve terminal glutamate within the rat mesolimbic system. Neuroscience 2001, 106, 15–25. [Google Scholar] [CrossRef]
- Kozell, B.; Meshul, K. Alterations in nerve terminal glutamate immunoreactivity in the nucleus accumbens and ventral tegmental area following single and repeated doses of cocaine. Psychopharmacology 2003, 165, 337–345. [Google Scholar] [CrossRef]
- Kozell, L.B.; Meshul, C.K. Nerve terminal glutamate immunoreactivity in the rat nucleus accumbens and ventral tegmental area after a short withdrawal from cocaine. Synapse 2004, 51, 224–232. [Google Scholar] [CrossRef]
- White, F.J.; Kalivas, P.W. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol. Depend. 1998, 51, 141–153. [Google Scholar] [CrossRef]
- Engblom, D.; Bilbao, A.; Sanchis-Segura, C.; Dahan, L.; Perreau-Lenz, S.; Balland, B.; Parkitna, J.R.; Lujan, R.; Halbout, B.; Mameli, M.; et al. Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron 2008, 59, 497–508. [Google Scholar] [CrossRef]
- Pan, B.; Hillard, C.J.; Liu, Q.S. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J. Neurosci. 2008, 28, 1385–1397. [Google Scholar] [CrossRef]
- Niehaus, J.L.; Murali, M.; Kauer, J.A. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur. J. Neurosci. 2010, 32, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.L.; Schiffer, W.K.; Horan, B.A.; Highfield, D.; Dewey, S.L.; Brodie, J.D.; Ashby, C.R., Jr. Gamma-vinyl GABA, an irreversible inhibitor of GABA transaminase, alters the acquisition and expression of cocaine-induced sensitization in male rats. Synapse 2002, 46, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, S.C.; Taylor, S.R.; Horton, M.L.; Barber, E.N.; Lyle, L.T.; Stobbs, S.H.; Allison, D.W. Cocaine disinhibits dopamine neurons in the ventral tegmental area via use-dependent blockade of GABA neuron voltage-sensitive sodium channels. Eur. J. Neurosci. 2008, 28, 2028–2040. [Google Scholar] [CrossRef] [PubMed]
- Lammel, S.; Lim, B.K.; Ran, C.; Huang, K.W.; Betley, M.J.; Tye, K.M.; Deisseroth, K.; Malenka, R.C. Input-specific control of reward and aversion in the ventral tegmental area. Nature 2012, 491, 212–217. [Google Scholar] [CrossRef]
- Bromberg-Martin, E.S.; Matsumoto, M.; Hikosaka, O. Dopamine in motivational control: Rewarding, aversive, and alerting. Neuron 2010, 68, 815–834. [Google Scholar] [CrossRef]
- Van Zessen, R.; Phillips, J.L.; Budygin, E.A.; Stuber, G.D. Activation of VTA GABA neurons disrupts reward consumption. Neuron 2012, 73, 1184–1194. [Google Scholar] [CrossRef]
- Margolis, E.B.; Lock, H.; Hjelmstad, G.O.; Fields, H.L. The ventral tegmental area revisited: Is there an electrophysiological marker for dopaminergic neurons? J. Physiol. 2006, 577, 907–924. [Google Scholar] [CrossRef]
- Sarti, F.; Borgland, S.L.; Kharazia, V.N.; Bonci, A. Acute cocaine exposure alters spine density and long-term potentiation in the ventral tegmental area. Eur. J. Neurosci. 2007, 26, 749–756. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).