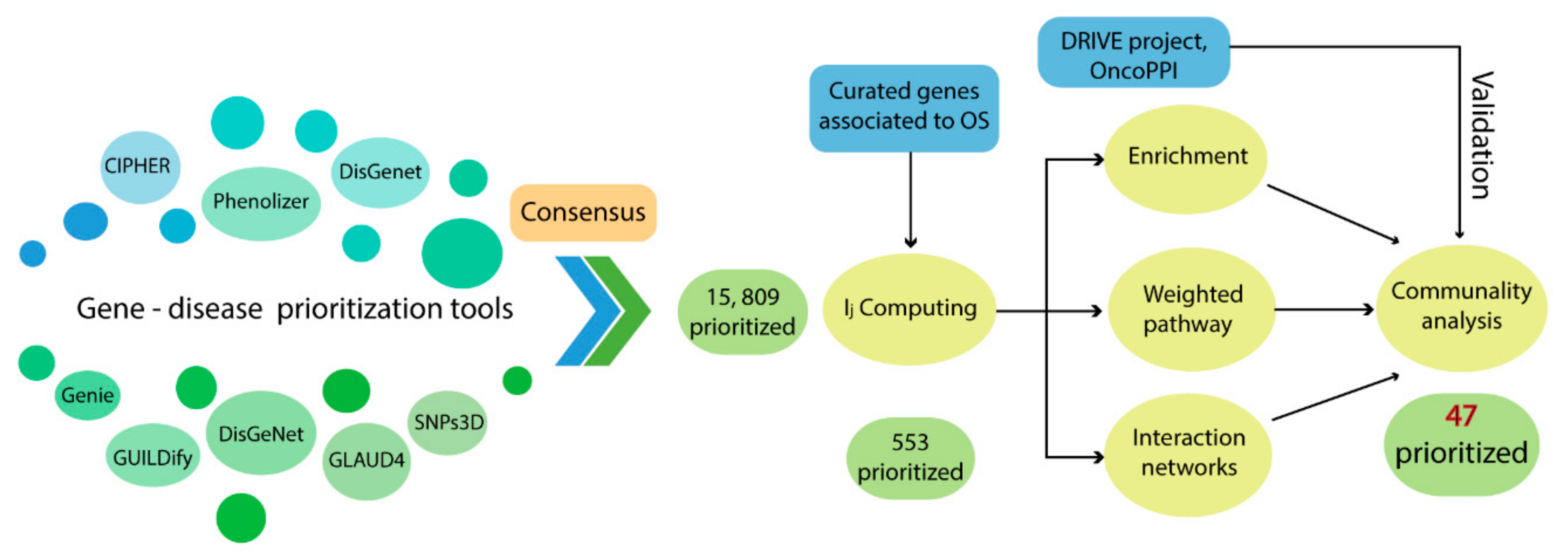
3. Discussion
As shown in
Table 1, the detection rate of our consensus prioritization strategy was higher than all the bioinformatics tools employed in this analysis. Moreover, the mean rank of the pathogenic genes detected in the top 1% of the list was 49.3.
Table 2 indicates that, on average, the 45 G1–G2 genes were located in the top 50 positions. These results confirm that this methodology does indeed improve the detection and prioritization of pathogenic genes, as had been previously described in other pathologies [
24,
25].
As a first approach, the prioritization strategy resulted in a consensus list of 553 genes and the 10 top-ranked genes were
TP53,
RB1,
CHEK2,
RUNX2,
E2F1,
MDM2,
CDKN1A,
JUN,
CCNA2 and
CDKN2A.
TP53,
RB1,
CHEK2, and
MDM2 were ranked in 1st, 2nd, 3rd, and 6th positions, respectively, and also the arrangement of the pathogenic genes in this list shows a distribution in the top positions. So far, the gene ranking along this prioritization reflects a proper gene weighting based mainly on this consensus strategy. These genes had been previously described in OS pathogenesis. Early studies focused on the molecular biology of OS were carried out on individuals with familial syndromes, which predisposed them to this tumor. Germline inactivation of
RB1 and
TP53 were initially described in patients with hereditary retinoblastoma and Li–Fraumeni syndrome, respectively [
26,
27], and subsequently in sporadic sarcomas [
28,
29]. Given that these two suppressors are central proteins in controlling the cell cycle, later studies briefly described many others that interacted with them. Mouse double minute 2 (
MDM2), for example, is a protein that binds to RB1 and inactivates TP53 [
30]. Its amplification is an event that occurs in primary OS (3–25%) and it is overexpressed in metastases and recurrences [
31,
32]. CHEK2 is another protein that is part of a DNA damage checkpoint, works as a stabilizer of TP53, and shows a 7% frequency of mutations in OS patients [
33,
34].
The biological processes derived from the GO analysis of the 553 genes describe TP53 as a principal signal transducer that mediates processes associated with cell cycle, DNA damage response, DNA replication and intrinsic/extrinsic apoptotic signaling regulation. Additionally, more specific biological processes were described, for instance, fibroblast proliferation, osteoblast differentiation and development, and mesenchymal cell proliferation and transition. In accordance with our results, previous studies have identified similar biological processes related to OS, where the following are considered OS-associated terms: cell cycle regulation (mainly mediated by RB1 and TP53), osteoblast differentiation (mediated by RUNX2), DNA damage, stress response, epigenetic processes, mitosis, cell motility functions, and members involved in OS cell proliferation (weighting NFKB signaling, NFKBIE, and RELA members) [
3,
35,
36,
37]. Taken together, these processes suggest that the consensus list is evidence of the genes associated with osteogenesis, cell differentiation, and transition to bone cell types. In addition, the terms derived from the pathway enrichment analysis (
Table 4) are in accordance with these biological processes.
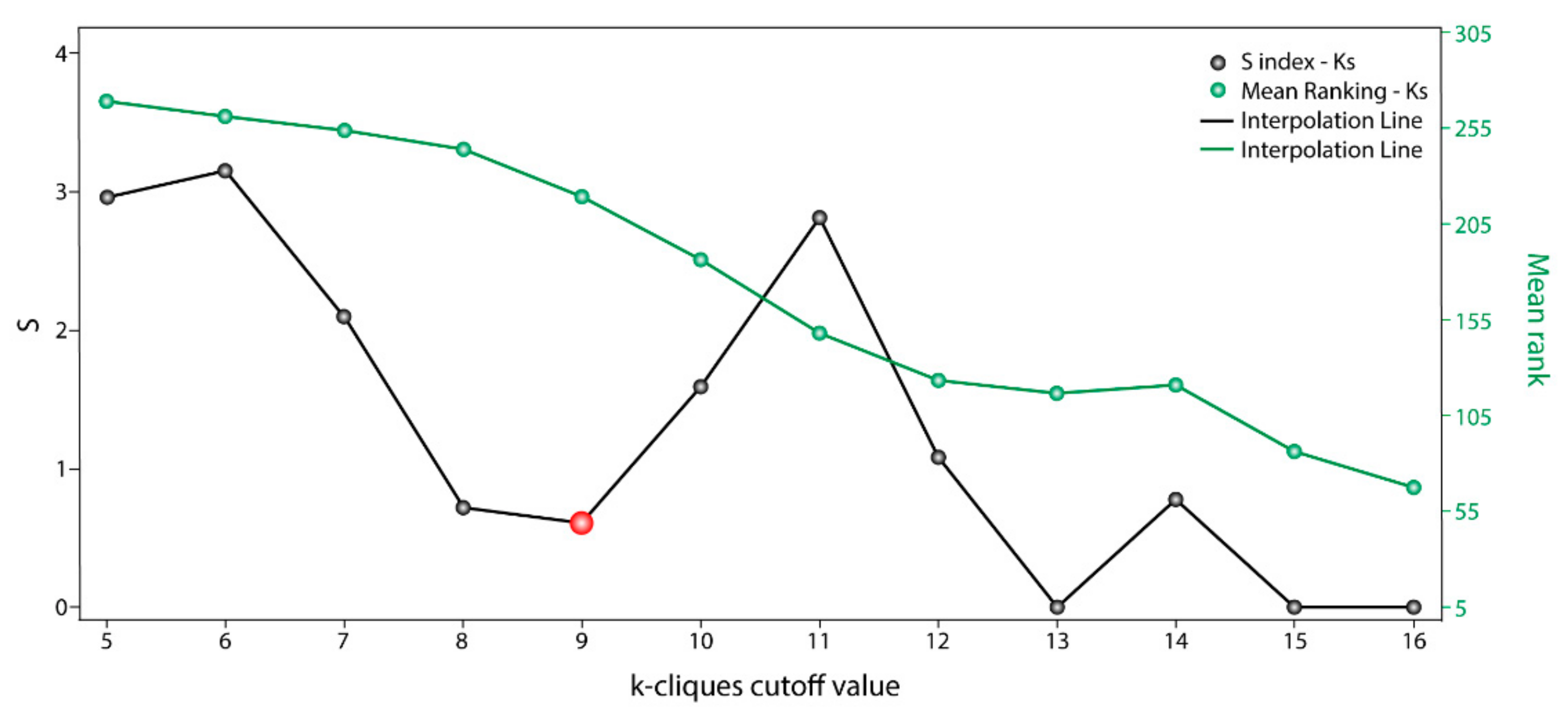
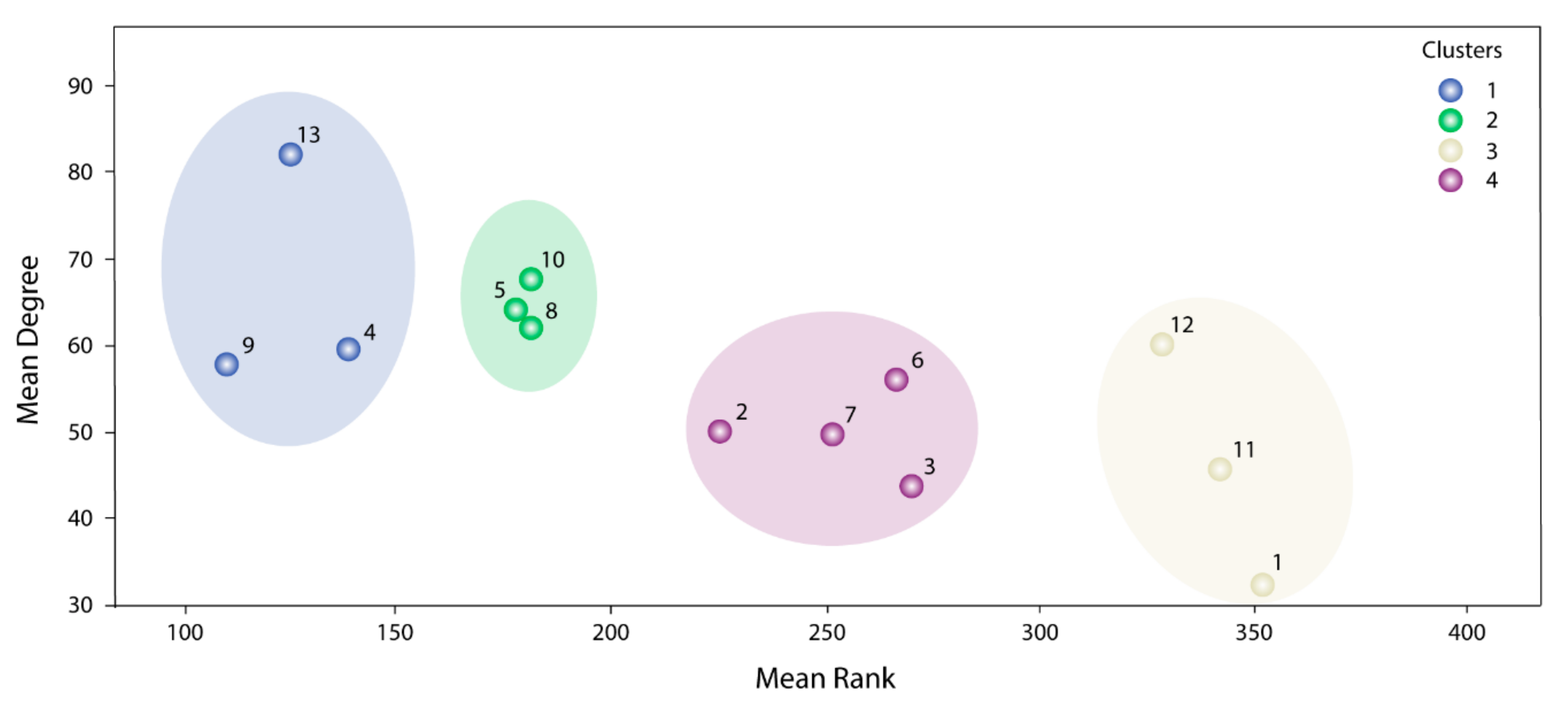
The information used by STRING allowed us to define the degree of physical interaction of the consensus list members and calculate their centrality index. This centrality index was used as a variable to evidence the contribution rate of the pathogenic genes to a common biological purpose. Thus, the greater the centrality for a node within the OS–PPI network, the greater the probability of its contributing to pathogenesis. This association was validated by analyzing the genes defined as pathogenic (G1–G2), in which significant differences were observed in comparison with the rest of the consensus genes (p < 0.0001). The centrality index calculated from the 503 nodes included in the protein–protein interaction network determined TP53 as the most central node, followed by AKT1, MYC, JUN, EP300, CREBBP, CCND1, CDKN1A, STAT3, and RB1. Furthermore, this degree allowed for the definition of more specific clusters and prioritization of gene communities associated with OS pathogenesis. Thus, k-9 was determined as the clique with the best gene distribution among all the resulting communities ( index 0.719) and Communities 4, 5, 8, 9, 10, and 13 as the most important groups of genes within our study.
The pathway enrichment analysis for the
k = 9-clique results in, almost in its entirety, the same terms obtained from the initial consensus list. This confirms that the gene filtered through the communality analysis comprised almost the same biological processes. Considering the
(
Table 5), the P53 signaling pathway and cell cycle are in the top positions. FOXO also increases its significance in this enrichment analysis. In different cancer types, PI3K/AKT, Ras-MEK-ERK, IKK, and AMPK are the most important signaling pathways interacting with FOXO [
38]. The gain of function of P13K and RAS, or PTEN disruption, are oncogenic events that promote a loss of function in the Forkhead Box transcription factors (
FOXO) [
39]. Interestingly, loss of its expression promotes impaired osteogenic differentiation, suggesting that
FOXO1 is involved in osteoblastogenesis and osteoclastogenesis [
40,
41,
42]. Moreover, FOXO members have an important role in cell fate decision, via triggering the expression of death receptor ligands like FASLG, TNF apoptosis ligand, and some BCL-2 family members (
BCL2L1,
BNIP3,
BCL2L11) [
43,
44,
45,
46]. FOXO expression in OS tumors is low or even lacking altogether, leading to tumor progression and cell cycle arrest [
47]. The fact that
FOXO enhances its weight within our enrichment analysis demonstrates its importance as a signaling pathway in the pathogenesis of OS. Furthermore, the close relationship between the FOXO signaling pathway and cell cycle, events of osteoclast differentiation and apoptosis via the TNF signaling pathway, is evidenced in the pathway enrichment analysis applied to the consensus list and the
k = 9 clique.
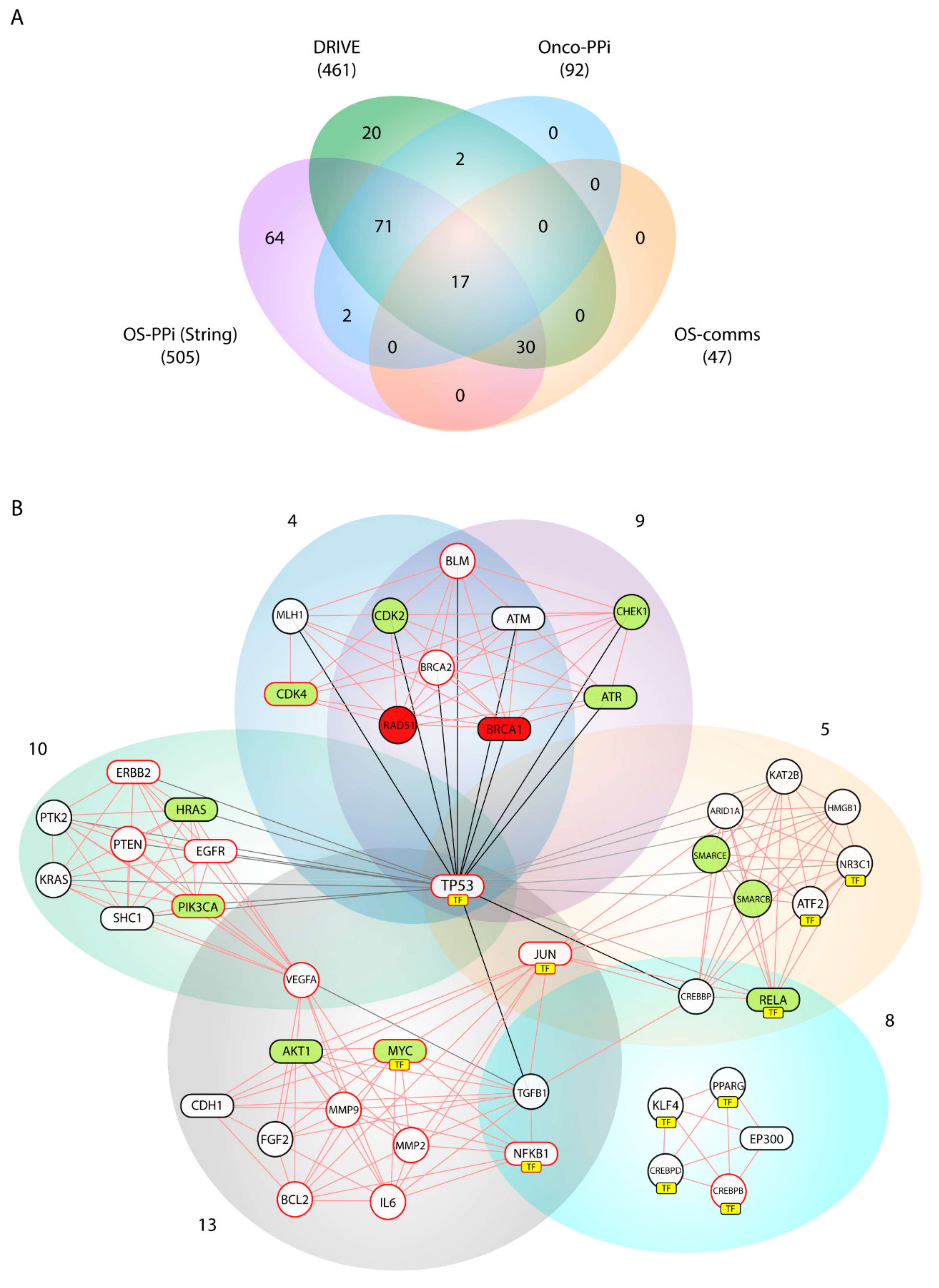
Our consensus strategy seeks to specify a group of genes that describe the molecular etiology of OS. In this sense, the use of all the strategies previously described prioritizes to a great extent the 47 genes arranged in Communities 4, 5, 8, 9, 10, and 13. From these six communities,
BRCA1,
AKT1,
ATR,
CDK4,
HRAS,
MYC,
PIK3CA,
RELA,
STAT3 are genes validated by DRIVE and Onco–PPI (19.1%),
RAD51,
CDK2,
CHEK1,
SMARCB1,
SMARCE1 are validated only by DRIVE (10.6%), and
ATM,
CDH1,
EGFR,
EP300,
ERBB2,
JUN,
NFKB1,
SHC1,
TP53,
SP1 by Onco–PPI (21.3%). The sub-network generated from these communities (OS–comms network) reflects closely interrelated genes at the cellular interaction level (
Figure 4B) and also groups of genes immersed in important oncological processes. Tamborero et al. [
48], from exome sequencing data of 3205 tumors in the Cancer Genome Atlas (TCGA) research network, proposed 291 high-confidence cancer driver genes acting on 12 different cancer types. Although in this study, data from samples of bone tumors were not taken into account, their results showed the members of the PI3K signaling pathway as central onco-drivers, ATR-BRCA1 as regulatory nodes of repair processes associated with TP53, CHEK1 and AKT as the main regulators of cell cycle in function of CDK1A, and CDK1B and activators for downstream pathways such as FOXO. This experimental data support our findings, where
PIK3CA,
AKT1,
PTEN,
HRAS and
SHC1 were nodes highly connected within our OS–comms network. Nodes that connect to Communities 10 and 13 describe genes representative of our weighted tumorigenic pathways, PI3K/AKT and MAPK/ERK.
The findings reported here suggest that PI3K/AKT and MAPK/ERK are the main signaling pathways deregulated for OS. Several reports have shown that these pathways are responsible for controlling cellular processes related to proliferation, growth, differentiation, and apoptosis [
49,
50]. In fact, the Ras/Raf/MEK/ERK pathway is hyperactivated in 30% of human cancers [
51] and nearly 67% of OS shows aberrant ERK activation [
52]. The extra cellular-signal-regulated kinases (ERK) promote cell proliferation, cell survival, and metastasis, particularly by its upstream activation from EGFR and the G protein-coupled receptor Ras [
53]. The presence of
SHC1,
EGFR,
HRAS,
PIK3CA,
ERBB2 within Community 10 support this scenario for OS. In addition, the high connectivity of the matrix metalloproteases,
MMP2 and
MMP9, in Community 13 suggests a metastasis event in the function of these signaling pathways.
Although the invasion of tumor cells is a general characteristic in carcinogenesis, metastasis to the lung is one of the main characteristics in patients with OS and one of the major causes of mortality [
54,
55], so this event is a hallmark for this sarcoma. Pathogenic events, including cellular detachment from primary tumors, matrix remodeling and invasion from tumor cells, angiogenesis, vascular dissemination, and proliferation at new sites, are involved in tumor metastasis [
56,
57]. Upstream regulators of MAP/ERK signaling such as
IL6,
VEGFA, and
FGFR1 demonstrate an important role in this process [
58,
59,
60,
61,
62] and are prioritized in our results. In addition, Community 13 shows the
MMP2 and
MMP9 genes with a high centrality index. A high expression of MMP9 was observed in metastatic OS samples [
63,
64], leading to speculation that this metalloproteinase can promote cell migration and invasion in OS by degradation components of the extracellular matrix. This evidence suggests that
MMP2 and
MMP9, together with upstream regulators of MAP/ERK signaling such as
IL6,
FGF2,
VEGFA,
EGFR and
ERBB2, are pathogenic nodes dependent on the centrality of PI3K/AKT and MAPK/ERK. This finding could be related to aspects of invasiveness and prognosis, mainly in tumors that present deregulation in these two signaling pathways.
In addition to evidencing the previous findings, Communities 4, 5 and 9 include genes widely described in processes of homologous recombination (HR), base excision repair, and chromatin modification. Cells DNA damage response principally involves maintaining chromosome integrity and genome stability and implies recognition of DNA lesions, followed by an activation of the checkpoints in the cell cycle that promotes cellular signaling cascades related to DNA repair. While the ATM-CHEK2 pathway is responsible for the initiation of cellular responses to double-strand breaks [
65,
66], ATR-CHEK1 responds to DNA replication stress by means of the phosphorylation of several substrates in response to agents such as UV and X-ray among others [
67].
ATM,
ATR, and
CHEK1 show a high centrality index in the OS–comms network, interacting in addition to
BRCA1 and
RAD51, described as essential genes, and with the cyclin-dependent kinases,
CDK2 and
CDK4, described as active ones according to the DRIVE validation. Checkpoint activation by ATM mainly controls G1/S, whereas ATM and ATR contribute to establishing and maintaining the S and G2/M checkpoints [
68]. Either by activation of ATR-CHEK1 or ATM-CHEK2, DNA damage signaling promotes inhibition of CDK activity and therefore the activation of G1/S, intra-S, and G2/M checkpoints [
69]. Consequently, it is likely that such nodes associated with DNA repair, such as
ATM,
ATR,
CHEK1,
BLM,
RAD51 and
MLH1 (as shown in our pathway enrichment analysis), together with those previously described (
BRCA1 and
BRCA2) from exome sequencing [
70], have important implications regarding the deregulation of the cell cycle evidenced in OS.
While it is true that the nodes described for Communities 4 and 9 are mainly related to repair and cell cycle control events, the HR repair complex is involved in a hallmark event for sarcomas, such as alternative telomere maintenance (ALT). Several molecular details of this mechanism still remain unknown; however, two distinctive telomere phenotypes are described for ALT in human telomerase-negative cells (ALT cells) such as long and heterogeneous telomere DNA and promyelocytic leukemia (PML) body [
71], together forming the ALT-associated promyelocytic leukemia body (APB). The PML body is a nuclear made up of proteins which form amongst the chromatin and is related to a wide range of cellular processes including tumors formation, cellular senescence, and DNA repair [
72,
73]. Numerous lines of evidence strongly suggest that the ALT pathway is dependent on HR since several proteins involved in DNA double-strand break (DSB) are localized at APBs [
74,
75,
76,
77]. It is significant that proteins localized at APBs, such as PML, DNA helicases of the RecQ family (
BLM,
WRN and
RECQL4),
RAD51 and
RAD52 (a member of the MNR complex), rank highly in our prioritization. In this sense, the members belonging to HR complexes are described as repair complexes in response to DNA damage. They are relevant to the pathogenesis of the OS, not only as factors immersed in cell cycle control, as previously discussed, but also because they are involved in processes of chromosome stability given by telomere maintenance [
78,
79,
80,
81]. Consistent with the literature, where bone tumors are termed as highly heterogeneous, highly mutable, and genetically unstable, members described in Communities 4 and 9 (
TP53,
ATM,
ATR,
CHEK1,
BLM,
BRCA1,
BRCA2,
RAD51,
MLH1,
CDK2,
CDK4) explain many of these key features within OS, and can also be associated with important clinical characteristics such as tumor aggressiveness, metastasis, and poor survival.
The use of the GTRD database allowed us to define the frequency of interaction of each TF with the 553 prioritized genes. It is worth noticing that more than half of the prioritized factors (103, 82.4%) interacted with more than half of all genes at the same time. This suggests that more than 80% of the genes defined as TFs actively regulated the genes associated with the pathogenesis of OS. The weight given to each one of these TFs through interaction analysis places the following genes on the top positions:
TP53,
E2F1,
JUN,
RUNX2,
FLI1,
YY1,
HIF1A,
MYC,
TP63,
ESR1,
WT1,
E2F4,
ATF2,
NFKB1,
AR,
SP1,
STAT1,
ERG,
CEBPB, and
TFAP2A. When compared to total prioritization, genes
E2F1,
JUN,
RUNX2,
FLI1,
YY1,
HIF1A,
MYC,
TP63,
ESR1,
WT1,
E2F4,
ATF2, and
NFKB1 significantly improved their ranking. During the G1 phase of the cell cycle, RB1 suppresses the function of the E2F1, E2F2, and E2F3 TFs. Sequential hypo phosphorylation of RB1 by cyclin-dependent kinases, CDK4 and CDK6, and CDK2, led in the release of E2F and transcription of genes necessary for cell cycle progression, including cyclins A, D, and E [
82]. The improved score of these TFs suggests that these deregulation events in the cell cycle are basal within the pathogenesis of the OS. Although this scenario is common for all types of cancer, a deeper study of the
E2F1 and
E2F4 genes, and depending on those prioritized in Communities 4 and 9 along with
TP53, would be necessary to define driver proteins in OS tumors.
We identified
TP53,
JUN,
MYC,
ATF2,
NFKB1,
SP1,
CEBPB,
STAT3,
KLF4,
RELA,
NR3C1,
CEBPD, and
PPARG as TFs (13 or the 47 nodes) in the OS–comms network. The new ranking calculated for each of them improved significantly when compared to the ranking of all OS genes (
Table S2). This suggests that their degree of regulation within this network is very significant and shows evidence of its importance as regulatory proteins within each prioritized cluster.
TFs were grouped over Communities 5, 8 and 13. With
TP53 as the central node,
JUN and
MYC are key factors in the pathogenesis of the OS that regulate signaling associated with the pathogenic pathways PI3K/AKT and MAPK/ERK. Furthermore, the prioritization of TFs evidenced
NFKB1 as a central node in these three communities. Nuclear factor-kappa B1 (
NFκB1) is a pleiotropic transcription factor that contributes to tumorigenesis in many types of cancer. It works as a key regulator of a variety of genes implicated in many biological events including cell survival, differentiation, apoptosis, and autophagy [
83]. When observing the OS–comms network, the high degree interaction of
AKT with respect to
JUN-
MYC,
TGFB1,
NFKB1, and
BCL2 suggests this cluster as an important group in the OS pathogenesis. GO terms listed in
Table 3 are in accordance with these findings since its activation promotes many types of downstream signaling including osteoblast differentiation via TGFB1 and NFK1 or apoptosis via BCL2 [
84,
85].
In conclusion, the use of a consensus strategy proved to be efficient when specifying a broad list of genes obtained from several bioinformatics prioritization tools. In addition, the combination of these strategies with a network enrichment analysis allowed us to show not only real interactions between specific genes but also to define internal interactions that explained cellular events associated with OS pathogenesis. Our results validate several studies that describe the signaling pathways PI3K/AKT and MAPK/ERK as oncological for OS. Nevertheless, given its centrality at the cellular signaling level, its deregulation can influence downstream specific pathways, such as FOXO, and promote tumorigenic scenarios like osteoblast undifferentiation via TGFB1 and NFK1, apoptosis via BCL2, and migration and metastasis mediated mainly by MMP2 and MMP9.
What is more, the gene composition of Communities 4 and 9, and more specifically to their ATM, ATR, CHEK1, and RAD51 genes, suggest that the HR repair complex is an important group of genes within the pathogenesis of the OS. Its deregulation can influence tumorigenic events characteristic of this sarcoma as generalized disruption in the cell cycle and ALT mechanisms. Hence, it is necessary to experimentally validate these results, taking into account not only the patient’s age group but also genetic factors that can influence the molecular behavior of these bone tumors, such as racial and ethnic factors. It should also be interesting to study genetic variants of the transcription factors identified and their relationship with possible disease prevalence.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}