The Degree of Cardiac Remodelling before Overload Relief Triggers Different Transcriptome and miRome Signatures during Reverse Remodelling (RR)—Molecular Signature Differ with the Extent of RR

 , , , , and
, , , , and
Abstract

1. Introduction
2. Results
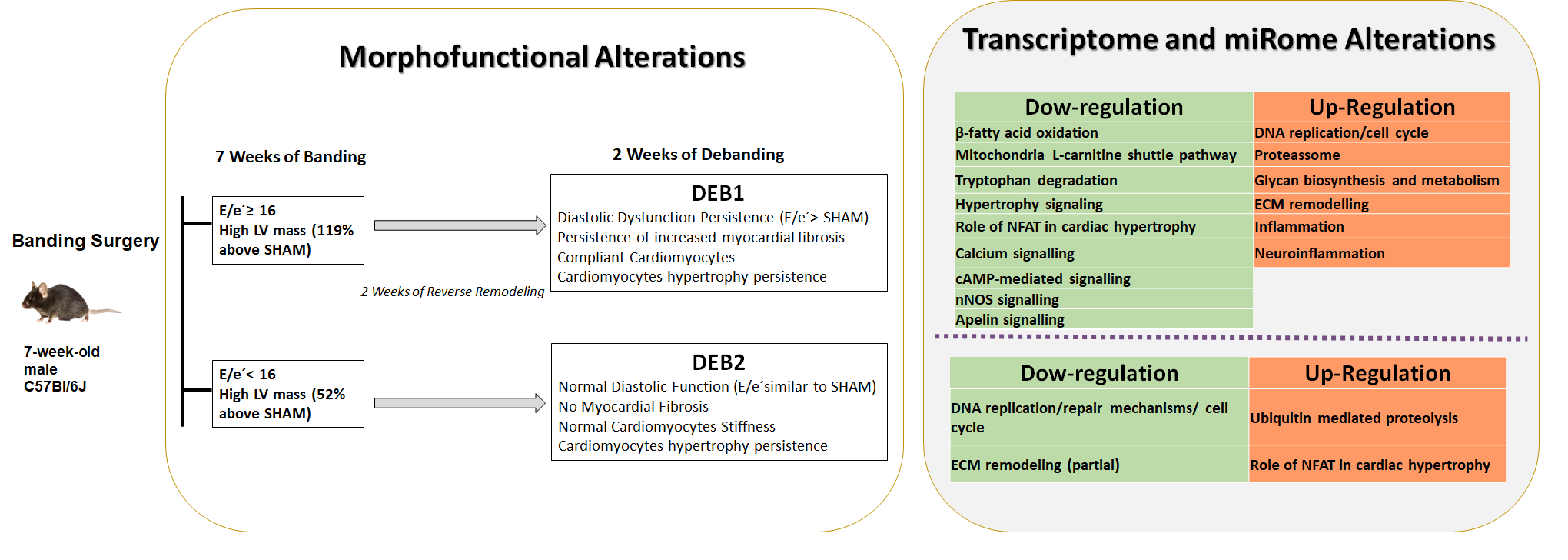
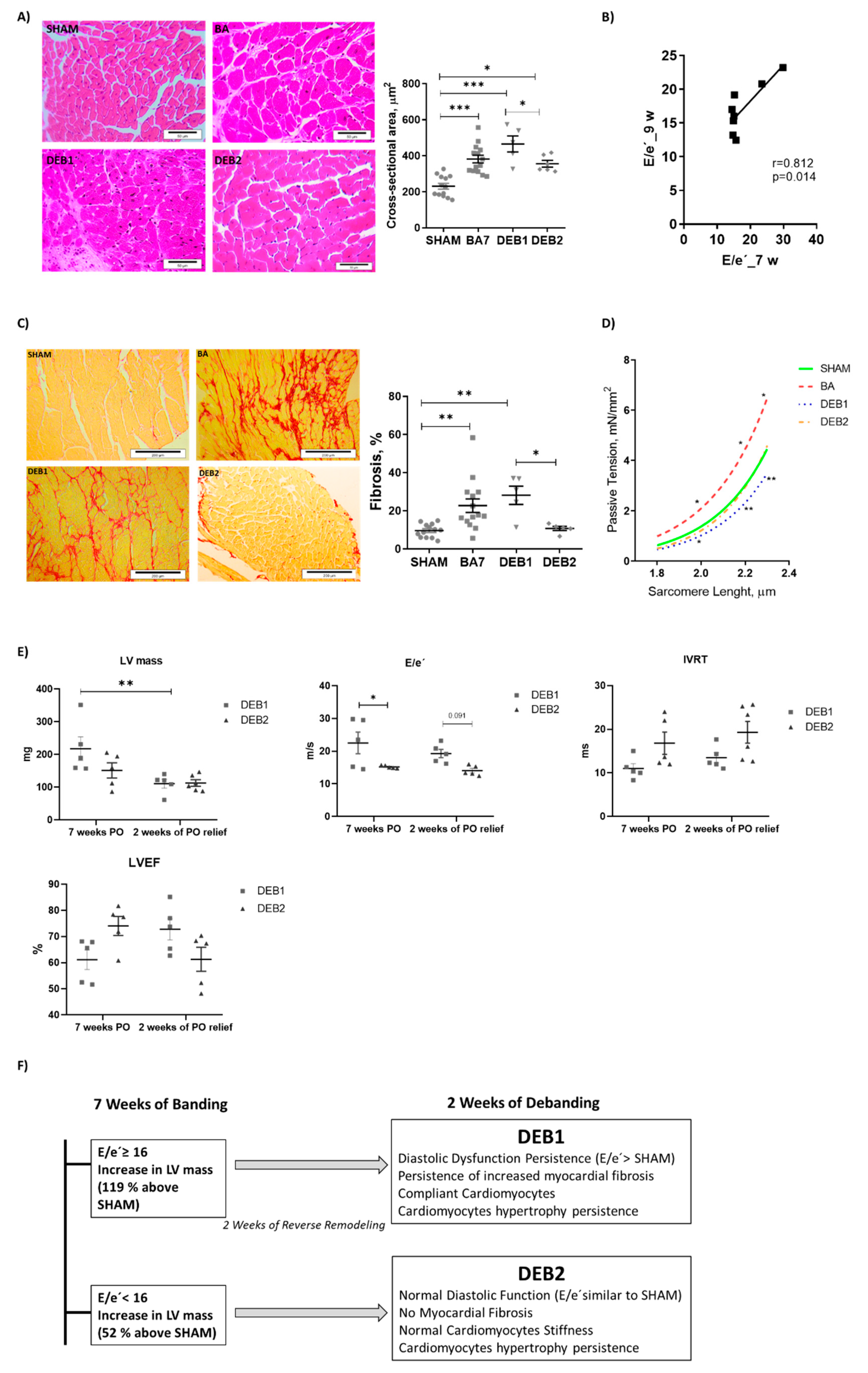
2.1. In Vivo and In Vitro Characterization of a Cardiac Reverse Remodelling Animal Model
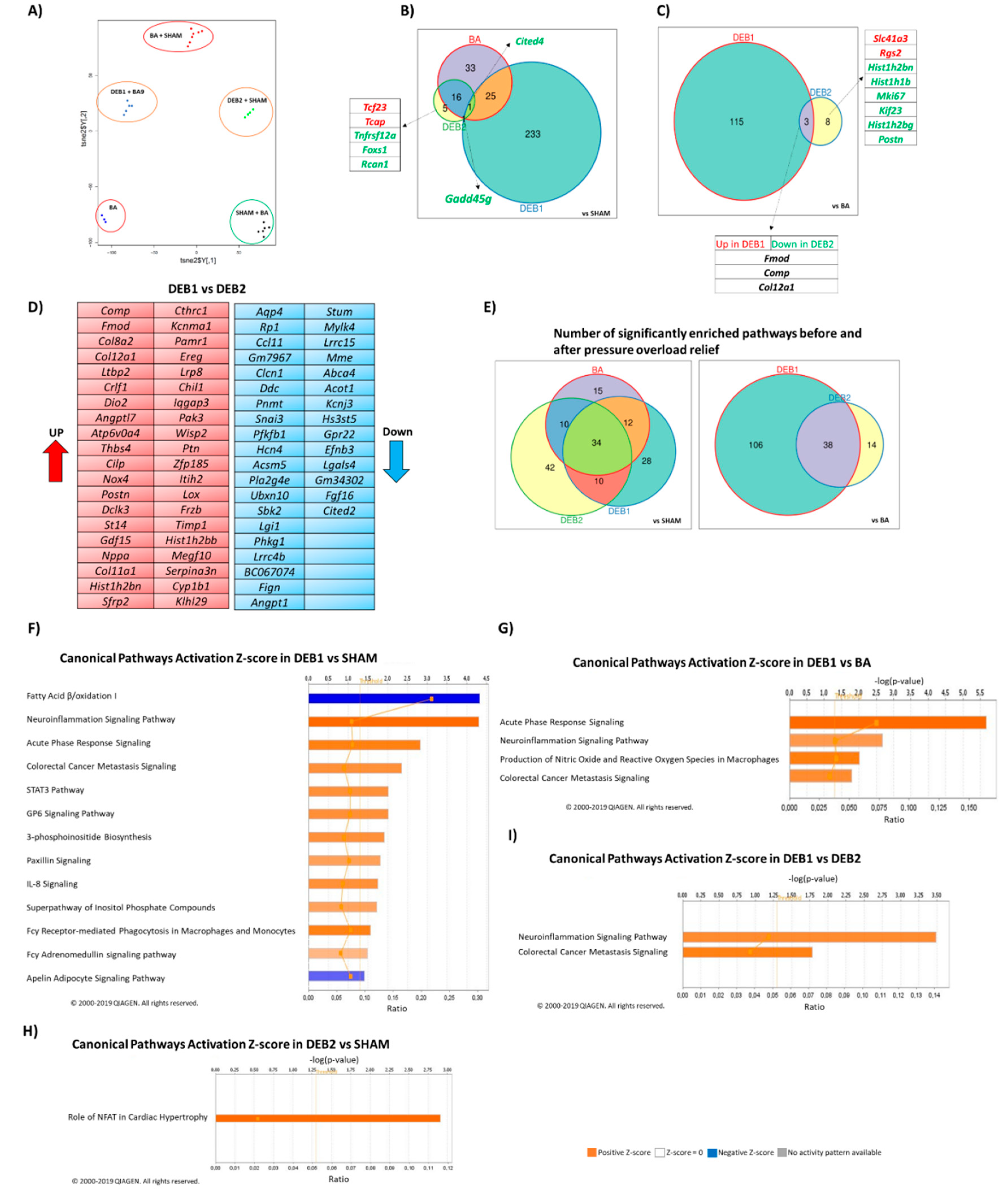
2.2. Transcriptome Profile in Experimental Cardiac Reverse Remodelling
2.2.1. Pressure Overload-Induced Remodelling (BA vs. SHAM)
2.2.2. Debanding-Induced Reverse Remodelling (DEB1 and DEB2 vs. BA)
2.3. Enrichment Analysis: Candidate Pathways
2.3.1. Pressure Overload-Induced Remodelling Fingerprint (BA vs. SHAM)
2.3.2. “Incomplete” Reverse Remodelling Fingerprint (DEB1)
2.3.3. “Complete” Reverse Remodelling Fingerprint (DEB2)
2.3.4. “Incomplete” vs. “Complete” Reverse Remodelling (DEB1 vs. DEB2)
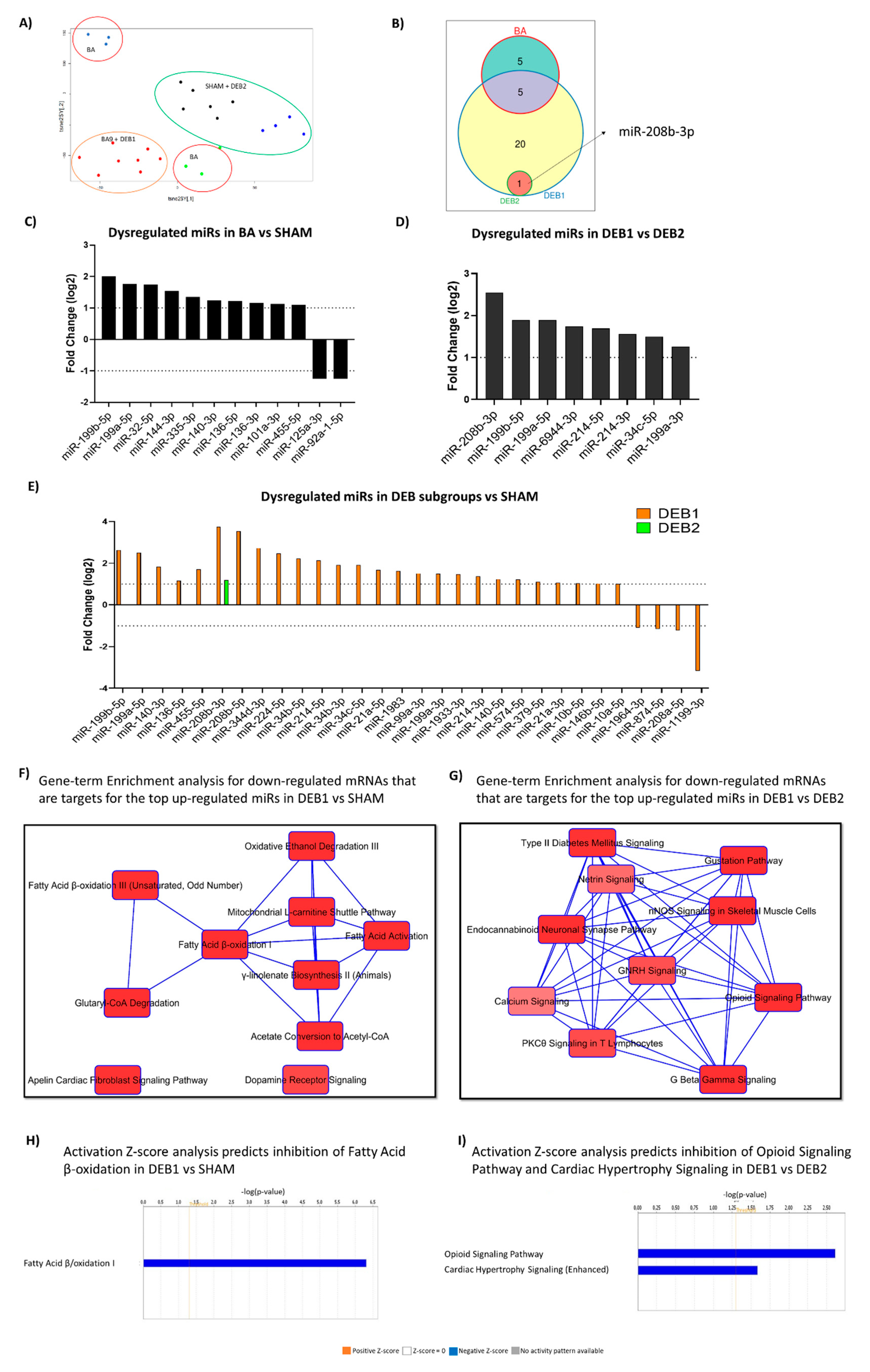
2.4. MiRome Profile in Experimental Cardiac Reverse Remodelling
2.5. Integrative mRNA/miR Expression Profiling
2.5.1. Pressure Overload-Induced Remodelling Fingerprint (BA vs. SHAM)
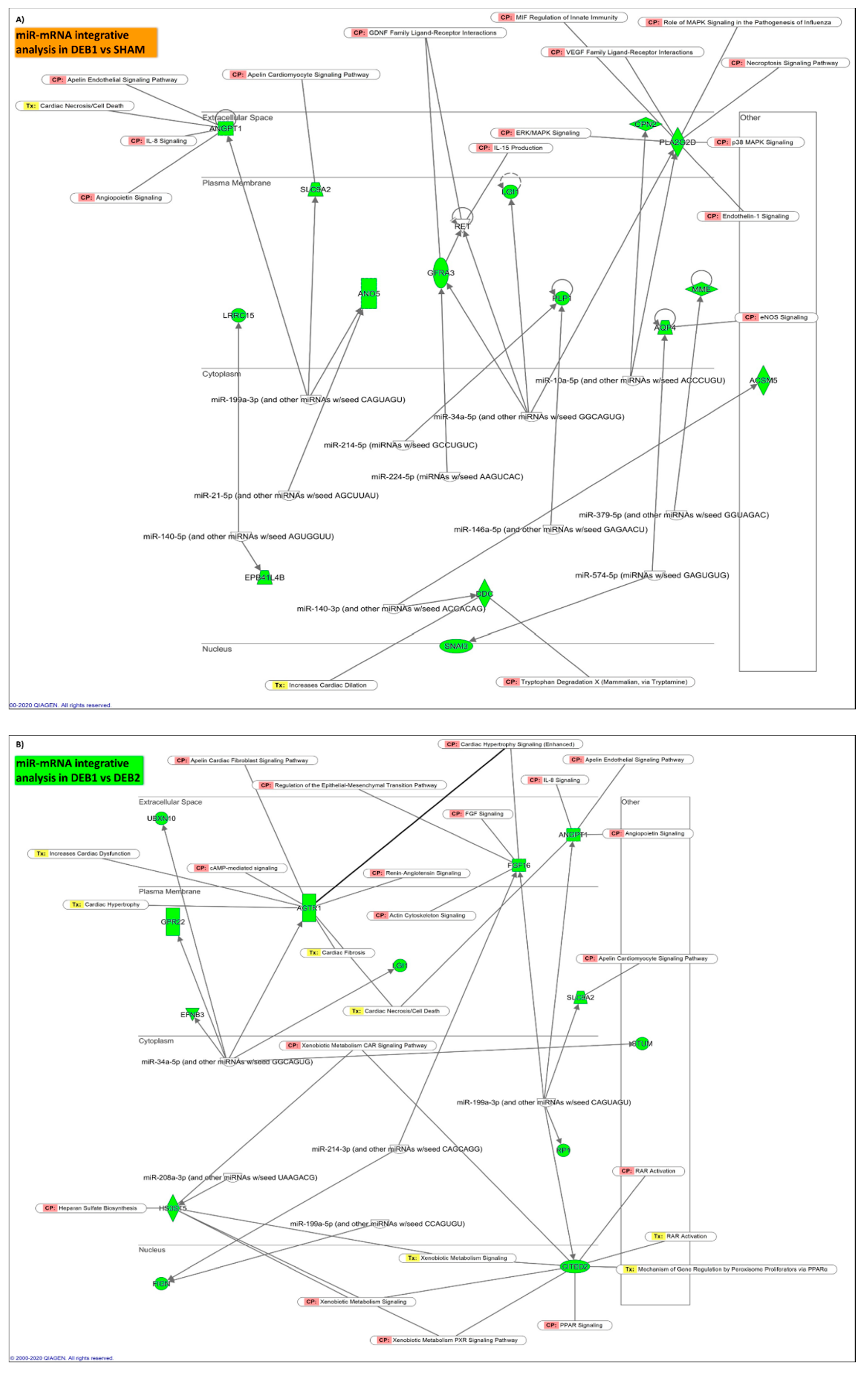
2.5.2. “Incomplete” Reverse Remodelling Fingerprint (DEB1 vs. SHAM)
2.5.3. “Incomplete” vs. “Complete” Reverse Remodelling (DEB1 vs. DEB2)
3. Discussion
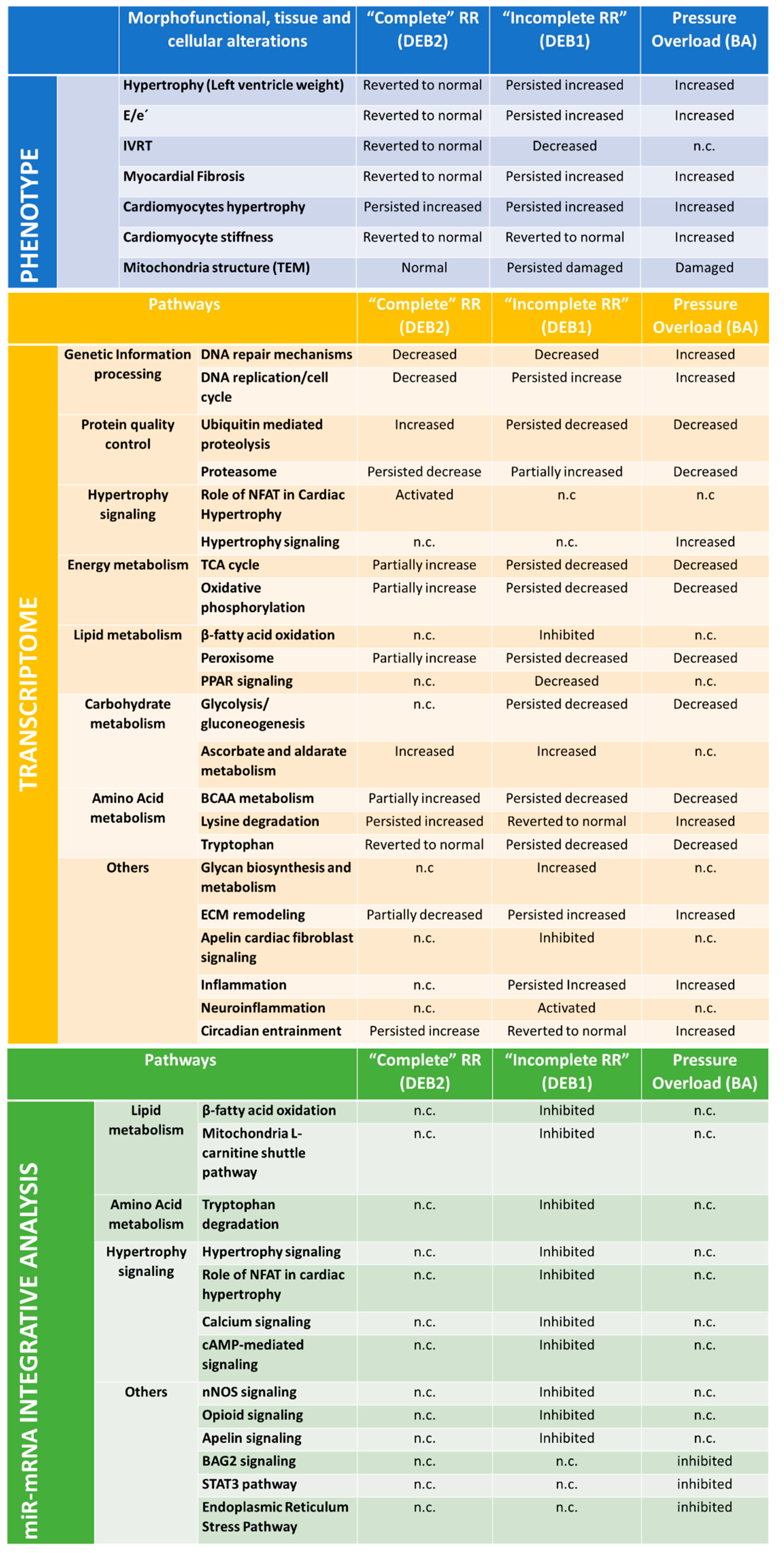
3.1. Processes Driving “Incomplete” and “Complete” Reverse Remodelling
3.2. Limitations
4. Materials and Methods
4.1. Ethics and Animal Care
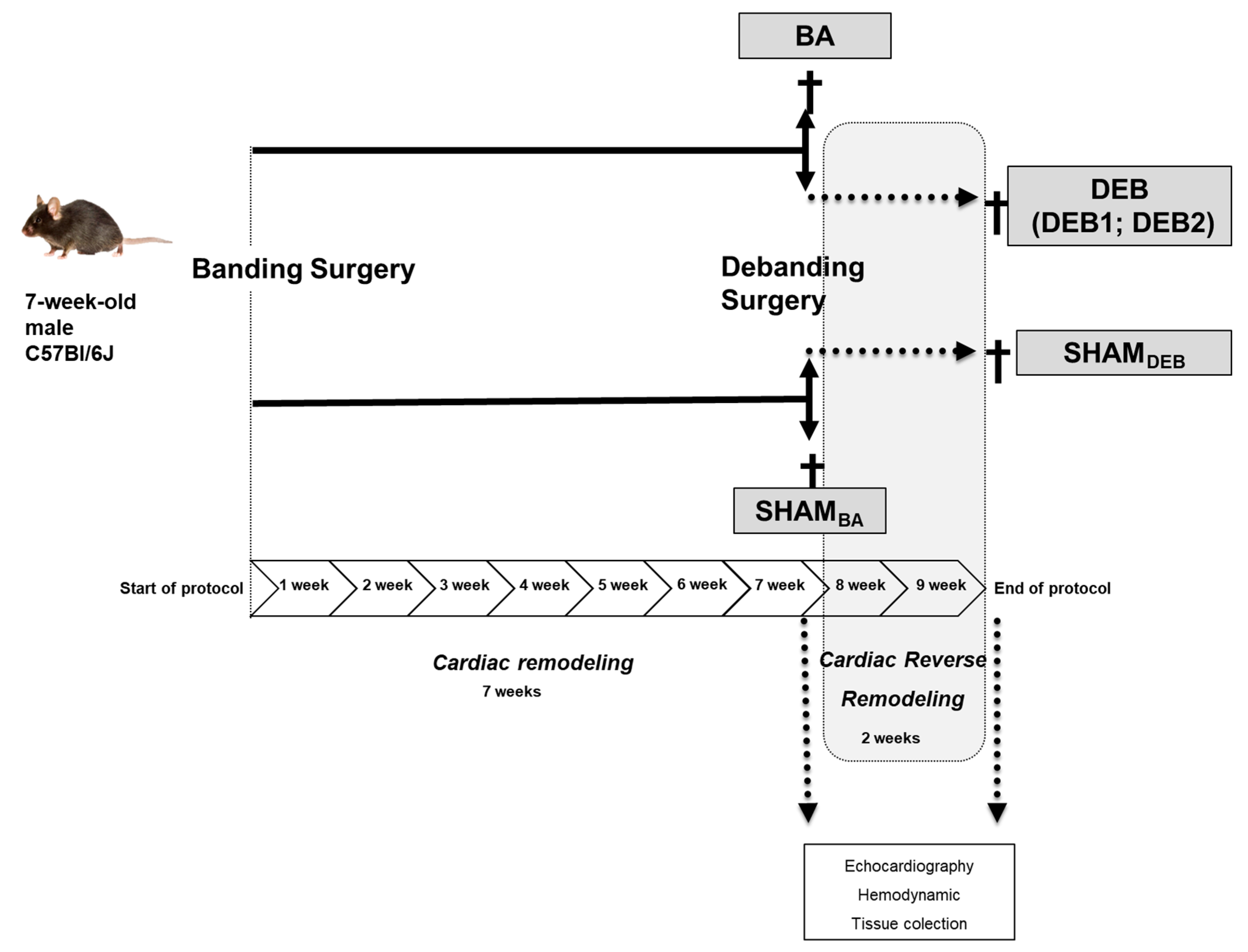
4.2. Animal Model of Reverse Remodelling
4.3. Animals’ Euthanasia
4.4. Echocardiography
4.5. Hemodynamic
4.6. Cardiomyocyte Sectional Area and Extracellular Matrix Alterations Evaluation
4.7. Isolated Cardiomyocyte Function Evaluation
4.8. Electron Microscopy
4.9. RNA Isolation
4.9.1. RNA Preparation for Sequencing
4.9.2. Bulk mRNA-Sequencing
4.9.3. Gene Set Enrichment Analysis
4.10. microRNA-Sequencing
4.11. mRNA-miR Integrative Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef]
- Bing, R.; Cavalcante, J.L.; Everett, R.J.; Clavel, M.A.; Newby, D.E.; Dweck, M.R. Imaging and Impact of Myocardial Fibrosis in Aortic Stenosis. JACC Cardiovasc. Imaging 2019, 12, 283–296. [Google Scholar] [CrossRef]
- Herrmann, S.; Fries, B.; Salinger, T.; Liu, D.; Hu, K.; Gensler, D.; Strotmann, J.; Christa, M.; Beer, M.; Gattenlohner, S.; et al. Myocardial Fibrosis Predicts 10-Year Survival in Patients Undergoing Aortic Valve Replacement. Circ. Cardiovasc. Imaging 2018, 11, e007131. [Google Scholar] [CrossRef]
- Chin, C.W.L.; Everett, R.J.; Kwiecinski, J.; Vesey, A.T.; Yeung, E.; Esson, G.; Jenkins, W.; Koo, M.; Mirsadraee, S.; White, A.C.; et al. Myocardial Fibrosis and Cardiac Decompensation in Aortic Stenosis. JACC Cardiovasc. Imaging 2017, 10, 1320–1333. [Google Scholar] [CrossRef]
- Galat, A.; Guellich, A.; Bodez, D.; Lipskaia, L.; Moutereau, S.; Bergoend, E.; Hue, S.; Ternacle, J.; Mohty, D.; Monin, J.L.; et al. Causes and consequences of cardiac fibrosis in patients referred for surgical aortic valve replacement. ESC Heart Fail. 2019, 6, 649–657. [Google Scholar] [CrossRef]
- Mannacio, V.; Di Tommaso, L.; Stassano, P.; De Amicis, V.; Vosa, C. Myocardial metabolism and diastolic function after aortic valve replacement for aortic stenosis: Influence of patient-prosthesis mismatch. Eur. J. Cardiothorac. Surg. 2012, 41, 316–321. [Google Scholar] [CrossRef]
- Heather, L.C.; Howell, N.J.; Emmanuel, Y.; Cole, M.A.; Frenneaux, M.P.; Pagano, D.; Clarke, K. Changes in cardiac substrate transporters and metabolic proteins mirror the metabolic shift in patients with aortic stenosis. PLoS ONE 2011, 6, e26326. [Google Scholar] [CrossRef]
- Coffey, S.; Williams, M.J.; Phillips, L.V.; Galvin, I.F.; Bunton, R.W.; Jones, G.T. Integrated microRNA and messenger RNA analysis in aortic stenosis. Sci. Rep. 2016, 6, 36904. [Google Scholar] [CrossRef]
- Shah, R.; Ziegler, O.; Yeri, A.; Liu, X.; Murthy, V.; Rabideau, D.; Xiao, C.Y.; Hanspers, K.; Belcher, A.; Tackett, M.; et al. MicroRNAs Associated With Reverse Left Ventricular Remodelling in Humans Identify Pathways of Heart Failure Progression. Circ. Heart Fail. 2018, 11, e004278. [Google Scholar] [CrossRef]
- Tobin, S.W.; Hashemi, S.; Dadson, K.; Turdi, S.; Ebrahimian, K.; Zhao, J.; Sweeney, G.; Grigull, J.; McDermott, J.C. Heart Failure and MEF2 Transcriptome Dynamics in Response to beta-Blockers. Sci. Rep. 2017, 7, 4476. [Google Scholar] [CrossRef]
- Leite-Moreira, A.M.; Lourenco, A.P.; Falcao-Pires, I.; Leite-Moreira, A.F. Pivotal role of microRNAs in cardiac physiology and heart failure. Drug Discov. Today 2013, 18, 1243–1249. [Google Scholar] [CrossRef]
- Condorelli, G.; Latronico, M.V.; Cavarretta, E. microRNAs in cardiovascular diseases: Current knowledge and the road ahead. Am. J. Cardiol. 2014, 63, 2177–2187. [Google Scholar] [CrossRef]
- Conceicao, G.; Heinonen, I.; Lourenco, A.P.; Duncker, D.J.; Falcao-Pires, I. Animal models of heart failure with preserved ejection fraction. Neth. Heart J. 2016, 24, 275–286. [Google Scholar] [CrossRef]
- Nomura, S.; Satoh, M.; Fujita, T.; Higo, T.; Sumida, T.; Ko, T.; Yamaguchi, T.; Tobita, T.; Naito, A.T.; Ito, M.; et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 2018, 9, 4435. [Google Scholar] [CrossRef]
- Lee, J.H.; Gao, C.; Peng, G.; Greer, C.; Ren, S.; Wang, Y.; Xiao, X. Analysis of transcriptome complexity through RNA sequencing in normal and failing murine hearts. Circ. Res. 2011, 109, 1332–1341. [Google Scholar] [CrossRef]
- Rosenberg, M.A.; Gottdiener, J.S.; Heckbert, S.R.; Mukamal, K.J. Echocardiographic diastolic parameters and risk of atrial fibrillation: The Cardiovascular Health Study. Eur. Heart J. 2012, 33, 904–912. [Google Scholar] [CrossRef]
- el Azzouzi, H.; Leptidis, S.; Dirkx, E.; Hoeks, J.; van Bree, B.; Brand, K.; McClellan, E.A.; Poels, E.; Sluimer, J.C.; van den Hoogenhof, M.M.; et al. The hypoxia-inducible microRNA cluster miR-199a∼214 targets myocardial PPARδ and impairs mitochondrial fatty acid oxidation. Cell Metab. 2013, 18, 341–354. [Google Scholar] [CrossRef]
- Stølen, T.O.; Høydal, M.A.; Ahmed, M.S.; Jørgensen, K.; Garten, K.; Hortigon-Vinagre, M.P.; Zamora, V.; Scrimgeour, N.R.; Berre, A.M.O.; Nes, B.M.; et al. Exercise training reveals micro-RNAs associated with improved cardiac function and electrophysiology in rats with heart failure after myocardial infarction. J. Mol. Cell. Cardiol. 2020, 148, 106–119. [Google Scholar] [CrossRef]
- Chandy, M. A tangled tale of microRNA and cardiac fibrosis. Clin. Sci. 2019, 133, 2217–2220. [Google Scholar] [CrossRef]
- Sucharov, C.C.; Kao, D.P.; Port, J.D.; Karimpour-Fard, A.; Quaife, R.A.; Minobe, W.; Nunley, K.; Lowes, B.D.; Gilbert, E.M.; Bristow, M.R. Myocardial microRNAs associated with reverse remodelling in human heart failure. JCI Insight 2017, 2, e89169. [Google Scholar] [CrossRef]
- Akat, K.M.; Moore-McGriff, D.; Morozov, P.; Brown, M.; Gogakos, T.; Correa Da Rosa, J.; Mihailovic, A.; Sauer, M.; Ji, R.; Ramarathnam, A.; et al. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc. Natl. Acad. Sci. USA 2014, 111, 11151–11156. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Gao, X.M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodelling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef]
- Dahl, J.S.; Barros-Gomes, S.; Videbæk, L.; Poulsen, M.K.; Issa, I.F.; Carter-Storch, R.; Christensen, N.L.; Kumme, A.; Pellikka, P.A.; Møller, J.E. Early Diastolic Strain Rate in Relation to Systolic and Diastolic Function and Prognosis in Aortic Stenosis. JACC Cardiovasc. Imaging 2016, 9, 519–528. [Google Scholar] [CrossRef]
- Seo, J.S.; Jang, M.K.; Lee, E.Y.; Yun, S.C.; Kim, D.H.; Song, J.M.; Kang, D.H.; Song, J.K. Evaluation of left ventricular diastolic function after valve replacement in aortic stenosis using exercise Doppler echocardiography. Circ. J. 2012, 76, 2792–2798. [Google Scholar] [CrossRef]
- Guo, Y.; Sun, M.; Chen, H.; Kong, D.; Shu, X.; Pan, C. Assessment of left ventricular diastolic function after Transcatheter aortic valve implantation in aortic stenosis patients by echocardiographic according to different guidelines. J. Cardiovasc. Ultrasound 2020, 18, 3. [Google Scholar] [CrossRef]
- Zacchigna, S.; Paldino, A.; Falcão-Pires, I.; Daskalopoulos, E.P.; Dal Ferro, M.; Vodret, S.; Lesizza, P.; Cannatà, A.; Miranda-Silva, D.; Lourenço, A.P.; et al. Toward standardization of echocardiography for the evaluation of left ventricular function in adult rodents: A position paper of the ESC Working Group on Myocardial Function. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Melleby, A.O.; Romaine, A.; Aronsen, J.M.; Veras, I.; Zhang, L.; Sjaastad, I.; Lunde, I.G.; Christensen, G. A novel method for high precision aortic constriction that allows for generation of specific cardiac phenotypes in mice. Cardiovasc. Res. 2018, 114, 1680–1690. [Google Scholar] [CrossRef]
- Bjornstad, J.L.; Skrbic, B.; Sjaastad, I.; Bjornstad, S.; Christensen, G.; Tonnessen, T. A mouse model of reverse cardiac remodelling following banding-debanding of the ascending aorta. Acta Physiol. 2012, 205, 92–102. [Google Scholar] [CrossRef]
- Merino, D.; Gil, A.; Gomez, J.; Ruiz, L.; Llano, M.; Garcia, R.; Hurle, M.A.; Nistal, J.F. Experimental modelling of cardiac pressure overload hypertrophy: Modified technique for precise, reproducible, safe and easy aortic arch banding-debanding in mice. Sci. Rep. 2018, 8, 3167. [Google Scholar] [CrossRef]
- Martini, E.; Kunderfranco, P.; Peano, C.; Carullo, P.; Cremonesi, M.; Schorn, T.; Carriero, R.; Termanini, A.; Colombo, F.S.; Jachetti, E.; et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation 2019, 140, 2089–2107. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, H.M.; Park, J.S.; Kim, Y.J. Differential Transcriptome Profile and Exercise Capacity in Cardiac Remodelling by Pressure Overload versus Volume Overload. Int. J. Cardiovasc. Imaging 2019, 27, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D. Parsing good versus bad signaling pathways in the heart: Role of calcineurin-nuclear factor of activated T-cells. Circ. Res. 2013, 113, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, E.; Sasaki, S.; Kinoshita, H.; Kito, T.; Ohta, H.; Konishi, M.; Kuwahara, K.; Nakao, K.; Itoh, N. Angiotensin II-induced cardiac hypertrophy and fibrosis are promoted in mice lacking Fgf16. Genes Cells 2013, 18, 544–553. [Google Scholar] [CrossRef]
- Wohlschlaeger, J.; Sixt, S.U.; Stoeppler, T.; Schmitz, K.J.; Levkau, B.; Tsagakis, K.; Vahlhaus, C.; Schmid, C.; Peters, J.; Schmid, K.W.; et al. Ventricular unloading is associated with increased 20s proteasome protein expression in the myocardium. J. Heart Lung Transplant. 2010, 29, 125–132. [Google Scholar] [CrossRef]
- Miranda-Silva, D.; Goncalves-Rodrigues, P.; Almeida-Coelho, J.; Hamdani, N.; Lima, T.; Conceicao, G.; Sousa-Mendes, C.; Claudia, M.; Gonzalez, A.; Diez, J.; et al. Characterization of biventricular alterations in myocardial (reverse) remodelling in aortic banding-induced chronic pressure overload. Sci. Rep. 2019, 9, 2956. [Google Scholar] [CrossRef]
- Aalaei-Andabili, S.H.; Bavry, A.A. Left Ventricular Diastolic Dysfunction and Transcatheter Aortic Valve Replacement Outcomes: A Review. Cardiol. Ther. 2019, 8, 21–28. [Google Scholar] [CrossRef]
- Villari, B.; Vassalli, G.; Monrad, E.S.; Chiariello, M.; Turina, M.; Hess, O.M. Normalization of diastolic dysfunction in aortic stenosis late after valve replacement. Circulation 1995, 91, 2353–2358. [Google Scholar] [CrossRef]
- Opie, L.H.; Commerford, P.J.; Gersh, B.J.; Pfeffer, M.A. Controversies in ventricular remodelling. Lancet 2006, 367, 356–367. [Google Scholar] [CrossRef]
- Weeks, K.L.; McMullen, J.R. The athlete’s heart vs. the failing heart: Can signaling explain the two distinct outcomes? Physiology 2011, 26, 97–105. [Google Scholar] [CrossRef]
- Rodrigues, P.G.; Leite-Moreira, A.F.; Falcao-Pires, I.A.-O. Myocardial reverse remodelling: How far can we rewind? Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1402–H1422. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.M.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004, 110, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Falcao-Pires, I.; Ladeiras-Lopes, R.; Leite-Moreira, A.F. The apelinergic system: A promising therapeutic target. Expert Opin. Ther. Targets 2010, 14, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.G.; Sopko, N.; Blech, L.; Popovic, Z.B.; Li, J.; Vasanji, A.; Drumm, C.; Krukovets, I.; Jain, M.K.; Penn, M.S.; et al. Thrombospondin-4 regulates fibrosis and remodelling of the myocardium in response to pressure overload. FASEB J. 2012, 26, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wu, H.; Xia, W.; Chen, X.; Zhu, S.; Zhang, S.; Shao, Y.; Ma, W.; Yang, D.; Zhang, J. Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J. Cardiol. 2014, 63, 373–378. [Google Scholar] [CrossRef]
- Lopez, B.; Gonzalez, A.; Lindner, D.; Westermann, D.; Ravassa, S.; Beaumont, J.; Gallego, I.; Zudaire, A.; Brugnolaro, C.; Querejeta, R.; et al. Osteopontin-mediated myocardial fibrosis in heart failure: A role for lysyl oxidase? Cardiovasc. Res. 2013, 99, 111–120. [Google Scholar] [CrossRef]
- Boyd, J.H.; Mathur, S.; Wang, Y.; Bateman, R.M.; Walley, K.R. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc. Res. 2006, 72, 384–393. [Google Scholar] [CrossRef]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodelling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef]
- Weinheimer, C.J.; Kovacs, A.; Evans, S.; Matkovich, S.J.; Barger, P.M.; Mann, D.L. Load-Dependent Changes in Left Ventricular Structure and Function in a Pathophysiologically Relevant Murine Model of Reversible Heart Failure. Circ. Heart Fail. 2018, 11, e004351. [Google Scholar] [CrossRef]
- Radley, G.; Pieper, I.L.; Ali, S.; Bhatti, F.; Thornton, C.A. The Inflammatory Response to Ventricular Assist Devices. Front. Immunol. 2018, 9, 2651. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Nakayama, H.; Otsu, K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem. J. 2018, 475, 839–852. [Google Scholar] [CrossRef]
- Byrne, N.J.; Levasseur, J.; Sung, M.M.; Masson, G.; Boisvenue, J.; Young, M.E.; Dyck, J.R. Normalization of cardiac substrate utilization and left ventricular hypertrophy precede functional recovery in heart failure regression. Cardiovasc. Res. 2016, 110, 249–257. [Google Scholar] [CrossRef]
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef]
- Uddin, G.M.; Zhang, L.; Shah, S.; Fukushima, A.; Wagg, C.S.; Gopal, K.; Al Batran, R.; Pherwani, S.; Ho, K.L.; Boisvenue, J.; et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc. Diabetol. 2019, 18, 86. [Google Scholar] [CrossRef]
- Pereyra, A.S.; Hasek, L.Y.; Harris, K.L.; Berman, A.G.; Damen, F.W.; Goergen, C.J.; Ellis, J.M. Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J. Biol. Chem. 2017, 292, 18443–18456. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Cockrill, T.C.; Frazier, O.H.; Taegtmeyer, H. Downregulation of myocardial myocyte enhancer factor 2C and myocyte enhancer factor 2C-regulated gene expression in diabetic patients with nonischemic heart failure. Circulation 2002, 106, 407–411. [Google Scholar] [CrossRef]
- Borbely, A.; van der Velden, J.; Papp, Z.; Bronzwaer, J.G.; Edes, I.; Stienen, G.J.; Paulus, W.J. Cardiomyocyte stiffness in diastolic heart failure. Circulation 2005, 111, 774–781. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Morphometry | Parameters | SHAM (n = 20) | BA (n = 12) | DEB1 (n = 5) | DEB2 (n = 5) |
| TL, cm | 1.79 ± 0.02 | 1.75 ± 0.02 | 1.80 ± 0.05 | 1.77 ± 0.02 | |
| Heart/TL, g/cm | 70.39 ± 1.72 | 89.96 ± 7.14 α | 91.16 ± 5.04 α | 70.45 ± 2.14 χ δ | |
| LV + Sp/TL, g/cm | 44.67 ± 2.29 | 60.03 ± 4.91 α | 57.01 ± 4.61 | 44.17 ± 2.83 | |
| Lungs/TL, g/cm | 91.25 ± 5.00 | 99.20 ± 7.88 | 105.30 ± 10.23 | 92.28 ± 7.16 | |
| Echocardiography | LV mass, mg | 99.6 ± 5.5 | 151.4 ± 9.6 ααα | 110.7 ± 13.4 | 113.0 ± 9.7 |
| LVEF, % | 68.55 ± 1.92 | 70.07 ± 3.46 | 72.81 ± 4.06 | 61.31 ± 4.59 | |
| EDV, uL | 65.34 ± 3.26 | 65.26 ± 4.88 | 62.97 ± 11.92 | 66.91 ± 4.47 | |
| IVRT, ms | 18.88 ± 1.05 | 17.03 ± 1.56 | 13.47 ± 1.18 | 19.32 ± 2.50 | |
| E/e’ | 13.57 ± 0.66 | 20.28 ± 1.94 αα | 19.27 ± 1.28 α | 14.07 ± 0.68 | |
| LAA, cm2 | 0.040 [0.039; 0.043] | 0.064 [0.040; 0.086] | 0.068 [0.060; 0.077] | 0.043 [0.041; 0.044] | |
| HR, bpm | 451.1 ± 22.6 | 434.8 ± 12.96 | 516.1 ± 19.11 | 492.5 ± 41.70 | |
| Haemodynamic | LVESP, mmHg | 80.70 ± 3.42 | 108.10 ± 8.84 α | 89.19 ± 3.67 | 87.69 ± 8.85 |
| LVEDP, mmHg | 4.75 ± 0.67 | 10.71 ± 2.47 α | 3.28 ± 0.93 χ | 4.06 ± 0.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, P.G.; Miranda-Silva, D.; Li, X.; Sousa-Mendes, C.; Martins-Ferreira, R.; Elbeck, Z.; Leite-Moreira, A.F.; Knöll, R.; Falcão-Pires, I. The Degree of Cardiac Remodelling before Overload Relief Triggers Different Transcriptome and miRome Signatures during Reverse Remodelling (RR)—Molecular Signature Differ with the Extent of RR. Int. J. Mol. Sci. 2020, 21, 9687. https://doi.org/10.3390/ijms21249687
Rodrigues PG, Miranda-Silva D, Li X, Sousa-Mendes C, Martins-Ferreira R, Elbeck Z, Leite-Moreira AF, Knöll R, Falcão-Pires I. The Degree of Cardiac Remodelling before Overload Relief Triggers Different Transcriptome and miRome Signatures during Reverse Remodelling (RR)—Molecular Signature Differ with the Extent of RR. International Journal of Molecular Sciences. 2020; 21(24):9687. https://doi.org/10.3390/ijms21249687
Chicago/Turabian StyleRodrigues, Patrícia G., Daniela Miranda-Silva, Xidan Li, Cláudia Sousa-Mendes, Ricardo Martins-Ferreira, Zaher Elbeck, Adelino F. Leite-Moreira, Ralph Knöll, and Inês Falcão-Pires. 2020. "The Degree of Cardiac Remodelling before Overload Relief Triggers Different Transcriptome and miRome Signatures during Reverse Remodelling (RR)—Molecular Signature Differ with the Extent of RR" International Journal of Molecular Sciences 21, no. 24: 9687. https://doi.org/10.3390/ijms21249687
APA StyleRodrigues, P. G., Miranda-Silva, D., Li, X., Sousa-Mendes, C., Martins-Ferreira, R., Elbeck, Z., Leite-Moreira, A. F., Knöll, R., & Falcão-Pires, I. (2020). The Degree of Cardiac Remodelling before Overload Relief Triggers Different Transcriptome and miRome Signatures during Reverse Remodelling (RR)—Molecular Signature Differ with the Extent of RR. International Journal of Molecular Sciences, 21(24), 9687. https://doi.org/10.3390/ijms21249687