Running to Stand Still: Naive CD8+ T Cells Actively Maintain a Program of Quiescence


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Chromatin Landscape Forms a Molecular Barrier Limiting Naive T Cell Activation
3. The Naïve Transcriptional Program Is Maintained by Permissive Chromatin Modifications and Is Shut Down upon Activation
4. Repressive Chromatin Modifications in Naïve Cd8+ T Cells Actively Restrains Effector Differentiation
5. Effector Differentiation-Associated Transcription Factors Are Poised in Naïve Cells
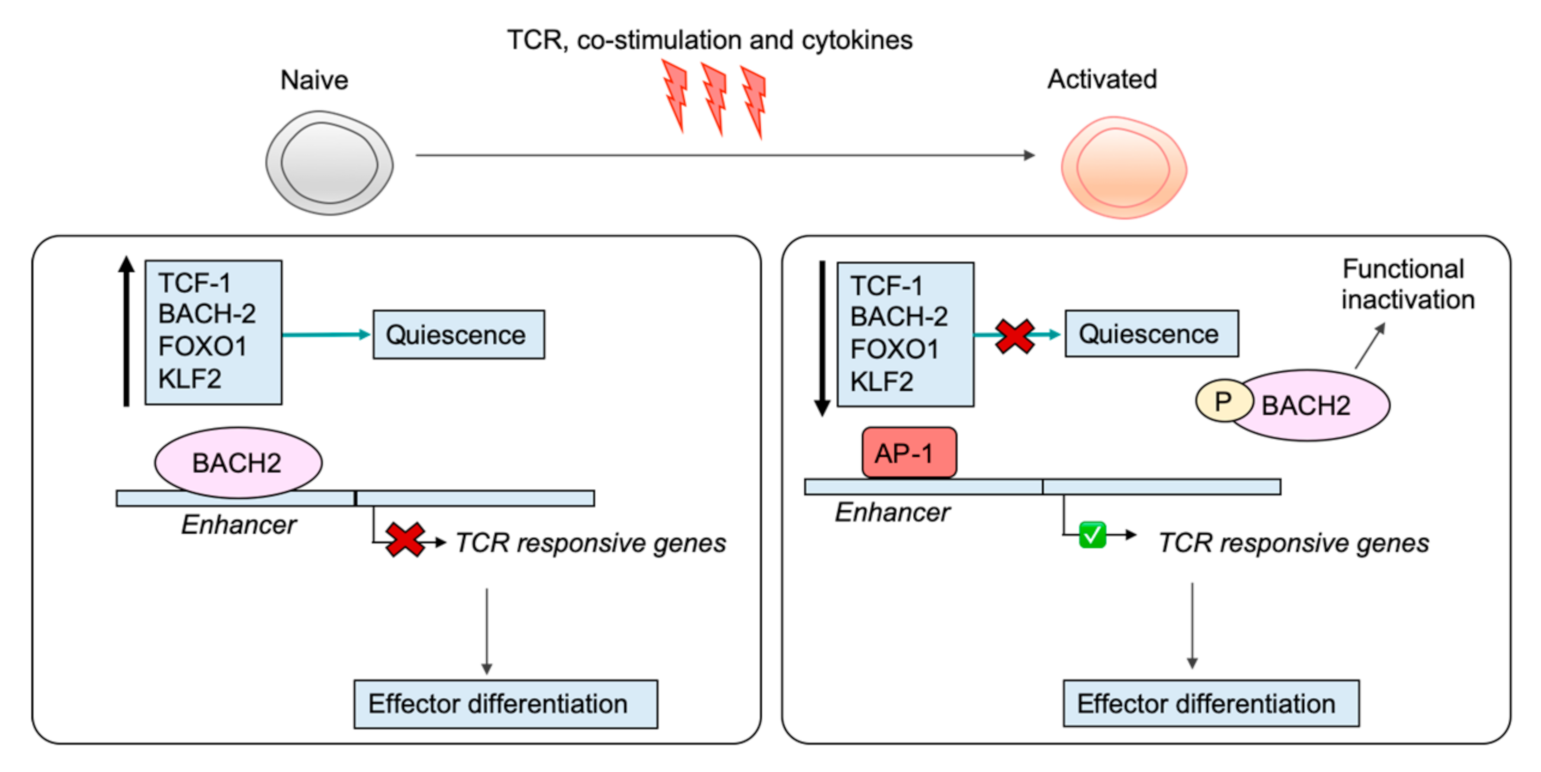
6. Shutting Down the Naïve T Cell Program
7. Engagement of the Effector CD8+ T Cell Program: Driven by Specific TFs
8. Regulation by Transcription Factors Restrains Activation in Naïve T Cells
9. At the Cell Surface: The Inhibitory Receptor VISTA Enforces T Cell Naivety
10. Regulation of RNA Degradation and Protein Abundance Are Crucial for Maintaining the Quiescence of Naïve T Cells
11. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| APC | Antigen-presenting cell |
| CTL | Cytotoxic T lymphocyte |
| IL-2 | Interleukin-2 |
| IFN-γ | Interferon gamma |
| MHC | Major histocompatibility complex |
| NK | Natural killer |
| PTM | Post-translational modification |
| TCR | T cell receptor |
| TE | Transcriptional enhancer |
| TF | Transcription factor |
| TNF | Tumour necrosis factor |
References
- Viola, A.; Lanzavecchia, A. T cell activation determined by T cell receptor number and tunable thresholds. Science 1996, 273, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, R.M.; Doherty, P.C. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 1974, 248, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Lins, D.C.; Mescher, M.F. Signal 3 determines tolerance versus full activation of naive CD8 T cells: Dissociating proliferation and development of effector function. J. Exp. Med. 2003, 197, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.R.; Mintern, J.; La Gruta, N.L.; Kedzierska, K.; Doherty, P.C.; Turner, S.J. Cell cycle-related acquisition of cytotoxic mediators defines the progressive differentiation to effector status for virus-specific CD8+ T cells. J. Immunol. 2008, 181, 3818–3822. [Google Scholar] [CrossRef]
- La Gruta, N.L.; Turner, S.J.; Doherty, P.C. Hierarchies in cytokine expression profiles for acute and resolving influenza virus-specific CD8+ T cell responses: Correlation of cytokine profile and TCR avidity. J. Immunol. 2004, 172, 5553–5560. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.R.; Kedzierska, K.; Doherty, P.C.; Turner, S.J. Heterogeneity of effector phenotype for acute phase and memory influenza A virus-specific CTL. J. Immunol. 2007, 179, 64–70. [Google Scholar] [CrossRef]
- Kaech, S.M.; Hemby, S.; Kersh, E.; Ahmed, R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell 2002, 111, 837–851. [Google Scholar] [CrossRef]
- Lalvani, A.; Brookes, R.; Hambleton, S.; Britton, W.J.; Hill, A.V.; McMichael, A.J. Rapid effector function in CD8+ memory T cells. J. Exp. Med. 1997, 186, 859–865. [Google Scholar] [CrossRef]
- Sridhar, S.; Begom, S.; Bermingham, A.; Hoschler, K.; Adamson, W.; Carman, W.; Bean, T.; Barclay, W.; Deeks, J.J.; Lalvani, A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013, 19, 1305–1312. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Stralin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell 2020, 183, 158–168. [Google Scholar] [CrossRef]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Carvalheiro, H.; da Silva, J.A.; Souto-Carneiro, M.M. Potential roles for CD8(+) T cells in rheumatoid arthritis. Autoimmun. Rev. 2013, 12, 401–409. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Schmidt, C.S.; Mondino, A.; Lins, D.C.; Kedl, R.M.; Jenkins, M.K.; Mescher, M.F. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J. Immunol. 1999, 162, 3256. [Google Scholar]
- Lenschow, D.J.; Walunas, T.L.; Bluestone, J.A. CD28/B7 System of T Cell costimulation. Annu. Rev. Immunol. 1996, 14, 233–258. [Google Scholar] [CrossRef]
- Hamilton, S.E.; Jameson, S.C. CD8 T cell quiescence revisited. Trends Immunol. 2012, 33, 224–230. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef]
- Orford, K.; Kharchenko, P.; Lai, W.; Dao, M.C.; Worhunsky, D.J.; Ferro, A.; Janzen, V.; Park, P.J.; Scadden, D.T. Differential H3K4 methylation identifies developmentally poised hematopoietic genes. Dev. Cell 2008, 14, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Krajewski, K.; Nady, N.; Tempel, W.; Xue, S.; Badeaux, A.I.; Barsyte-Lovejoy, D.; Martinez, J.Y.; Bedford, M.T.; Fuchs, S.M.; et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012, 19, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Denton, A.E.; Russ, B.E.; Doherty, P.C.; Rao, S.; Turner, S.J. Differentiation-dependent functional and epigenetic landscapes for cytokine genes in virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 15306–15311. [Google Scholar] [CrossRef]
- Russ, B.E.; Olshanksy, M.; Smallwood, H.S.; Li, J.; Denton, A.E.; Prier, J.E.; Stock, A.T.; Croom, H.A.; Cullen, J.G.; Nguyen, M.L.; et al. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8(+) T cell differentiation. Immunity 2014, 41, 853–865. [Google Scholar] [CrossRef]
- Gray, S.M.; Kaech, S.M.; Staron, M.M. The interface between transcriptional and epigenetic control of effector and memory CD8(+) T-cell differentiation. Immunol. Rev. 2014, 261, 157–168. [Google Scholar] [CrossRef]
- Scharer, C.D.; Barwick, B.G.; Youngblood, B.A.; Ahmed, R.; Boss, J.M. Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol. 2013, 191, 3419–3429. [Google Scholar] [CrossRef]
- Youngblood, B.; Oestreich, K.J.; Ha, S.J.; Duraiswamy, J.; Akondy, R.S.; West, E.E.; Wei, Z.; Lu, P.; Austin, J.W.; Riley, J.L.; et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity 2011, 35, 400–412. [Google Scholar] [CrossRef]
- Youngblood, B.; Hale, J.S.; Kissick, H.T.; Ahn, E.; Xu, X.; Wieland, A.; Araki, K.; West, E.E.; Ghoneim, H.E.; Fan, Y.; et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 2017, 552, 404–409. [Google Scholar] [CrossRef]
- Chen, Y.; Zander, R.; Khatun, A.; Schauder, D.M.; Cui, W. Transcriptional and epigenetic regulation of effector and memory CD8 T cell differentiation. Front. Immunol. 2018, 9, 2826. [Google Scholar] [CrossRef] [PubMed]
- Russ, B.E.; Denton, A.E.; Hatton, L.; Croom, H.; Olson, M.R.; Turner, S.J. Defining the molecular blueprint that drives CD8(+) T cell differentiation in response to infection. Front. Immunol. 2012, 3, 371. [Google Scholar] [CrossRef] [PubMed]
- Henning, A.N.; Roychoudhuri, R.; Restifo, N.P. Epigenetic control of CD8(+) T cell differentiation. Nat. Rev. Immunol. 2018, 18, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef]
- Yasui, D.; Miyano, M.; Cai, S.; Varga-Weisz, P.; Kohwi-Shigematsu, T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 2002, 419, 641–645. [Google Scholar] [CrossRef]
- Cai, S.; Lee, C.C.; Kohwi-Shigematsu, T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat. Genet. 2006, 38, 1278–1288. [Google Scholar] [CrossRef]
- Stephen, T.L.; Payne, K.K.; Chaurio, R.A.; Allegrezza, M.J.; Zhu, H.; Perez-Sanz, J.; Perales-Puchalt, A.; Nguyen, J.M.; Vara-Ailor, A.E.; Eruslanov, E.B.; et al. SATB1 expression governs epigenetic repression of PD-1 in tumor-reactive T Cells. Immunity 2017, 46, 51–64. [Google Scholar] [CrossRef]
- Nussing, S.; Koay, H.F.; Sant, S.; Loudovaris, T.; Mannering, S.I.; Lappas, M.; d′Udekem, Y.; Konstantinov, I.E.; Berzins, S.P.; Rimmelzwaan, G.F.; et al. Divergent SATB1 expression across human life span and tissue compartments. Immunol. Cell Biol. 2019, 97, 498–511. [Google Scholar] [CrossRef]
- Tiemessen, M.M.; Baert, M.R.; Kok, L.; van Eggermond, M.C.; van den Elsen, P.J.; Arens, R.; Staal, F.J. T Cell factor 1 represses CD8+ effector T cell formation and function. J. Immunol. 2014, 193, 5480–5487. [Google Scholar] [CrossRef]
- Ladle, B.H.; Li, K.P.; Phillips, M.J.; Pucsek, A.B.; Haile, A.; Powell, J.D.; Jaffee, E.M.; Hildeman, D.A.; Gamper, C.J. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proc. Natl. Acad. Sci. USA 2016, 113, 10631–10636. [Google Scholar] [CrossRef]
- Crompton, J.G.; Narayanan, M.; Cuddapah, S.; Roychoudhuri, R.; Ji, Y.; Yang, W.; Patel, S.J.; Sukumar, M.; Palmer, D.C.; Peng, W.; et al. Lineage relationship of CD8(+) T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell Mol. Immunol. 2016, 13, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Russ, B.E.; Olshansky, M.; Li, J.; Nguyen, M.L.T.; Gearing, L.J.; Nguyen, T.H.O.; Olson, M.R.; McQuilton, H.A.; Nussing, S.; Khoury, G.; et al. Regulation of H3K4me3 at transcriptional enhancers characterizes acquisition of virus-specific CD8(+) T Cell-lineage-specific function. Cell Rep. 2017, 21, 3624–3636. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L. Metabolism in T cell activation and differentiation. Curr. Opin. Immunol. 2010, 22, 314–320. [Google Scholar] [CrossRef]
- Phan, A.T.; Goldrath, A.W.; Glass, C.K. Metabolic and epigenetic coordination of T Cell and macrophage immunity. Immunity 2017, 46, 714–729. [Google Scholar] [CrossRef]
- Lawrence, C.W.; Braciale, T.J. Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J. Immunol. 2004, 173, 1209–1218. [Google Scholar] [CrossRef]
- Araki, Y.; Wang, Z.; Zang, C.; Wood, W.H., III; Schones, D.; Cui, K.; Roh, T.Y.; Lhotsky, B.; Wersto, R.P.; Peng, W.; et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 2009, 30, 912–925. [Google Scholar] [CrossRef]
- Kersh, E.N.; Fitzpatrick, D.R.; Murali-Krishna, K.; Shires, J.; Speck, S.H.; Boss, J.M.; Ahmed, R. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J. Immunol. 2006, 176, 4083–4093. [Google Scholar] [CrossRef]
- Carty, S.A.; Gohil, M.; Banks, L.B.; Cotton, R.M.; Johnson, M.E.; Stelekati, E.; Wells, A.D.; Wherry, E.J.; Koretzky, G.A.; Jordan, M.S. The loss of TET2 promotes CD8(+) T Cell memory differentiation. J. Immunol. 2018, 200, 82–91. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- LaMere, S.A.; Thompson, R.C.; Meng, X.; Komori, H.K.; Mark, A.; Salomon, D.R. H3K27 methylation dynamics during CD4 T Cell activation: Regulation of JAK/STAT and IL12RB2 expression by JMJD3. J. Immunol. 2017, 199, 3158–3175. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zou, J.; Wang, M.; Ding, X.; Chepelev, I.; Zhou, X.; Zhao, W.; Wei, G.; Cui, J.; Zhao, K.; et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat. Commun. 2014, 5, 5780. [Google Scholar] [CrossRef] [PubMed]
- Aranda, S.; Mas, G.; Di Croce, L. Regulation of gene transcription by Polycomb proteins. Sci. Adv. 2015, 1, e1500737. [Google Scholar] [CrossRef] [PubMed]
- Aloia, L.; Di Stefano, B.; Di Croce, L. Polycomb complexes in stem cells and embryonic development. Development 2013, 140, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Cui, X.; Huie, P.; Cleary, M.L. Set domain-dependent regulation of transcriptional silencing and growth control by SUV39H1, a mammalian ortholog of Drosophila Su (var) 3-9. Mol. Cell. Biol. 2000, 20, 4900–4909. [Google Scholar] [CrossRef]
- Pace, L.; Goudot, C.; Zueva, E.; Gueguen, P.; Burgdorf, N.; Waterfall, J.J.; Quivy, J.P.; Almouzni, G.; Amigorena, S. The epigenetic control of stemness in CD8(+) T cell fate commitment. Science 2018, 359, 177–186. [Google Scholar] [CrossRef]
- Yang, X.P.; Jiang, K.; Hirahara, K.; Vahedi, G.; Afzali, B.; Sciume, G.; Bonelli, M.; Sun, H.W.; Jankovic, D.; Kanno, Y.; et al. EZH2 is crucial for both differentiation of regulatory T cells and T effector cell expansion. Sci. Rep. 2015, 5, 10643. [Google Scholar] [CrossRef]
- Zhang, Y.; Kinkel, S.; Maksimovic, J.; Bandala-Sanchez, E.; Tanzer, M.C.; Naselli, G.; Zhang, J.G.; Zhan, Y.; Lew, A.M.; Silke, J.; et al. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood 2014, 124, 737–749. [Google Scholar] [CrossRef]
- Man, K.; Miasari, M.; Shi, W.; Xin, A.; Henstridge, D.C.; Preston, S.; Pellegrini, M.; Belz, G.T.; Smyth, G.K.; Febbraio, M.A.; et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat. Immunol. 2013, 14, 1155–1165. [Google Scholar] [CrossRef]
- Iwata, A.; Durai, V.; Tussiwand, R.; Briseno, C.G.; Wu, X.; Grajales-Reyes, G.E.; Egawa, T.; Murphy, T.L.; Murphy, K.M. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat. Immunol. 2017, 18, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, M.; Barnitz, R.A.; Yosef, N.; Odorizzi, P.M.; DiIorio, M.A.; Lemieux, M.E.; Yates, K.; Godec, J.; Klatt, M.G.; Regev, A.; et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 2014, 15, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.M.; Juedes, A.; Szabo, S.J.; von Herrath, M.; Glimcher, L.H. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc. Natl. Acad. Sci. USA 2003, 100, 15818–15823. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Guilloty, F.; Pipkin, M.E.; Djuretic, I.M.; Levanon, D.; Lotem, J.; Lichtenheld, M.G.; Groner, Y.; Rao, A. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 2009, 206, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.N.; Chi, A.W.; Chavez, A.; Yashiro-Ohtani, Y.; Yang, Q.; Shestova, O.; Bhandoola, A. A critical role for TCF-1 in T-lineage specification and differentiation. Nature 2011, 476, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Jeannet, G.; Boudousquie, C.; Gardiol, N.; Kang, J.; Huelsken, J.; Held, W. Essential role of the WNT pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. USA 2010, 107, 9777–9782. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, S.; Zhao, D.M.; Harty, J.T.; Badovinac, V.P.; Xue, H.H. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 2010, 33, 229–240. [Google Scholar] [CrossRef]
- Danilo, M.; Chennupati, V.; Silva, J.G.; Siegert, S.; Held, W. Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T Cell differentiation. Cell Rep. 2018, 22, 2107–2117. [Google Scholar] [CrossRef]
- Scharer, C.D.; Bally, A.P.; Gandham, B.; Boss, J.M. Cutting edge: Chromatin accessibility programs CD8 T Cell memory. J. Immunol. 2017, 198, 2238–2243. [Google Scholar] [CrossRef]
- Roychoudhuri, R.; Clever, D.; Li, P.; Wakabayashi, Y.; Quinn, K.M.; Klebanoff, C.A.; Ji, Y.; Sukumar, M.; Eil, R.L.; Yu, Z.; et al. BACH2 regulates CD8(+) T cell differentiation by controlling access of AP-1 factors to enhancers. Nat. Immunol. 2016, 17, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Kerdiles, Y.M.; Beisner, D.R.; Tinoco, R.; Dejean, A.S.; Castrillon, D.H.; DePinho, R.A.; Hedrick, S.M. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat. Immunol. 2009, 10, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.F.; Kuo, C.T.; Leiden, J.M. Transcription factor LKLF is sufficient to program T cell quiescence via a c-Myc-dependent pathway. Nat. Immunol. 2001, 2, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, S.; Binh Giang, T.; Kan, A.; Marchingo, J.M.; Lye, B.K.; Corcoran, L.M.; Hodgkin, P.D. A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nat. Immunol. 2017, 18, 96–103. [Google Scholar] [CrossRef] [PubMed]
- El Tanbouly, M.A.; Zhao, Y.; Nowak, E.; Li, J.; Schaafsma, E.; Le Mercier, I.; Ceeraz, S.; Lines, J.L.; Peng, C.; Carriere, C.; et al. VISTA is a checkpoint regulator for naïve T cell quiescence and peripheral tolerance. Science 2020, 367, eaay0524. [Google Scholar] [CrossRef]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Wang, L.; Le Mercier, I.; Putra, J.; Chen, W.; Liu, J.; Schenk, A.D.; Nowak, E.C.; Suriawinata, A.A.; Li, J.; Noelle, R.J. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc. Natl. Acad. Sci. USA 2014, 111, 14846–14851. [Google Scholar] [CrossRef]
- Flies, D.B.; Han, X.; Higuchi, T.; Zheng, L.; Sun, J.; Ye, J.J.; Chen, L. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J. Clin. Investig. 2014, 124, 1966–1975. [Google Scholar] [CrossRef]
- Flies, D.B.; Higuchi, T.; Chen, L. Mechanistic assessment of PD-1H coinhibitory receptor-induced T Cell tolerance to allogeneic antigens. J. Immunol. 2015, 194, 5294–5304. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2020, 20, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; Loos, J.A. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes: II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp. Cell Res. 1973, 77, 127–135. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Neale, G.; Green, D.R.; He, W.; Chi, H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat. Immunol. 2011, 12, 888–897. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, Y.; Chen, C.; Ikenoue, T.; Qiao, Y.; Li, C.-S.; Li, W.; Guan, K.-L.; Liu, Y.; Zheng, P. The tuberous sclerosis complex-mammalian target of rapamycin pathway maintains the quiescence and survival of naive T cells. J. Immunol. 2011, 187, 1106–1112. [Google Scholar] [CrossRef]
- Hwang, S.S.; Lim, J.; Yu, Z.; Kong, P.; Sefik, E.; Xu, H.; Harman, C.C.D.; Kim, L.K.; Lee, G.R.; Li, H.B.; et al. mRNA destabilization by BTG1 and BTG2 maintains T cell quiescence. Science 2020, 367, 1255–1260. [Google Scholar] [CrossRef]
- Wolf, T.; Jin, W.; Zoppi, G.; Vogel, I.A.; Akhmedov, M.; Bleck, C.K.E.; Beltraminelli, T.; Rieckmann, J.C.; Ramirez, N.J.; Benevento, M.; et al. Dynamics in protein translation sustaining T cell preparedness. Nat. Immunol. 2020, 21, 927–937. [Google Scholar] [CrossRef]
- Lau, C.M.; Adams, N.M.; Geary, C.D.; Weizman, O.E.; Rapp, M.; Pritykin, Y.; Leslie, C.S.; Sun, J.C. Epigenetic control of innate and adaptive immune memory. Nat. Immunol. 2018, 19, 963–972. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Uldrich, A.P.; McCluskey, J.; Rossjohn, J.; Moody, D.B. The burgeoning family of unconventional T cells. Nat. Immunol. 2015, 16, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Treiner, E.; Duban, L.; Bahram, S.; Radosavljevic, M.; Wanner, V.; Tilloy, F.; Affaticati, P.; Gilfillan, S.; Lantz, O. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 2003, 422, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Motoki, K.; Ueno, H.; Nakagawa, R.; Sato, H.; Kondo, E.; et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 1997, 278, 1626–1629. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bennett, T.J.; Udupa, V.A.V.; Turner, S.J. Running to Stand Still: Naive CD8+ T Cells Actively Maintain a Program of Quiescence. Int. J. Mol. Sci. 2020, 21, 9773. https://doi.org/10.3390/ijms21249773
Bennett TJ, Udupa VAV, Turner SJ. Running to Stand Still: Naive CD8+ T Cells Actively Maintain a Program of Quiescence. International Journal of Molecular Sciences. 2020; 21(24):9773. https://doi.org/10.3390/ijms21249773
Chicago/Turabian StyleBennett, Taylah J., Vibha A. V. Udupa, and Stephen J. Turner. 2020. "Running to Stand Still: Naive CD8+ T Cells Actively Maintain a Program of Quiescence" International Journal of Molecular Sciences 21, no. 24: 9773. https://doi.org/10.3390/ijms21249773
APA StyleBennett, T. J., Udupa, V. A. V., & Turner, S. J. (2020). Running to Stand Still: Naive CD8+ T Cells Actively Maintain a Program of Quiescence. International Journal of Molecular Sciences, 21(24), 9773. https://doi.org/10.3390/ijms21249773