Cell Death in Liver Diseases: A Review



Abstract
1. Introduction
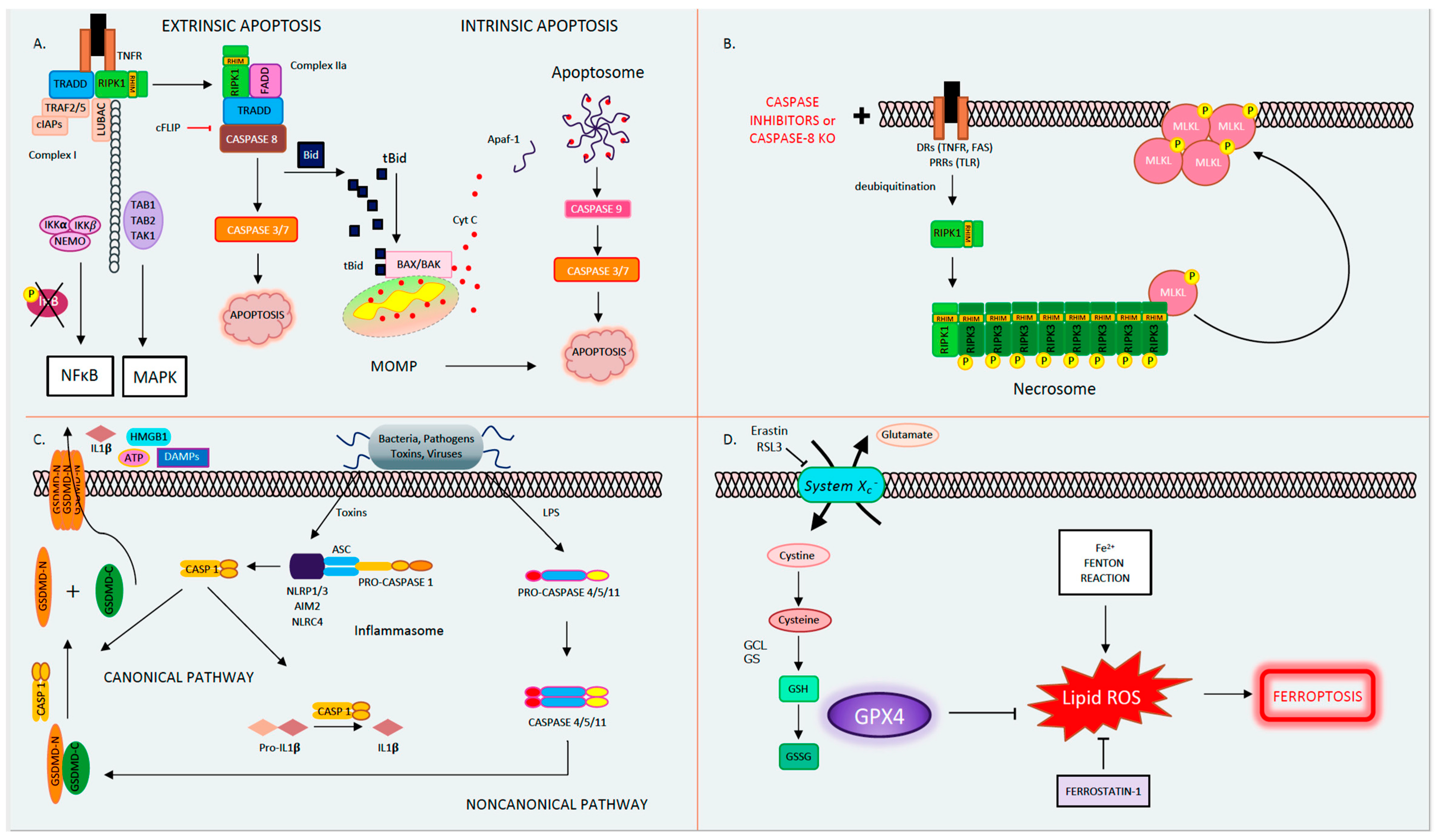
1.1. Apoptosis
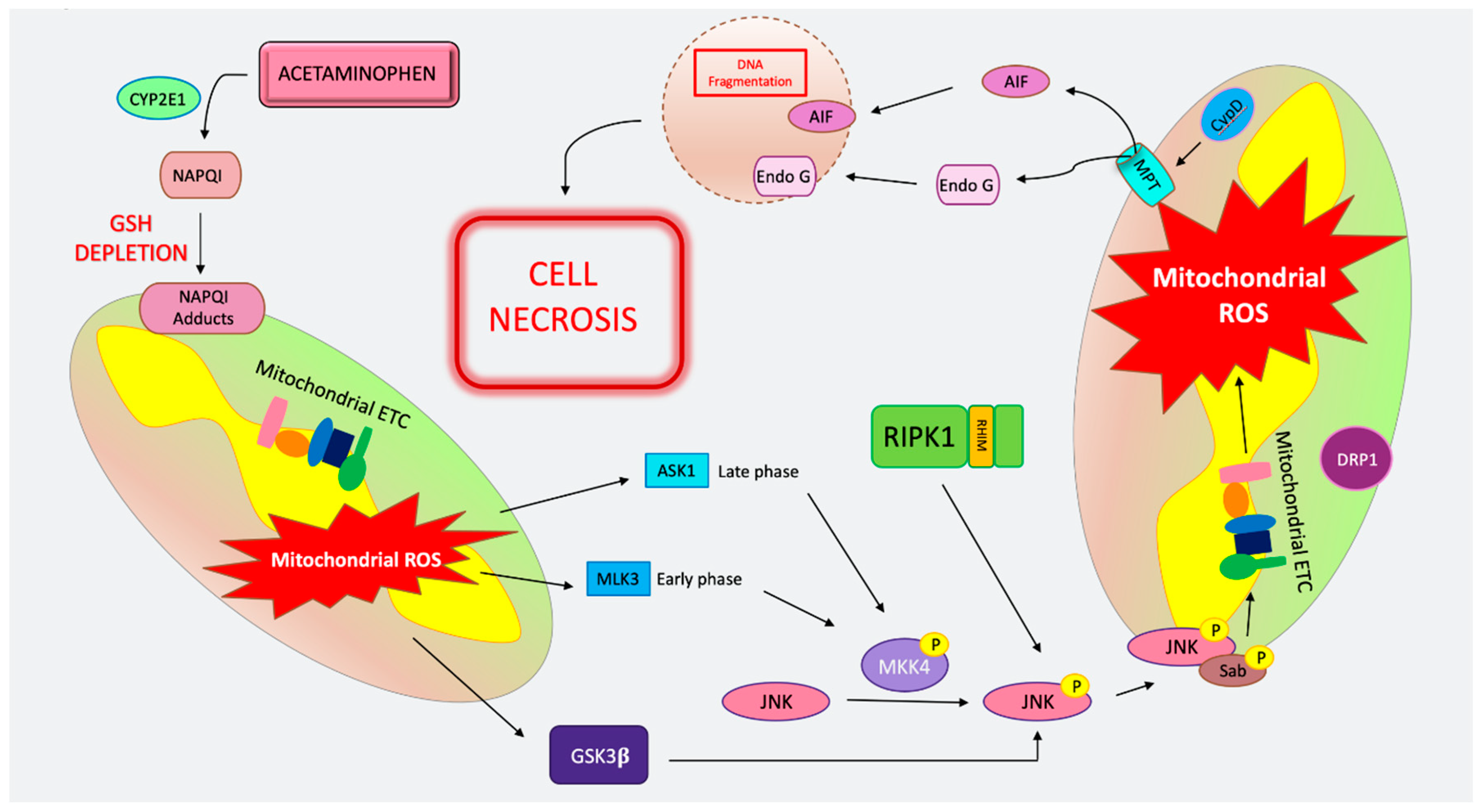
1.2. Mitochondrial Permeability Transition-Driven Necrosis
1.3. Necroptosis
1.4. Autophagy and Autophagy-Dependent Cell Death
1.5. Pyroptosis
1.6. Ferroptosis
1.7. Other Modes of Cell Death
2. Cell Death in Alcoholic Liver Disease (ALD)
2.1. Apoptosis in ALD
2.2. Necroptosis in ALD
2.3. Autophagy in ALD
2.4. Pyroptosis in ALD
2.5. Ferroptosis in ALD
3. Cell Death in Nonalcoholic Fatty Liver Disease NASH/NAFLD
3.1. Apoptosis in NASH/NAFLD
3.2. MPT-Mediated Necrosis and Necroptosis in NASH/NAFLD
3.3. Pyroptosis in NASH/NAFLD
3.4. Ferroptosis in NASH/NAFLD
4. Cell Death in Acetaminophen Toxicity
4.1. MPT-Mediated Regulated Necrosis in APAP
4.2. Apoptosis in APAP
4.3. Necroptosis in APAP
4.4. Autophagy in APAP
4.5. Pyroptosis in APAP
4.6. Ferroptosis in APAP
5. Cell Death in Autoimmune Hepatitis (AIH)
5.1. Apoptosis in AIH
5.2. Necroptosis in AIH
5.3. Pyroptosis in AIH
5.4. Ferroptosis in AIH
6. Cell Death in Cholestatic Liver Diseases
6.1. Apoptosis in Cholestatic Liver Diseases
6.2. Necroptosis and Necrosis in Cholestatic Liver Disease
6.3. Pyroptosis in Cholestatic Liver Disease
7. Cell Death in Viral Hepatitis
7.1. Apoptosis in Viral Hepatitis
7.2. Necroptosis in Viral Hepatitis
7.3. Pyroptosis in Viral Hepatitis
8. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AH | alcoholic hepatitis |
| AIF | apoptosis inducing factor |
| AIH | autoimmune hepatitis |
| AIM2 | absent in melanoma 2 |
| ALD | alcoholic liver disease |
| ALT | alanine aminotransferase |
| AMAP | acetyl-metal-aminophenol |
| AMP | adenosine monophosphate |
| AMPK | adenosine monophosphate–activated protein kinase |
| APAF1 | apoptotic peptidase activating factor 1 |
| APAP | acetaminophen |
| ASC | apoptosis-associated speck-like protein containing a CARD |
| ASK1 | apoptosis signal-regulating kinase 1 |
| AST | aspartate aminotransferase |
| ATG | autophagy-related gene |
| BAK | BCL2 antagonist/killer |
| Bax | BCL-2-like protein 4 |
| Bcl-2 | B-cell lymphoma 2 |
| BDL | bile duct ligation |
| BID | BH3 interacting domain death agonist |
| CASP | caspase |
| Cav-1 | Caveolin-1 |
| CCL4 | carbon tetra chloride |
| CDCA | chenodeoxycholic acid |
| CDE | choline-deficient, ethionine-supplemented diet |
| CDK2AP1 | cyclin-dependent kinase 2-associated protein 1 |
| cFLAR | CASP8 and FADD Like Apoptosis Regulator |
| cFLIP | cellular FLICE inhibitory protein |
| cIAP1/2 | cellular inhibitor of apoptosis 1/2 |
| CMA | chaperone-mediated autophagy |
| ConA | concanavalin A |
| CsA | cyclosporin A |
| CYLD | cylindromatosis |
| CypD | cyclophilin D |
| DAMP | danger-associated molecular patterns |
| DDC | d 3,5-diethoxycarbonyl-1,4-dihydrocollidine |
| DILI | drug-induced liver injury |
| DISC | death-inducing signaling complex |
| DR | death receptor |
| DRP1 | dynamin-related protein-1 |
| EndoG | endonuclease G |
| ER | endoplasmic reticulum |
| ESCRT | endosomal sorting complexes required for transport |
| ETC | electron transport chain |
| FADD | Fas-associated via death domain |
| Fer-1 | ferrostatin 1 |
| FFC | Fat, fructose, cholesterol |
| FLICE | Fas-associating protein with death domain-like interleukin-1 β-converting enzyme |
| FoxO1 | forkhead box protein O1 |
| GCDCA | glycochenodeoxycholic acid |
| GPX4 | glutathione peroxidase 4 |
| GSDMD | gasdermin D |
| GSH | glutathione |
| GSK3β | glycogen synthase kinase 3 beta |
| HBV | hepatitis B virus |
| HCV | hepatitis C virus |
| HFD | high fat diet |
| HMGB1 | high mobility group box 1 |
| HSC | hepatic stellate cell |
| IDO1 | indoleamine 2,3-dioxygenase |
| IFN | interferon |
| IL | interleukin |
| IRF3 | interferon regulatory factor3 |
| JNK | cJun-N-terminal |
| KC | kupffer cell |
| KO | knockout |
| LAMP 2A | lysosome-associated membrane protein type 2A |
| LPS | lipopolysaccharide |
| MAPK | mitogen activated protein kinase |
| MCD | methionine and choline deficient diet |
| MCL1 | myeloid cell leukemia 1 |
| MDR2 | multidrug resistance 2 |
| MHV3 | murine hepatitis virus type 3 |
| miR-148a | microRNA 148a |
| MKK4 | mitogen activated protein kinase 4 |
| MLKL | pseudokinase mixed lineage domain-like |
| MLKL3 | mixed lineage kinase protein 3 |
| MOMP | mitochondrial outer membrane pore formation |
| MTP | mitochondrial permeability transition |
| MPTP | mitochondrial permeability transition pore |
| MyD88 | myeloid differentiation primary response protein |
| NAFLD | non-alcoholic fatty liver disease |
| NAPQI | N-acetyl-p-benzoquinone imine |
| NAS | NAFLD activity score |
| NASH | non-alcoholic steatohepatitis |
| Nec-1 | necrostatin-1 |
| NEMO | NF kappa B essential modulator |
| NF-κΒ | Nuclear factor-κΒ |
| NLRP1/3 | NOD-like receptor family, pyrin domain-containing 1/3 |
| NPC | non-parenchymal cells |
| PAMP | pathogen-associated molecular patterns |
| PBC | primary biliary cirrhosis |
| PBMC | peripheral blood mononuclear cells |
| Pink1 | PTEN-induced putative kinase 1 |
| PIP | phosphatidylinositol phosphates |
| PRR | pattern Recognition Receptors |
| PSC | primary sclerosing cholangitis |
| PTEN | phosphatase and Tensin homolog |
| PUMA | p53 upregulated modulator of apoptosis |
| RCD | regulated cell death |
| RCT | randomized clinical trials |
| rhIL-1Ra | recombinant human interleukin-1 receptor antagonist |
| RHIM | RIP homology interaction motif |
| RIPK1/3 | receptor-interacting protein kinase 1/3 |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SHB1 | SH2 phosphatase 1 |
| SIRT1 | Sirtuin 1 |
| Smac/DIABLO | second mitochondria-derived activator of caspase |
| SNX10 | Sorting nexin 10 |
| SS | sodium selenite |
| STING | stimulator of interferon genes |
| TAK1 | transforming growth factor-activated kinase-1 |
| tBID | cleaved BID |
| TG | triglyceride |
| TLR | Toll Like Receptors |
| TNF | tumor necrosis factor |
| TNFAIP3 | tumor necrosis factor-alpha-induced protein 3 |
| TNFR | tumor Necrosis Factor Receptor |
| TRADD | TNFR-associated death domain |
| TRAF | TNF receptor-associated factors |
| TUDCA | taurine-conjugate of Ursodeoxycholic acid |
| UDCA | ursodeoxycholic acid |
| UPR | unfolded protein response |
| WAT | White Adipose Tissue |
| WT | Wild Type |
| XIAP | x-linked inhibitor of apoptosis |
| ZBP1/DAI | Z-DNA binding protein 1 |
| α-GalCer | α-galactosylceramide |
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 Family Proteins: Changing Partners in the Dance towards Death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Riedl, S.J.; Salvesen, G.S. The Apoptosome: Signalling Platform of Cell Death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.L.; et al. Linear Ubiquitination Prevents Inflammation and Regulates Immune Signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.A.; Legarda-Addison, D.; Skountzos, P.; Yeh, W.C.; Ting, A.T. Ubiquitination of RIP1 Regulates an NF-ΚB-Independent Cell-Death Switch in TNF Signaling. Curr. Biol. 2007, 17, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Salvesen, G. Regulated Cell Death: Signaling and Mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 337–356. [Google Scholar] [CrossRef]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of Tumour Necrosis Factor Signalling: Live or Let Die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Sun, S.C. CYLD: A Tumor Suppressor Deubiquitinase Regulating NF-B Activation and Diverse Biological Processes. Cell Death Differ. 2010, 17, 25–34. [Google Scholar] [CrossRef]
- Iorga, A.; Dara, L.; Kaplowitz, N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. Int. J. Mol. Sci. 2017, 18, 1018. [Google Scholar] [CrossRef]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.R.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-Operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular Mechanisms of Necroptosis: An Ordered Cellular Explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus Accessory Aspects of Cell Death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef]
- Giorgio, V.; Von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in Mitochondrial Pathophysiology. Biochim. Biophys. Acta Bioenerg. 2010, 1113–1118. [Google Scholar] [CrossRef]
- Ramachandran, A.; Lebofsky, M.; Baines, C.P.; Lemasters, J.J.; Jaeschke, H. Cyclophilin D Deficiency Protects against Acetaminophen-Induced Oxidant Stress and Liver Injury. Free Radic. Res. 2011, 45, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Du, H.; Shao, S.; Bo, T.; Yu, C.; Chen, W.; Zhao, L.; Li, Q.; Wang, L.; Liu, X.; et al. Cyclophilin D Deficiency Attenuates Mitochondrial Perturbation and Ameliorates Hepatic Steatosis. Hepatology 2018, 68, 62–77. [Google Scholar] [CrossRef]
- Song, K.; Wang, S.; Qi, D. Effects of Cyclosporine on Reperfusion Injury in Patients: A Meta-Analysis of Randomized Controlled Trials. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of Caspases Increases the Sensitivity of L929 Cells to Necrosis Mediated by Tumor Necrosis Factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Chen, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 Kinase as a Specific Cellular Target of Necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Teng, X.; Degterev, A.; Jagtap, P.; Xing, X.; Choi, S.; Denu, R.; Yuan, J.; Cuny, G.D. Structure-Activity Relationship Study of Novel Necroptosis Inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5039–5044. [Google Scholar] [CrossRef]
- Murphy, J.M.; Vince, J.E. Post-Translational Control of RIPK3 and MLKL Mediated Necroptotic Cell Death. F1000Research 2015. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Newton, K.; Manning, G. Necroptosis and Inflammation. Annu. Rev. Biochem. 2016, 85, 743–763. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The Pseudokinase MLKL Mediates Necroptosis via a Molecular Switch Mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by Caspase-8 Is Crucial for Limiting Apoptosis and Necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef]
- O’Donnell, M.A.; Perez-Jimenez, E.; Oberst, A.; Ng, A.; Massoumi, R.; Xavier, R.; Green, D.R.; Ting, A.T. Caspase 8 Inhibits Programmed Necrosis by Processing CYLD. Nat. Cell Biol. 2011, 13, 1437–1442. [Google Scholar] [CrossRef]
- Dara, L.; Liu, Z.X.; Kaplowitz, N. Questions and Controversies: The Role of Necroptosis in Liver Disease. Cell Death Discov. 2016. [Google Scholar] [CrossRef]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the Treatment of Human Diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef]
- Dara, L. The Receptor Interacting Protein Kinases in the Liver. Semin. Liver Dis. 2018, 38, 73–86. [Google Scholar] [CrossRef]
- Dara, L.; Johnson, H.; Suda, J.; Win, S.; Gaarde, W.; Han, D.; Kaplowitz, N. Receptor Interacting Protein Kinase 1 Mediates Murine Acetaminophen Toxicity Independent of the Necrosome and Not through Necroptosis. Hepatology 2015, 62, 1847–1857. [Google Scholar] [CrossRef]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein That Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65.e7. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Vaux, D.L.; Murphy, J.M.; Vince, J.E.; Lindqvist, L.M. Activated MLKL Attenuates Autophagy Following Its Translocation to Intracellular Membranes. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N.; Win, S.; Than, T.A.; Liu, Z.X.; Dara, L. Targeting Signal Transduction Pathways which Regulate Necrosis in Acetaminophen Hepatotoxicity. J. Hepatol. 2015, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Kasof, G.M.; Prosser, J.C.; Liu, D.; Lorenzi, M.V.; Gomes, B.C. The RIP-like Kinase, RIP3, Induces Apoptosis and NF-ΚB Nuclear Translocation and Localizes to Mitochondria. FEBS Lett. 2000, 473, 285–291. [Google Scholar] [CrossRef]
- Xiaoqing, S.; James, L.; Navas, T.; Baldwin, D.T.; Stewart, T.A.; Dixit, V.M. RIP3, a Novel Apoptosis-Inducing Kinase. J. Biol. Chem. 1999, 274, 16871–16875. [Google Scholar] [CrossRef]
- Günther, C.; He, G.W.; Kremer, A.E.; Murphy, J.M.; Petrie, E.J.; Amann, K.; Vandenabeele, P.; Linkermann, A.; Poremba, C.; Schleicher, U.; et al. The Pseudokinase MLKL Mediates Programmed Hepatocellular Necrosis Independently of RIPK3 during Hepatitis. J. Clin. Investig. 2016, 126, 4346–4360. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome Formation: Core Machinery and Adaptations. Nat. Cell Biol. 2007, 1102–1109. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular Definitions of Cell Death Subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 107–120. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-Consumption: The Interplay of Autophagy and Apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 81–94. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy 2016, 1425–1428. [Google Scholar] [CrossRef]
- Yin, X.M. Autophagy in Liver Diseases: A Matter of What to Remove and Whether to Keep. Liver Res. 2018, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen Blockade of TAK1 Triggers Caspase-8–Dependent Cleavage of Gasdermin D and Cell Death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Regulation of Inflammasome Activation. Immunol. Rev. 2015, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Aachoui, Y.; Sagulenko, V.; Miao, E.A.; Stacey, K.J. Inflammasome-Mediated Pyroptotic and Apoptotic Cell Death, and Defense against Infection. Curr. Opin. Microbiol. 2013, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Beier, J.I.; Banales, J.M. Pyroptosis: An Inflammatory Link between NAFLD and NASH with Potential Therapeutic Implications. J. Hepatol. 2018, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D Plays a Key Role as a Pyroptosis Executor of Non-Alcoholic Steatohepatitis in Humans and Mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef]
- Wree, A.; McGeough, M.D.; Inzaugarat, M.E.; Eguchi, A.; Schuster, S.; Johnson, C.D.; Peña, C.A.; Geisler, L.J.; Papouchado, B.G.; Hoffman, H.M.; et al. NLRP3 Inflammasome Driven Liver Injury and Fibrosis: Roles of IL-17 and TNF in Mice. Hepatology 2018, 67, 736–749. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 165–176. [Google Scholar] [CrossRef]
- Mao, L.; Zhao, T.; Song, Y.; Lin, L.; Fan, X.; Cui, B.; Feng, H.; Wang, X.; Yu, Q.; Zhang, J.; et al. The Emerging Role of Ferroptosis in Non-Cancer Liver Diseases: Hype or Increasing Hope? Cell Death Dis. 2020. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020. [Google Scholar] [CrossRef]
- Doll, S.; Conrad, M. Iron and Ferroptosis: A Still Ill-Defined Liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef]
- Capelletti, M.M.; Manceau, H.; Puy, H.; Peoc’h, K. Ferroptosis in Liver Diseases: An Overview. Int. J. Mol. Sci. 2020, 21, 4908. [Google Scholar] [CrossRef]
- Yang, W.S.; Sriramaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Wang, S.; Pacher, P.; De Lisle, R.C.; Huang, H.; Ding, W.X. A Mechanistic Review of Cell Death in Alcohol-Induced Liver Injury. Alcohol. Clin. Exp. Res. 2016, 1215–1223. [Google Scholar] [CrossRef]
- Williams, J.A.; Ding, W.X. Role of Autophagy in Alcohol and Drug-Induced Liver Injury. Food Chem. Toxicol. 2020, 136. [Google Scholar] [CrossRef]
- Zhou, Z.; Ye, T.J.; Bonavita, G.; Daniels, M.; Kainrad, N.; Jogasuria, A.; You, M. Adipose-Specific Lipin-1 Overexpression Renders Hepatic Ferroptosis and Exacerbates Alcoholic Steatohepatitis in Mice. Hepatol. Commun. 2019, 3, 656–669. [Google Scholar] [CrossRef]
- Cahill, A.; Cunningham, C.C.; Adachi, M.; Ishii, H.; Bailey, S.M.; Fromenty, B.; Davies, A. Effects of Alcohol and Oxidative Stress on Liver Pathology: The Role of the Mitochondrion. Alcohol. Clin. Exp. Res. 2002, 26, 907–915. [Google Scholar] [CrossRef]
- Adachi, M.; Higuchi, H.; Miura, S.; Azuma, T.; Inokuchi, S.; Saito, H.; Kato, S.; Ishii, H. Bax Interacts with the Voltage-Dependent Anion Channel and Mediates Ethanol-Induced Apoptosis in Rat Hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287. [Google Scholar] [CrossRef]
- Malhi, H.; Gores, G.J. Cellular and Molecular Mechanisms of Liver Injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar] [CrossRef]
- Ji, C.; Mehrian-Shai, R.; Chan, C.; Hsu, Y.H.; Kaplowitz, N. Role of CHOP in Hepatic Apoptosis in the Murine Model of Intragastric Ethanol Feeding. Alcohol. Clin. Exp. Res. 2005, 29, 1496–1503. [Google Scholar] [CrossRef]
- Ji, C.; Kaplowitz, N. Betaine Decreases Hyperhomocysteinemia, Endoplasmic Reticulum Stress, and Liver Injury in Alcohol-Fed Mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Dara, L.; Ji, C.; Kaplowitz, N. The Contribution of Endoplasmic Reticulum Stress to Liver Diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 Pathway Links Endoplasmic Reticulum Stress with Hepatocyte Apoptosis in Early Alcoholic Liver Disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; McVicker, D.L.; Zetterman, R.K.; MacDonald, R.G.; Donohue, T.M. Flow Cytometric Analysis of Vesicular PH in Rat Hepatocytes after Ethanol Administration. Hepatology 1997, 26, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Zhou, Z.; Kang, Y.J. Suppression of Fas-Mediated Signaling Pathway Is Involved in Zinc Inhibition of Ethanol-Induced Liver Apoptosis. Exp. Biol. Med. 2003, 228, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.E.; Ding, W.X.; Cresci, G.; Saikia, P.; Shah, V.H. Linking Pathogenic Mechanisms of Alcoholic Liver Disease with Clinical Phenotypes. Gastroenterology 2016, 150, 1756–1768. [Google Scholar] [CrossRef]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic Liver Disease: The Gut Microbiome and Liver Cross Talk. Alcohol. Clin. Exp. Res. 2015, 763–775. [Google Scholar] [CrossRef]
- Natori, S.; Rust, C.; Stadheim, L.M.; Srinivasan, A.; Burgart, L.J.; Gores, G.J. Hepatocyte Apoptosis Is a Pathologic Feature of Human Alcoholic Hepatitis. J. Hepatol. 2001, 34, 248–253. [Google Scholar] [CrossRef]
- Naveau, S.; Emilie, D.; Balian, A.; Grangeot-Keros, L.; Borotto, E.; Portier, A.; Giraud, V.; Capron, F.; Galanaud, P.; Chaput, J.C. Plasma Levels of Soluble Tumor Necrosis Factor Receptors P55 and P75 in Patients with Alcoholic Liver Disease of Increasing Severity. J. Hepatol. 1998, 28, 778–784. [Google Scholar] [CrossRef]
- Naveau, S.; Chollet-Martin, S.; Dharancy, S.; Mathurin, P.; Jouet, P.; Piquet, M.A.; Davion, T.; Oberti, F.; Broët, P.; Emilie, D. A Double-Blind Randomized Controlled Trial of Infliximab Associated with Prednisolone in Acute Alcoholic Hepatitis. Hepatology 2004, 39, 1390–1397. [Google Scholar] [CrossRef]
- Hao, F.; Cubero, F.J.; Ramadori, P.; Liao, L.; Haas, U.; Lambertz, D.; Sonntag, R.; Bangen, J.M.; Gassler, N.; Hoss, M.; et al. Inhibition of Caspase-8 Does Not Protect from Alcohol-Induced Liver Apoptosis but Alleviates Alcoholic Hepatic Steatosis in Mice. Cell Death Dis. 2017, 8, e3152. [Google Scholar] [CrossRef]
- Wilson, C.H.; Kumar, S. Caspases in Metabolic Disease and Their Therapeutic Potential. Cell Death Differ. 2018, 1010–1024. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Chiang, D.J.; Mandal, P.; McMullen, M.R.; Liu, X.; Cohen, J.I.; Pollard, J.; Feldstein, A.E.; Nagy, L.E. Inhibition of Apoptosis Protects Mice from Ethanol-Mediated Acceleration of Early Markers of CCl4-Induced Fibrosis but Not Steatosis or Inflammation. Alcohol. Clin. Exp. Res. 2012, 36, 1139–1147. [Google Scholar] [CrossRef]
- Sebastian, B.M.; Roychowdhury, S.; Tang, H.; Hillian, A.D.; Feldstein, A.E.; Stahl, G.L.; Takahashi, K.; Nagy, L.E. Identification of a Cytochrome P4502E1/Bid/C1q-Dependent Axis Mediating Inflammation in Adipose Tissue after Chronic Ethanol Feeding to Mice. J. Biol. Chem. 2011, 286, 35989–35997. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Mcmullen, M.R.; Pisano, S.G.; Liu, X.; Nagy, L.E. Absence of Receptor Interacting Protein Kinase 3 Prevents Ethanol-Induced Liver Injury. Hepatology 2013, 57, 1773–1783. [Google Scholar] [CrossRef]
- Wang, S.; Ni, H.M.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Nawabi, A.; Komatsu, M.; Huang, H.; Ding, W.X. Increased Hepatic Receptor Interacting Protein Kinase 3 Expression Due to Impaired Proteasomal Functions Contributes to Alcohol-Induced Steatosis and Liver Injury. Oncotarget 2016, 7, 17681–17698. [Google Scholar] [CrossRef]
- Ding, W.; Li, M.; Chen, X.; Ni, H.; Lin, C.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X. Autophagy Reduces Acute Ethanol-Induced Hepatotoxicity and Steatosis in Mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef]
- Lin, C.W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.M. Pharmacological Promotion of Autophagy Alleviates Steatosis and Injury in Alcoholic and Non-Alcoholic Fatty Liver Conditions in Mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 Promotes the Development of Fibrosis and Tumorigenesis in Mice with Defective Hepatic Autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Sorting, Recognition and Activation of the Misfolded Protein Degradation Pathways through Macroautophagy and the Proteasome. Autophagy 2008, 141–150. [Google Scholar] [CrossRef]
- Khambu, B.; Wang, L.; Zhang, H.; Yin, X.-M. The Activation and Function of Autophagy in Alcoholic Liver Disease. Curr. Mol. Pharmacol. 2017, 10, 165–171. [Google Scholar] [CrossRef]
- Eid, N.; Ito, Y.; Maemura, K.; Otsuki, Y. Elevated Autophagic Sequestration of Mitochondria and Lipid Droplets in Steatotic Hepatocytes of Chronic Ethanol-Treated Rats: An Immunohistochemical and Electron Microscopic Study. J. Mol. Histol. 2013, 44, 311–326. [Google Scholar] [CrossRef]
- Williams, J.A.; Ni, H.M.; Haynes, A.; Manley, S.; Li, Y.; Jaeschke, H.; Ding, W.X. Chronic Deletion and Acute Knockdown of Parkin Have Differential Responses to Acetaminophen-Induced Mitophagy and Liver Injury in Mice. J. Biol. Chem. 2015, 290, 10934–10946. [Google Scholar] [CrossRef]
- Williams, J.A.; Ding, W.X. Targeting Pink1-Parkin-Mediated Mitophagy for Treating Liver Injury. Pharmacol. Res. 2015, 102, 264–269. [Google Scholar] [CrossRef]
- Williams, J.A.; Ni, H.M.; Ding, Y.; Ding, W.X. Parkin Regulates Mitophagy and Mitochondrial Function to Protect against Alcohol-Induced Liver Injury and Steatosis in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G324–G340. [Google Scholar] [CrossRef]
- You, Y.; Li, W.Z.; Zhang, S.; Hu, B.; Li, Y.X.; Li, H.D.; Tang, H.H.; Li, Q.W.; Guan, Y.Y.; Liu, L.X.; et al. SNX10 Mediates Alcohol-Induced Liver Injury and Steatosis by Regulating the Activation of Chaperone-Mediated Autophagy. J. Hepatol. 2018, 69, 129–141. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 280–293. [Google Scholar] [CrossRef]
- Wu, J.; Lin, S.; Wan, B.; Velani, B.; Zhu, Y. Pyroptosis in Liver Disease: New Insights into Disease Mechanisms. Aging Dis. 2019, 10, 1094–1108. [Google Scholar] [CrossRef]
- Petrasek, J.; Iracheta-Vellve, A.; Saha, B.; Satishchandran, A.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Metabolic Danger Signals, Uric Acid and ATP, Mediate Inflammatory Cross-Talk between Hepatocytes and Immune Cells in Alcoholic Liver Disease. J. Leukoc. Biol. 2015, 98, 249–256. [Google Scholar] [CrossRef]
- Heo, M.J.; Kim, T.H.; You, J.S.; Blaya, D.; Sancho-Bru, P.; Kim, S.G. Alcohol Dysregulates MiR-148a in Hepatocytes through FoxO1, Facilitating Pyroptosis via TXNIP Overexpression. Gut 2019, 68, 708–720. [Google Scholar] [CrossRef]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 Receptor Antagonist Ameliorates Inflammasome-Dependent Alcoholic Steatohepatitis in Mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Roh, Y.S.; Seki, E. Toll-like Receptors in Alcoholic Liver Disease, Non-Alcoholic Steatohepatitis and Carcinogenesis. J. Gastroenterol. Hepatol. 2013, 38–42. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-Like Receptors. Curr. Protoc. Immunol. 2015, 21, 335–376. [Google Scholar] [CrossRef]
- Zhou, H.; Yu, M.; Roychowdhury, S.; Sanz-Garcia, C.; Pollard, K.A.; McMullen, M.R.; Liu, X.; Li, X.; Nagy, L.E. Myeloid-MyD88 Contributes to Ethanol-Induced Liver Injury in Mice Linking Hepatocellular Death to Inflammation. Alcohol. Clin. Exp. Res. 2017, 41, 719–726. [Google Scholar] [CrossRef][Green Version]
- Khanova, E.; Wu, R.; Wang, W.; Yan, R.; Chen, Y.; French, S.W.; Llorente, C.; Pan, S.Q.; Yang, Q.; Li, Y.; et al. Pyroptosis by Caspase11/4-Gasdermin-D Pathway in Alcoholic Hepatitis in Mice and Patients. Hepatology 2018, 67, 1737–1753. [Google Scholar] [CrossRef]
- You, M.; Jogasuria, A.; Taylor, C.; Wu, J. Sirtuin 1 Signaling and Alcoholic Fatty Liver Disease. Hepatobiliary Surg. Nutr. 2015, 4, 88–100. [Google Scholar] [CrossRef]
- Zhou, Z.; Ye, T.J.; DeCaro, E.; Buehler, B.; Stahl, Z.; Bonavita, G.; Daniels, M.; You, M. Intestinal SIRT1 Deficiency Protects Mice from Ethanol-Induced Liver Injury by Mitigating Ferroptosis. Am. J. Pathol. 2020, 190, 82–92. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte Apoptosis and Fas Expression Are Prominent Features of Human Nonalcoholic Steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Cazanave, S.C.; Gores, G.J. Mechanisms and Clinical Implications of Hepatocyte Lipoapoptosis. Clin. Lipidol. 2010, 71–85. [Google Scholar] [CrossRef]
- Maher, J.J. Modeling Fatty Liver Disease in Animals: Is There an Optimal Approach, and Is the Effort Worthwhile? Hepatology 2016, 1398–1400. [Google Scholar] [CrossRef] [PubMed]
- Soon, R.K.; Yan, J.S.; Grenert, J.P.; Maher, J.J. Stress Signaling in the Methionine-Cholinedeficient Model of Murine Fatty Liver Disease. Gastroenterology 2010, 139. [Google Scholar] [CrossRef] [PubMed]
- Imajo, K.; Yoneda, M.; Kessoku, T.; Ogawa, Y.; Maeda, S.; Sumida, Y.; Hyogo, H.; Eguchi, Y.; Wada, K.; Nakajima, A. Rodent Models of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2013, 14, 21833–21857. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical Models of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Thapaliya, S.; Wree, A.; Povero, D.; Inzaugarat, M.E.; Berk, M.; Dixon, L.; Papouchado, B.G.; Feldstein, A.E. Caspase 3 Inactivation Protects against Hepatic Cell Death and Ameliorates Fibrogenesis in a Diet-Induced NASH Model. Dig. Dis. Sci. 2014, 59, 1197–1206. [Google Scholar] [CrossRef]
- Hatting, M.; Zhao, G.; Schumacher, F.; Sellge, G.; Al Masaoudi, M.; Gaßler, N.; Boekschoten, M.; Müller, M.; Liedtke, C.; Cubero, F.J.; et al. Hepatocyte Caspase-8 Is an Essential Modulator of Steatohepatitis in Rodents. Hepatology 2013, 57, 2189–2201. [Google Scholar] [CrossRef]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The Pan-Caspase Inhibitor Emricasan (IDN-6556) Decreases Liver Injury and Fibrosis in a Murine Model of Non-Alcoholic Steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef]
- Witek, R.P.; Stone, W.C.; Karaca, F.G.; Syn, W.K.; Pereira, T.A.; Agboola, K.M.; Omenetti, A.; Jung, Y.; Teaberry, V.; Choi, S.S.; et al. Pan-Caspase Inhibitor VX-166 Reduces Fibrosis in an Animal Model of Nonalcoholic Steatohepatitis. Hepatology 2009, 50, 1421–1430. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Concas, D.; Kudo, H.; Levene, A.; Pollard, J.; Charlton, P.; Thomas, H.C.; Thursz, M.R.; Goldin, R.D. Impact of Pan-Caspase Inhibition in Animal Models of Established Steatosis and Non-Alcoholic Steatohepatitis. J. Hepatol. 2010, 53, 542–550. [Google Scholar] [CrossRef]
- Ratziu, V.; Sheikh, M.Y.; Sanyal, A.J.; Lim, J.K.; Conjeevaram, H.; Chalasani, N.; Abdelmalek, M.; Bakken, A.; Renou, C.; Palmer, M.; et al. A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study of GS-9450 in Subjects with Nonalcoholic Steatohepatitis. Hepatology 2012, 55, 419–428. [Google Scholar] [CrossRef]
- Shiffman, M.; Freilich, B.; Vuppalanchi, R.; Watt, K.; Chan, J.L.; Spada, A.; Hagerty, D.T.; Schiff, E. Randomised Clinical Trial: Emricasan versus Placebo Significantly Decreases ALT and Caspase 3/7 Activation in Subjects with Non-Alcoholic Fatty Liver Disease. Aliment. Pharmacol. Ther. 2019, 49, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Goodman, Z.; Jabbar, A.; Vemulapalli, R.; Younes, Z.H.; Freilich, B.; Sheikh, M.Y.; Schattenberg, J.M.; Kayali, Z.; Zivony, A.; et al. A Randomized, Placebo-Controlled Trial of Emricasan in Patients with NASH and F1-F3 Fibrosis. J. Hepatol. 2020, 72, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G.; Bosch, J.; Kayali, Z.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.; Satapathy, S.K.; Ghabril, M.; Shiffman, M.L.; Younes, Z.H.; et al. Randomized Placebo-Controlled Trial of Emricasan for Non-Alcoholic Steatohepatitis-Related Cirrhosis with Severe Portal Hypertension. J. Hepatol. 2020, 72, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; Wong, K.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK–Caspase-6 Axis Controls Liver Damage in Nonalcoholic Steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.X.; Ji, Y.X.; Zhang, X.J.; Zhao, L.P.; Yan, Z.Z.; Zhang, P.; Shen, L.J.; Yang, X.; Fang, J.; Tian, S.; et al. Targeting CASP8 and FADD-like Apoptosis Regulator Ameliorates Nonalcoholic Steatohepatitis in Mice and Nonhuman Primates. Nat. Med. 2017, 23, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, P.X.; Zhao, L.P.; Zhang, X.; Ji, Y.X.; Zhang, X.J.; Fang, C.; Lu, Y.X.; Yang, X.; Gao, M.M.; et al. The Deubiquitinating Enzyme TNFAIP3 Mediates Inactivation of Hepatic ASK1 and Ameliorates Nonalcoholic Steatohepatitis. Nat. Med. 2018, 24, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Maiers, J.L.; Malhi, H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin. Liver Dis. 2019, 39, 235–248. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef]
- Roh, Y.S.; Kim, J.W.; Park, S.; Shon, C.; Kim, S.; Eo, S.K.; Kwon, J.K.; Lim, C.W.; Kim, B. Toll-Like Receptor-7 Signaling Promotes Nonalcoholic Steatohepatitis by Inhibiting Regulatory T Cells in Mice. Am. J. Pathol. 2018, 188, 2574–2588. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like Receptor-4 Signaling and Kupffer Cells Play Pivotal Roles in the Pathogenesis of Non-Alcoholic Steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef]
- Kuo, J.; Bobardt, M.; Chatterji, U.; Mayo, P.R.; Trepanier, D.J.; Foster, R.T.; Gallay, P.; Ure, D.R. A Pan-Cyclophilin Inhibitor, CRV431, Decreases Fibrosis and Tumor Development in Chronic Liver Disease Models. J. Pharmacol. Exp. Ther. 2019, 371, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Dolganiuc, A.; Kodys, K.; Nath, B.; Petrasek, J.; Bala, S.; Lippai, D.; Szabo, G. Mitochondrial Antiviral Signaling Protein Defect Links Impaired Antiviral Response and Liver Injury in Steatohepatitis in Mice. Hepatology 2011, 53, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Neumann, U.P.; et al. A Positive Feedback Loop between RIP 3 and JNK Controls Non-alcoholic Steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; McCullough, R.L.; Sanz-Garcia, C.; Saikia, P.; Alkhouri, N.; Matloob, A.; Pollard, K.A.; McMullen, M.R.; Croniger, C.M.; Nagy, L.E. Receptor Interacting Protein 3 Protects Mice from High-Fat Diet-Induced Liver Injury. Hepatology 2016, 64, 1518–1533. [Google Scholar] [CrossRef]
- Gautheron, J.; Vucur, M.; Schneider, A.T.; Severi, I.; Roderburg, C.; Roy, S.; Bartneck, M.; Schrammen, P.; Diaz, M.B.; Ehling, J.; et al. The Necroptosis-Inducing Kinase RIPK3 Dampens Adipose Tissue Inflammation and Glucose Intolerance. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Carvalho, T.; Caridade, M.; Borralho, P.; Cortez-Pinto, H.; Castro, R.E.; Rodrigues, C.M.P. Necroptosis Is a Key Pathogenic Event in Human and Experimental Murine Models of Non-Alcoholic Steatohepatitis. Clin. Sci. 2015, 129, 721–739. [Google Scholar] [CrossRef]
- Xu, H.; Du, X.; Liu, G.; Huang, S.; Du, W.; Zou, S.; Tang, D.; Fan, C.; Xie, Y.; Wei, Y.; et al. The Pseudokinase MLKL Regulates Hepatic Insulin Sensitivity Independently of Inflammation. Mol. Metab. 2019, 23, 14–23. [Google Scholar] [CrossRef]
- Majdi, A.; Aoudjehane, L.; Ratziu, V.; Islam, T.; Afonso, M.B.; Conti, F.; Mestiri, T.; Lagouge, M.; Foufelle, F.; Ballenghien, F.; et al. Inhibition of Receptor-Interacting Protein Kinase 1 Improves Experimental Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2020, 72, 627–635. [Google Scholar] [CrossRef]
- Wu, X.; Poulsen, K.L.; Sanz-Garcia, C.; Huang, E.; McMullen, M.R.; Roychowdhury, S.; Dasarathy, S.; Nagy, L.E. MLKL-Dependent Signaling Regulates Autophagic Flux in a Murine Model of Non-Alcohol-Associated Fatty Liver and Steatohepatitis. J. Hepatol. 2020, 73, 616–627. [Google Scholar] [CrossRef]
- Newton, K. RIPK1 and RIPK3: Critical Regulators of Inflammation and Cell Death. Trends Cell Biol. 2015, 347–353. [Google Scholar] [CrossRef]
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-Mcnulty, B.; Carano, R.A.D.; Cao, T.C.; Van Bruggen, N.; Bernstein, L.; et al. RIPK3 Deficiency or Catalytically Inactive RIPK1 Provides Greater Benefit than MLKL Deficiency in Mouse Models of Inflammation and Tissue Injury. Cell Death Differ. 2016, 23, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Petrasek, J. Inflammasome Activation and Function in Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z. The Inflammasome in Liver Injury and Non-Alcoholic Fatty Liver Disease. Dig. Dis. 2014, 32, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-Mediated Dysbiosis Regulates Progression of NAFLD and Obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental NASH in Mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Gautheron, J.; Gores, G.J.; Rodrigues, C.M.P. Lytic Cell Death in Metabolic Liver Disease. J. Hepatol. 2020, 394–408. [Google Scholar] [CrossRef]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V. Relationship between the Pattern of Hepatic Iron Deposition and Histological Severity in Nonalcoholic Fatty Liver Disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Valenti, L.; Moscatiello, S.; Vanni, E.; Fracanzani, A.L.; Bugianesi, E.; Fargion, S.; Marchesini, G. Venesection for Non-Alcoholic Fatty Liver Disease Unresponsive to Lifestyle Counselling-a Propensity Score-Adjusted Observational Study. QJM 2011, 104, 141–149. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation–Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic Ferroptosis Plays an Important Role as the Trigger for Initiating Inflammation in Nonalcoholic Steatohepatitis. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, T.X.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.L.; Xiong, Y.; Liu, J.; Yuan, H.X. Targeting Ferroptosis Alleviates Methionine-Choline Deficient (MCD)-Diet Induced NASH by Suppressing Liver Lipotoxicity. Liver Int. 2020, 40, 1378–1394. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Liu, Z.X.; Kaplowitz, N. Mechanisms of Adaptation and Progression in Idiosyncratic Drug Induced Liver Injury, Clinical Implications. Liver Int. 2016, 36, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Dara, L. Cell Death in Drug-Induced Liver Injury. Adv. Pharmacol. 2019, 85, 31–74. [Google Scholar] [CrossRef]
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.S.; Reisch, J.S.; Schiødt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-Induced Acute Liver Failure: Results of a United States Multicenter, Prospective Study. Hepatology 2005, 42, 1364–1372. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ramachandran, A.; Chao, X.; Ding, W.X. Emerging and Established Modes of Cell Death during Acetaminophen-Induced Liver Injury. Arch. Toxicol. 2019, 3491–3502. [Google Scholar] [CrossRef]
- Dara, L.; Kaplowitz, N. Drug-Induced Liver Injury. In The Liver; Arias, I.M., Alter, H.J., Boyer, J.L., Cohen, D.E., Shafritz, D.A., Thorgeirsson, S.S., Wolkoff, A.W., Eds.; Wiley: Hoboken, NJ, USA, 2020; pp. 701–713. [Google Scholar] [CrossRef]
- Jollow, D.J.; Thorgeirsson, S.S.; Potter, W.Z.; Hashimoto, M.; Mitchell, J.R. Acetaminophen Induced Hepatic Necrosis. VI. Metabolic Disposition of Toxic and Nontoxic Doses of Acetaminophen. Pharmacology 1974, 12, 251–271. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Gillette, J.R.; Brodie, B.B. Acetaminophen Induced Hepatic Necrosis. IV. Protective Role of Glutathione. J. Pharmacol. Exp. Ther. 1973, 187, 211–217. [Google Scholar]
- Pumford, N.R.; Hinson, J.A.; Wayne Benson, R.; Roberts, D.W. Immunoblot Analysis of Protein Containing 3-(Cystein-S-Yl)Acetaminophen Adducts in Serum and Subcellular Liver Fractions from Acetaminophen-Treated Mice. Toxicol. Appl. Pharmacol. 1990, 104, 521–532. [Google Scholar] [CrossRef]
- Peterson, R.G.; Rumack, B.H. Treating Acute Acetaminophen Poisoning with Acetylcysteine. JAMA J. Am. Med. Assoc. 1977, 237, 2406–2407. [Google Scholar] [CrossRef]
- Ni, H.M.; McGill, M.R.; Chao, X.; Du, K.; Williams, J.A.; Xie, Y.; Jaeschke, H.; Ding, W.X. Removal of Acetaminophen Protein Adducts by Autophagy Protects against Acetaminophen-Induced Liver Injury in Mice. J. Hepatol. 2016, 65, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Hepatotoxicity. Semin. Liver Dis. 2019, 39, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Moles, A.; Torres, S.; Baulies, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial-Lysosomal Axis in Acetaminophen Hepatotoxicity. Front. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, B.K.; Liu, Z.X.; Han, D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N. C-Jun N-Terminal Kinase Plays a Major Role in Murine Acetaminophen Hepatotoxicity. Gastroenterology 2006, 131, 165–178. [Google Scholar] [CrossRef]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK Translocation to Mitochondria Leading to Inhibition of Mitochondria Bioenergetics in Acetaminophen-Induced Liver Injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef]
- Sharma, M.; Gadang, V.; Jaeschke, A. Critical Role for Mixed-Lineage Kinase 3 in Acetaminophen-Induced Hepatotoxicity. Mol. Pharmacol. 2012, 82, 1001–1007. [Google Scholar] [CrossRef]
- Nakagawa, H.; Maeda, S.; Hikiba, Y.; Ohmae, T.; Shibata, W.; Yanai, A.; Sakamoto, K.; Ogura, K.; Noguchi, T.; Karin, M.; et al. Deletion of Apoptosis Signal-Regulating Kinase 1 Attenuates Acetaminophen-Induced Liver Injury by Inhibiting c-Jun N-Terminal Kinase Activation. Gastroenterology 2008, 135, 1311–1321. [Google Scholar] [CrossRef]
- Zhang, J.; Min, R.W.M.; Le, K.; Zhou, S.; Aghajan, M.; Than, T.A.; Win, S.; Kaplowitz, N. The Role of MAP2 Kinases and P38 Kinase in Acute Murine Liver Injury Models. Cell Death Dis. 2017, 8, e2903. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Min, R.W.M.; Aghajan, M.; Kaplowitz, N. C-Jun N-Terminal Kinase Mediates Mouse Liver Injury through a Novel Sab (SH3BP5)-Dependent Pathway Leading to Inactivation of Intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef]
- Bajt, M.L.; Cover, C.; Lemasters, J.J.; Jaeschke, H. Nuclear Translocation of Endonuclease G and Apoptosis-Inducing Factor during Acetaminophen-Induced Liver Cell Injury. Toxicol. Sci. 2006, 94, 217–225. [Google Scholar] [CrossRef]
- Bajt, M.L.; Ramachandran, A.; Yan, H.M.; Lebofsky, M.; Farhood, A.; Lemasters, J.J.; Jaeschke, H. Apoptosis-Inducing Factor Modulates Mitochondrial Oxidant Stress in Acetaminophen Hepatotoxicity. Toxicol. Sci. 2011, 122, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Kon, K.; Kim, J.S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial Permeability Transition in Acetaminophen-Induced Necrosis and Apoptosis of Cultured Mouse Hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Suda, C.; Horie, T. Involvement of Mitochondrial Permeability Transition in Acetaminophen-Induced Liver Injury in Mice. J. Hepatol. 2005, 42, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Mcgill, M.R.; Xie, Y.; Ni, H.M.; Ding, W.X.; Jaeschke, H. Receptor Interacting Protein Kinase 3 Is a Critical Early Mediator of Acetaminophen-Induced Hepatocyte Necrosis in Mice. Hepatology 2013, 58, 2099–2108. [Google Scholar] [CrossRef]
- Shinohara, M.; Ybanez, M.D.; Win, S.; Than, T.A.; Jain, S.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Silencing Glycogen Synthase Kinase-3β Inhibits Acetaminophen Hepatotoxicity and Attenuates JNK Activation and Loss of Glutamate Cysteine Ligase and Myeloid Cell Leukemia Sequence. J. Biol. Chem. 2010, 285, 8244–8255. [Google Scholar] [CrossRef]
- Chen, D.; Ni, H.M.; Wang, L.; Ma, X.; Yu, J.; Ding, W.X.; Zhang, L. P53 Up-Regulated Modulator of Apoptosis Induction Mediates Acetaminophen-Induced Necrosis and Liver Injury in Mice. Hepatology 2019, 69, 2164–2179. [Google Scholar] [CrossRef]
- Knight, T.R.; Jaeschke, H. Acetaminophen-Induced Inhibition of Fas Receptor-Mediated Liver Cell Apoptosis: Mitochondrial Dysfunction versus Glutathione Depletion. Toxicol. Appl. Pharmacol. 2002, 181, 133–141. [Google Scholar] [CrossRef]
- Cover, C.; Mansouri, A.; Knight, T.R.; Bajt, M.L.; Lemasters, J.J.; Pessayre, D.; Jaeschke, H. Peroxynitrite-Induced Mitochondrial and Endonuclease-Mediated Nuclear DNA Damage in Acetaminophen Hepatotoxicity. J. Pharmacol. Exp. Ther. 2005, 315, 879–887. [Google Scholar] [CrossRef]
- El-Hassan, H.; Anwar, K.; Macanas-Pirard, P.; Crabtree, M.; Chow, S.C.; Johnson, V.L.; Lee, P.C.; Hinton, R.H.; Price, S.C.; Kass, G.E.N. Involvement of Mitochondria in Acetaminophen-Induced Apoptosis and Hepatic Injury: Roles of Cytochrome c, Bax, Bid, and Caspases. Toxicol. Appl. Pharmacol. 2003, 191, 118–129. [Google Scholar] [CrossRef]
- Jaeschke, H.; Cover, C.; Bajt, M.L. Role of Caspases in Acetaminophen-Induced Liver Injury. Life Sci. 2006, 78, 1670–1676. [Google Scholar] [CrossRef]
- Hu, B.; Colletti, L.M. CXC Receptor-2 Knockout Genotype Increases X-Linked Inhibitor of Apoptosis Protein and Protects Mice from Acetaminophen Hepatotoxicity. Hepatology 2010, 52, 691–702. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Toxicity: Novel Insights into Mechanisms and Future Perspectives. Gene Expr. 2018, 38, 19–30. [Google Scholar] [CrossRef]
- Jaeschke, H.; Duan, L.; Akakpo, J.Y.; Farhood, A.; Ramachandran, A. The Role of Apoptosis in Acetaminophen Hepatotoxicity. Food Chem. Toxicol. 2018, 709–718. [Google Scholar] [CrossRef]
- Volkmann, X.; Anstaett, M.; Hadem, J.; Stiefel, P.; Bahr, M.J.; Lehner, F.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Caspase Activation Is Associated with Spontaneous Recovery from Acute Liver Failure. Hepatology 2008, 47, 1624–1633. [Google Scholar] [CrossRef]
- Leist, M.; Single, B.; Castoldi, A.F.; Kühnle, S.; Nicotera, P. Intracellular Adenosine Triphosphate (ATP) Concentration: A Switch in the Decision between Apoptosis and Necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef]
- Jaeschke, H.; Williams, C.D.; Farhood, A. No Evidence for Caspase-Dependent Apoptosis in Acetaminophen Hepatotoxicity. Hepatology 2011, 718–719. [Google Scholar] [CrossRef]
- Adams, M.L.; Pierce, R.H.; Vail, M.E.; White, C.C.; Tonge, R.P.; Kavanagh, T.J.; Fausto, N.; Nelson, S.D.; Bruschi, S.A. Enhanced Acetaminophen Hepatotoxicity in Transgenic Mice Overexpressing BCL-2. Mol. Pharmacol. 2001, 60, 907–915. [Google Scholar] [CrossRef]
- Takahashi, N.; Duprez, L.; Grootjans, S.; Cauwels, A.; Nerinckx, W.; Duhadaway, J.B.; Goossens, V.; Roelandt, R.; Van Hauwermeiren, F.; Libert, C.; et al. Necrostatin-1 Analogues: Critical Issues on the Specificity, Activity and in Vivo Use in Experimental Disease Models. Cell Death Dis. 2012, 3. [Google Scholar] [CrossRef]
- Park, Y.; Smith, R.D.; Combs, A.B.; Kehrer, J.P. Prevention of Acetaminophen-Induced Hepatotoxicity by Dimethyl Sulfoxide. Toxicology 1988, 52, 165–175. [Google Scholar] [CrossRef]
- Deutsch, M.; Graffeo, C.S.; Rokosh, R.; Pansari, M.; Ochi, A.; Levie, E.M.; Van Heerden, E.; Tippens, D.M.; Greco, S.; Barilla, R.; et al. Divergent Effects of RIP1 or RIP3 Blockade in Murine Models of Acute Liver Injury. Cell Death Dis. 2015, 6. [Google Scholar] [CrossRef]
- Ni, H.M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.X. Activation of Autophagy Protects against Acetaminophen-Induced Hepatotoxicity. Hepatology 2012, 55, 222–232. [Google Scholar] [CrossRef]
- Hu, C.; Zhao, L.; Shen, M.; Wu, Z.; Li, L. Autophagy Regulation Is an Effective Strategy to Improve the Prognosis of Chemically Induced Acute Liver Injury Based on Experimental Studies. J. Cell. Mol. Med. 2020, 8315–8325. [Google Scholar] [CrossRef]
- Lin, Z.; Wu, F.; Lin, S.; Pan, X.; Jin, L.; Lu, T.; Shi, L.; Wang, Y.; Xu, A.; Li, X. Adiponectin Protects against Acetaminophen-Induced Mitochondrial Dysfunction and Acute Liver Injury by Promoting Autophagy in Mice. J. Hepatol. 2014, 61, 825–831. [Google Scholar] [CrossRef]
- Baulies, A.; Ribas, V.; Núñez, S.; Torres, S.; Alarcón-Vila, C.; Martínez, L.; Suda, J.; Ybanez, M.D.; Kaplowitz, N.; García-Ruiz, C.; et al. Lysosomal Cholesterol Accumulation Sensitizes to Acetaminophen Hepatotoxicity by Impairing Mitophagy. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Rashed, M.S.; Myers, T.G.; Nelson, S.D. Hepatic Protein Arylation, Glutathione Depletion, and Metabolite Profiles of Acetaminophen and a Non-Hepatotoxic Regioisomer, 3′-Hydroxyacetanilide, in the Mouse. Drug Metab. Dispos. 1990, 18, 765–770. [Google Scholar]
- Tirmenstein, M.A.; Nelson, S.D. Subcellular Binding and Effects on Calcium Homeostasis Produced by Acetaminophen and a Nonhepatotoxic Regioisomer, 3′-Hydroxyacetanilide, in Mouse Liver. J. Biol. Chem. 1989, 264, 9814–9819. [Google Scholar]
- Ni, H.M.; Williams, J.A.; Jaeschke, H.; Ding, W.X. Zonated Induction of Autophagy and Mitochondrial Spheroids Limits Acetaminophen-Induced Necrosis in the Liver. Redox Biol. 2013, 427–432. [Google Scholar] [CrossRef]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-Induced Hepatotoxicity in Mice Is Dependent on Tlr9 and the Nalp3 Inflammasome. J. Clin. Investig. 2009, 119, 305–314. [Google Scholar] [CrossRef]
- Williams, C.D.; Farhood, A.; Jaeschke, H. Role of Caspase-1 and Interleukin-1β in Acetaminophen-Induced Hepatic Inflammation and Liver Injury. Toxicol. Appl. Pharmacol. 2010, 247, 169–178. [Google Scholar] [CrossRef]
- Williams, C.D.; Antoine, D.J.; Shaw, P.J.; Benson, C.; Farhood, A.; Williams, D.P.; Kanneganti, T.D.; Park, B.K.; Jaeschke, H. Role of the Nalp3 Inflammasome in Acetaminophen-Induced Sterile Inflammation and Liver Injury. Toxicol. Appl. Pharmacol. 2011, 252, 289–297. [Google Scholar] [CrossRef]
- Zhang, C.; Feng, J.; Du, J.; Zhuo, Z.; Yang, S.; Zhang, W.; Wang, W.; Zhang, S.; Iwakura, Y.; Meng, G.; et al. Macrophage-Derived IL-1α Promotes Sterile Inflammation in a Mouse Model of Acetaminophen Hepatotoxicity. Cell. Mol. Immunol. 2018, 15, 973–982. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ramachandran, A. Mechanisms and Pathophysiological Significance of Sterile Inflammation during Acetaminophen Hepatotoxicity. Food Chem. Toxicol. 2020, 138. [Google Scholar] [CrossRef]
- Yang, C.; Sun, P.; Deng, M.; Loughran, P.; Li, W.; Yi, Z.; Li, S.; Zhang, X.; Fan, J.; Billiar, T.R.; et al. Gasdermin D Protects against Noninfectious Liver Injury by Regulating Apoptosis and Necroptosis. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- C57BL/6N-Gsdmd<em4Fcw>/J. Available online: https://www.jax.org/strain/032410 (accessed on 3 November 2020).
- Bourdi, M.; Davies, J.S.; Pohl, L.R. Mispairing C57BL/6 Substrains of Genetically Engineered Mice and Wild-Type Controls Can Lead to Confounding Results as It Did in Studies of JNK2 in Acetaminophen and Concanavalin a Liver Injury. Chem. Res. Toxicol. 2011, 24, 794–796. [Google Scholar] [CrossRef]
- Krenkel, O.; Mossanen, J.C.; Tacke, F. Immune Mechanisms in Acetaminophen-Induced Acute Liver Failure. Hepatobiliary Surg. Nutr. 2014, 3, 331–33143. [Google Scholar] [CrossRef]
- Wang, M.; Liu, C.Y.; Wang, T.; Yu, H.M.; Ouyang, S.H.; Wu, Y.P.; Gong, H.B.; Ma, X.H.; Jiao, G.L.; Fu, L.L.; et al. (+)-Clausenamide Protects against Drug-Induced Liver Injury by Inhibiting Hepatocyte Ferroptosis. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Lőrincz, T.; Jemnitz, K.; Kardon, T.; Mandl, J.; Szarka, A. Ferroptosis Is Involved in Acetaminophen Induced Cell Death. Pathol. Oncol. Res. 2015, 21, 1115–1121. [Google Scholar] [CrossRef]
- Schnellmann, J.G.; Pumford, N.R.; Kusewitt, D.F.; Bucci, T.J.; Hinson, J.A. Deferoxamine Delays the Development of the Hepatotoxicity of Acetaminophen in Mice. Toxicol. Lett. 1999, 106, 79–88. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis Driven by Radical Oxidation of N-6 Polyunsaturated Fatty Acids Mediates Acetaminophen-Induced Acute Liver Failure. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Takahashi, M. Role of Ferroptosis in Acetaminophen-Induced Hepatotoxicity. Arch. Toxicol. 2020, 1769–1770. [Google Scholar] [CrossRef]
- Czaja, A.J. Targeting Apoptosis in Autoimmune Hepatitis. Dig. Dis. Sci. 2014, 59, 2890–2904. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Hamesch, K.; Weiskirchen, R.; Tacke, F. The Concanavalin a Model of Acute Hepatitis in Mice. Lab. Anim. 2015, 49 (Suppl. 1), 12–20. [Google Scholar] [CrossRef] [PubMed]
- Suda, J.; Dara, L.; Yang, L.; Aghajan, M.; Song, Y.; Kaplowitz, N.; Liu, Z.-X. Knockdown of RIPK1 Markedly Exacerbates Murine Immune-Mediated Liver Injury through Massive Apoptosis of Hepatocytes, Independent of Necroptosis and Inhibition of NF-ΚB. J. Immunol. 2016, 197, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- Trautwein, C.; Rakemann, T.; Brenner, D.A.; Streetz, K.; Licato, L.; Manns, M.P.; Tiegs, G. Concanavalin A-Induced Liver Cell Damage: Activation of Intracellular Pathways Triggered by Tumor Necrosis Factor in Mice. Gastroenterology 1998, 114, 1035–1045. [Google Scholar] [CrossRef]
- Faletti, L.; Peintner, L.; Neumann, S.; Sandler, S.; Grabinger, T.; Mac Nelly, S.; Merfort, I.; Huang, C.H.; Tschaharganeh, D.; Kang, T.W.; et al. TNFα Sensitizes Hepatocytes to FasL-Induced Apoptosis by NFκB-Mediated Fas Upregulation. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.K.; Furtwaengler, A.; Nepomuceno, R.R.; Martinez, O.M.; Krams, S.M. Apoptotic Pathways in Primary Biliary Cirrhosis and Autoimmune Hepatitis. Liver 2001, 21, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Künstle, G.; Hentze, H.; Germann, P.G.; Tiegs, G.; Meergans, T.; Wendel, A. Concanavalin a Hepatotoxicity in Mice: Tumor Necrosis Factor-Mediated Organ Failure Independent of Caspase-3-like Protease Activation. Hepatology 1999, 30, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL Induces Necroptosis Involving RIPK1/RIPK3-Dependent PARP-1 Activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.I.; Piquet-Pellorce, C.; Filliol, A.; L’Helgoualc’h, A.; Lucas-Clerc, C.; Jouan-Lanhouet, S.; Dimanche-Boitrel, M.T.; Samson, M. The Chemical Inhibitors of Cellular Death, PJ34 and Necrostatin-1, down-Regulate IL-33 Expression in Liver. J. Mol. Med. 2015, 93, 867–878. [Google Scholar] [CrossRef]
- Zhou, Y.; Dai, W.; Lin, C.; Wang, F.; He, L.; Shen, M.; Chen, P.; Wang, C.; Lu, J.; Xu, L.; et al. Protective Effects of Necrostatin-1 against Concanavalin A-Induced Acute Hepatic Injury in Mice. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef]
- Dara, L.; Liu, Z.X.; Kaplowitz, N. A Murder Mystery in the Liver: Who Done It and How? J. Clin. Investig. 2016, 4068–4071. [Google Scholar] [CrossRef]
- Hamon, A.; Piquet-Pellorce, C.; Dimanche-Boitrel, M.T.; Samson, M.; Le Seyec, J. Intrahepatocytic Necroptosis Is Dispensable for Hepatocyte Death in Murine Immune-Mediated Hepatitis. J. Hepatol. 2020, 699–701. [Google Scholar] [CrossRef]
- Filliol, A.; Piquet-Pellorce, C.; Raguénès-Nicol, C.; Dion, S.; Farooq, M.; Lucas-Clerc, C.; Vandenabeele, P.; Bertrand, M.J.M.; Le Seyec, J.; Samson, M. RIPK1 Protects Hepatocytes from Kupffer Cells-Mediated TNF-Induced Apoptosis in Mouse Models of PAMP-Induced Hepatitis. J. Hepatol. 2017, 66, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Filliol, A.; Piquet-Pellorce, C.; Le Seyec, J.; Farooq, M.; Genet, V.; Lucas-Clerc, C.; Bertin, J.; Gough, P.J.; Dimanche-Boitrel, M.T.; Vandenabeele, P.; et al. RIPK1 Protects from TNF-α-Mediated Liver Damage during Hepatitis. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Bang, B.R.; Han, K.H.; Hong, L.; Shim, E.J.; Ma, J.; Lerner, R.A.; Otsuka, M. Regulation of NKT Cell-Mediated Immune Responses to Tumours and Liver Inflammation by Mitochondrial PGAM5-Drp1 Signalling. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Cohen, D.E.; El-Omar, E.M.; Jalan, R.; Low, M.J.; Nathanson, M.H.; Peek, R.M.; Turner, J.R. Not All Mice Are the Same: Standardization of Animal Research Data Presentation. Gastroenterology 2016, 1503–1504. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Zhang, X.; Wang, S.; Li, Y.; Fan, J.; Chen, W.; Zai, W.; Wang, S.; Wang, Y.; Chen, M.; et al. NOD-like Receptor Protein 3 Inflammasome-Dependent IL-1β Accelerated ConA-Induced Hepatitis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Wang, J.; Ren, H.; Yuan, X.; Ma, H.; Shi, X.; Ding, Y. Interleukin-10 Secreted by Mesenchymal Stem Cells Attenuates Acute Liver Failure through Inhibiting Pyroptosis. Hepatol. Res. 2018, 48, E194–E202. [Google Scholar] [CrossRef]
- Lan, P.; Fan, Y.; Zhao, Y.; Lou, X.; Monsour, H.P.; Zhang, X.; Choi, Y.; Dou, Y.; Ishii, N.; Ghobrial, R.M.; et al. TNF Superfamily Receptor OX40 Triggers Invariant NKT Cell Pyroptosis and Liver Injury. J. Clin. Investig. 2017, 127, 2222–2234. [Google Scholar] [CrossRef]
- Zeng, T.; Deng, G.; Zhong, W.; Gao, Z.; Ma, S.; Mo, C.; Li, Y.; Huang, S.; Zhou, C.; Lai, Y.; et al. Indoleamine 2,3-Dioxygenase 1enhanceshepatocytes Ferroptosis in Acute Immune Hepatitis Associated with Excess Nitrative Stress. Free Radic. Biol. Med. 2020, 152, 668–679. [Google Scholar] [CrossRef]
- Deng, G.; Li, Y.; Ma, S.; Gao, Z.; Zeng, T.; Chen, L.; Ye, H.; Yang, M.; Shi, H.; Yao, X.; et al. Caveolin-1 Dictates Ferroptosis in the Execution of Acute Immune-Mediated Hepatic Damage by Attenuating Nitrogen Stress. Free Radic. Biol. Med. 2020, 148, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Faubion, W.A.; Guicciardi, M.E.; Miyoshi, H.; Bronk, S.F.; Roberts, P.J.; Svingen, P.A.; Kaufmann, S.H.; Gores, G.J. Toxic Bile Salts Induce Rodent Hepatocyte Apoptosis via Direct Activation of Fas. J. Clin. Investig. 1999, 103, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ozaki, S.; Gershwin, M.E.; Nakanuma, Y. Enhanced Apoptosis Relates to Bile Duct Loss in Primary Biliary Cirrhosis. Hepatology 1997, 26, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Harada, K.; Hiramatsu, K.; Tsuneyama, K.; Kaneko, S.; Kobayashi, K.; Nakanuma, Y. Fas Ligand Expressing Mononuclear Cells around Intrahepatic Bile Ducts Co-Express CD68 in Primary Biliary Cirrhosis. Liver 2000, 20, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Rust, C.; Roberts, P.J.; Burgart, L.J.; Gores, G.J. Hepatocyte Apoptosis after Bile Duct Ligation in the Mouse Involves Fas. Gastroenterology 1999, 117, 669–677. [Google Scholar] [CrossRef]
- Takeda, K.; Kojima, Y.; Ikejima, K.; Harada, K.; Yamashina, S.; Okumura, K.; Aoyama, T.; Frese, S.; Ikeda, H.; Haynes, N.M.; et al. Death Receptor 5 Mediated-Apoptosis Contributes to Cholestatic Liver Disease. Proc. Natl. Acad. Sci. USA 2008, 105, 10895–10900. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Bile Acid-Mediated Hepatocyte Apoptosis and Cholestatic Liver Disease. Dig. Liver Dis. 2002, 34, 387–392. [Google Scholar] [CrossRef]
- Schoemaker, M.H.; Conde De La Rosa, L.; Buist-Homan, M.; Vrenken, T.E.; Havinga, R.; Poelstra, K.; Haisma, H.J.; Jansen, P.L.M.; Moshage, H. Tauroursodeoxycholic Acid Protects Rat Hepatocytes from Bile Acid-Induced Apoptosis via Activation of Survival Pathways. Hepatology 2004, 39, 1563–1573. [Google Scholar] [CrossRef]
- Azad, A.I.; Krishnan, A.; Troop, L.; Li, Y.; Katsumi, T.; Pavelko, K.; Kostallari, E.; Guicciardi, M.E.; Gores, G.J. Targeted Apoptosis of Ductular Reactive Cells Reduces Hepatic Fibrosis in a Mouse Model of Cholestasis. Hepatology 2020, 72, 1013–1028. [Google Scholar] [CrossRef]
- Cubero, F.J.; Peng, J.; Liao, L.; Su, H.; Zhao, G.; Eugenio Zoubek, M.; Macías-Rodríguez, R.; Ruiz-Margain, A.; Reißing, J.; Zimmermann, H.W.; et al. Inactivation of Caspase 8 in Liver Parenchymal Cells Confers Protection against Murine Obstructive Cholestasis. J. Hepatol. 2018, 69, 1326–1334. [Google Scholar] [CrossRef]
- Canbay, A.; Feldstein, A.; Baskin-Bey, E.; Bronk, S.F.; Gores, G.J. The Caspase Inhibitor IDN-6556 Attenuates Hepatic Injury and Fibrosis in the Bile Duct Ligated Mouse. J. Pharmacol. Exp. Ther. 2004, 308, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Koyama, Y.; Wree, A.; Johnson, C.D.; Nakamura, R.; Povero, D.; Kneiber, D.; Tameda, M.; Contreras, P.; Spada, A.; et al. Emricasan, a Pan-Caspase Inhibitor, Improves Survival and Portal Hypertension in a Murine Model of Common Bile-Duct Ligation. J. Mol. Med. 2018, 96, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Dorko, K.; Antoine, D.J.; Clarke, J.I.; Gholami, P.; Li, F.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Fan, F.; et al. Bile Acid-Induced Necrosis in Primary Human Hepatocytes and in Patients with Obstructive Cholestasis. Toxicol. Appl. Pharmacol. 2015, 283, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Inflammation and Cell Death during Cholestasis: The Evolving Role of Bile Acids. Gene Expr. J. Liver Res. 2019, 215–228. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell Death and Cell Death Responses in Liver Disease: Mechanisms and Clinical Relevance. Gastroenterology 2014, 765–783.e4. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Simão, A.L.; Ofengeim, D.; Carvalho, T.; Amaral, J.D.; Gaspar, M.M.; Cortez-Pinto, H.; Castro, R.E.; Yuan, J.; et al. Activation of Necroptosis in Human and Experimental Cholestasis. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Simão, A.L.; Gaspar, M.M.; Carvalho, T.; Borralho, P.; Bañales, J.M.; Castro, R.E.; Rodrigues, C.M.P. MiRNA-21 Ablation Protects against Liver Injury and Necroptosis in Cholestasis. Cell Death Differ. 2018, 25, 857–872. [Google Scholar] [CrossRef]
- Maroni, L.; Agostinelli, L.; Saccomanno, S.; Pinto, C.; Giordano, D.M.; Rychlicki, C.; De Minicis, S.; Trozzi, L.; Banales, J.M.; Melum, E.; et al. Nlrp3 Activation Induces Il-18 Synthesis and Affects the Epithelial Barrier Function in Reactive Cholangiocytes. Am. J. Pathol. 2017, 187, 366–376. [Google Scholar] [CrossRef]
- Gong, Z.; Zhou, J.; Zhao, S.; Tian, C.; Wang, P.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Chenodeoxycholic Acid Activates NLRP3 Inflammasome and Contributes to Cholestatic Liver Fibrosis. Oncotarget 2016, 7, 83951–83963. [Google Scholar] [CrossRef]
- González, M.I.; Vannan, D.; Eksteen, B.; Reyes, J.L. NLRP3 Receptor Contributes to Protection against Experimental Antigen-Mediated Cholangitis. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Skeldon, A.; Saleh, M. The Inflammasomes: Molecular Effectors of Host Resistance against Bacterial, Viral, Parasitic, and Fungal Infections. Front. Microbiol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Schneider, K.M.; Galvez, E.J.C.; Frissen, M.; Marschall, H.U.; Su, H.; Hatting, M.; Wahlström, A.; Haybaeck, J.; Puchas, P.; et al. Intestinal Dysbiosis Augments Liver Disease Progression via NLRP3 in a Murine Model of Primary Sclerosing Cholangitis. Gut 2019, 68, 1477–1492. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, L.; Barikbin, R.; Schiller, B.; Ravichandran, G.; Krech, T.; Neumann, K.; Sass, G.; Tiegs, G. Deletion of Tumour Necrosis Factor α Receptor 1 Elicits an Increased TH17 Immune Response in the Chronically Inflamed Liver. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.F.; Zhang, Q.; Ding, C.J.; Sun, H.Y.; Che, Y.; Huang, H.; Wang, Y.; Wu, J.W.; Hao, H.P.; Cao, L.J. Gasdermin E-Derived Caspase-3 Inhibitors Effectively Protect Mice from Acute Hepatic Failure. Acta Pharmacol. Sin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.Y.N.; Xie, X.; Lai, M.M.C.; Wu, P.C. Apoptosis and Viral Hepatitis. Semin. Liver Dis. 1998, 18, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Zavos, C.; Chatzopoulos, D. Apoptosis in Hepatitis C. J. Viral Hepat. 2003, 335–342. [Google Scholar] [CrossRef]
- Ehrmann, J.; Galuszková, D.; Ehrmann, J.; Krè, I.; Jezdinská, V.; Vojtì Ek, B.; Murray, P.G.; Koláø, Z. Apoptosis-Related Proteins, BCL-2, BAX, FAS, FAS-L and PCNA in Liver Biopsies of Patients with Chronic Hepatitis B Virus Infection. Pathol. Oncol. Res. 2000, 6, 130–135. [Google Scholar] [CrossRef]
- Mundt, B.; Kühnel, F.; Zender, L.; Paul, Y.; Tillmann, H.; Trautwein, C.; Manns, M.P.; Kubicka, S. Involvement of TRAIL and Its Receptors in Viral Hepatitis. FASEB J. 2003, 17, 94–96. [Google Scholar] [CrossRef]
- Song, L.H.; Binh, V.Q.; Duy, D.N.; Bock, T.C.; Kremsner, P.G.; Luty, A.J.F.; Mavoungou, E. Variations in the Serum Concentrations of Soluble Fas and Soluble Fas Ligand in Vietnamese Patients Infected with Hepatitis B Virus. J. Med. Virol. 2004, 73, 244–249. [Google Scholar] [CrossRef]
- Streetz, K.; Leifeld, L.; Grundmann, D.; Ramakers, J.; Eckert, K.; Spengler, U.; Brenner, D.; Manns, M.; Trautwein, C. Tumor Necrosis Factor α in the Pathogenesis of Human and Murine Fulminant Hepatic Failure. Gastroenterology 2000, 119, 446–460. [Google Scholar] [CrossRef]
- Mita, E.; Hayashi, N.; Iio, S.; Takehara, T.; Hijioka, T.; Kasahara, A.; Fusamoto, H.; Kamada, T. Role of Fas Ligand in Apoptosis Induced by Hepatitis C Virus Infection. Biochem. Biophys. Res. Commun. 1994, 204, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.X.; Zhu, Y.F.; Zhang, L.X.; He, H.T.; Wang, X.S.; Zhang, L. In Situ Investigation of Fas/FasL Expression in Chronic Hepatitis B Infection and Related Liver Diseases. J. Viral Hepat. 1997, 4, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Galle, P.R.; Hofmann, W.J.; Walczak, H.; Schaller, H.; Otto, G.; Stremmel, W.; Krammer, P.H.; Runkel, L. Involvement of the CD95 (APO-1/Fas) Receptor and Ligand in Liver Damage. J. Exp. Med. 1995, 182, 1223–1230. [Google Scholar] [CrossRef]
- Asselah, T.; Bièche, I.; Mansouri, A.; Laurendeau, I.; Cazals-Hatem, D.; Feldmann, G.; Bedossa, P.; Paradis, V.; Martinot-Peignoux, M.; Lebrec, D.; et al. In Vivo Hepatic Endoplasmic Reticulum Stress in Patients with Chronic Hepatitis C. J. Pathol. 2010, 221, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Lügering, A.; Poremba, C.; Lügering, N.; Held, J.; Domschke, W.; Schulze-Osthoff, K. Caspase Activation Correlates with the Degree of Inflammatory Liver Injury in Chronic Hepatitis C Virus Infection. Hepatology 2001, 34, 758–767. [Google Scholar] [CrossRef]
- Pockros, P.J.; Schiff, E.R.; Shiffman, M.L.; McHutchison, J.G.; Gish, R.G.; Afdhal, N.H.; Makhviladze, M.; Huyghe, M.; Hecht, D.; Oltersdorf, T.; et al. Oral IDN-6556, an Antiapoptotic Caspase Inhibitor, May Lower Aminotransferase Activity in Patients with Chronic Hepatitis C. Hepatology 2007, 46, 324–329. [Google Scholar] [CrossRef]
- Shiffman, M.L.; Pockros, P.; McHutchison, J.G.; Schiff, E.R.; Morris, M.; Burgess, G. Clinical Trial: The Efficacy and Safety of Oral PF-03491390, a Pancaspase Inhibitor—A Randomized Placebo-Controlled Study in Patients with Chronic Hepatitis C. Aliment. Pharmacol. Ther. 2010, 31, 969–978. [Google Scholar] [CrossRef]
- Toubi, E.; Kessel, A.; Goldstein, L.; Slobodin, G.; Sabo, E.; Shmuel, Z.; Zuckerman, E. Enhanced Peripheral T-Cell Apoptosis in Chronic Hepatitis C Virus Infection: Association with Liver Disease Severity. J. Hepatol. 2001, 35, 774–780. [Google Scholar] [CrossRef]
- Chiou, H.L.; Hsieh, Y.S.; Hsieh, M.R.; Chen, T.Y. HCV E2 May Induce Apoptosis of Huh-7 Cells via a Mitochondrial-Related Caspase Pathway. Biochem. Biophys. Res. Commun. 2006, 345, 453–458. [Google Scholar] [CrossRef]
- Ciccaglione, A.R.; Marcantonio, C.; Costantino, A.; Equestre, M.; Rapicetta, M. Expression of HCV E1 Protein in Baculovirus-Infected Cells: Effects on Cell Viability and Apoptosis Induction. Intervirology 2003, 46, 121–126. [Google Scholar] [CrossRef]
- Kato, N.; Yoshida, H.; Kioko Ono-Nita, S.; Kato, J.; Goto, T.; Otsuka, M.; Lan, K.H.; Matsushima, K.; Shiratori, Y.; Omata, M. Activation of Intracellular Signaling by Hepatitis B and C Viruses: C-Viral Core Is the Most Potent Signal Inducer. Hepatology 2000, 32, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.M. Hepatitis Viruses and Signal Transduction: True to the Core? Hepatology 2000, 32, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Benedict Yen, T.S. Nuclear Factor ΚB and Hepatitis C—Is There a Connection? Hepatology 2000, 785–787. [Google Scholar] [CrossRef]
- Jahan, S.; Ashfaq, U.A.; Khaliq, S.; Samreen, B.; Afzal, N. Dual Behavior of HCV Core Gene in Regulation of Apoptosis Is Important in Progression of HCC. Infect. Genet. Evol. 2012, 236–239. [Google Scholar] [CrossRef]
- Muratori, L.; Gibellini, D. A New Route to Apoptosis in Hepatitis C Virus Infection. J. Hepatol. 2001, 814–815. [Google Scholar] [CrossRef]
- Farooq, M.; Filliol, A.; Simoes Eugénio, M.; Piquet-Pellorce, C.; Dion, S.; Raguenes-Nicol, C.; Jan, A.; Dimanche-Boitrel, M.T.; Le Seyec, J.; Samson, M. Depletion of RIPK1 in Hepatocytes Exacerbates Liver Damage in Fulminant Viral Hepatitis. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Chen, L.; Cao, Z.; Yan, L.; Ding, Y.; Shen, X.; Liu, K.; Xiang, X.; Xie, Q.; Zhu, C.; Bao, S.; et al. Circulating Receptor-Interacting Protein Kinase 3 Are Increased in HBV Patients with Acute-on-Chronic Liver Failure and Are Associated with Clinical Outcome. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Han, L.; Teng, Y.; Fan, Y.; Gao, S.; Li, F.; Wang, K. Receptor-Interacting Protein Kinase 3 (RIPK3) MRNA Levels Are Elevated in Blood Mononuclear Cells of Patients with Poor Prognosis of Acute-on-Chronic Hepatitis B Liver Failure. Tohoku J. Exp. Med. 2019, 247, 237–245. [Google Scholar] [CrossRef]
- Kofahi, H.M.; Taylor, N.G.A.; Hirasawa, K.; Grant, M.D.; Russell, R.S. Hepatitis C Virus Infection of Cultured Human Hepatoma Cells Causes Apoptosis and Pyroptosis in Both Infected and Bystander Cells. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Serti, E.; Werner, J.M.; Chattergoon, M.; Cox, A.L.; Lohmann, V.; Rehermann, B. Monocytes Activate Natural Killer Cells via Inflammasome-Induced Interleukin 18 in Response to Hepatitis C Virus Replication. Gastroenterology 2014, 147. [Google Scholar] [CrossRef]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.Y.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β Production through the NLRP3 Inflammasome by Hepatic Macrophages Links Hepatitis C Virus Infection with Liver Inflammation and Disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef]
- Manigold, T.; Böcker, U.; Chen, J.; Gundt, J.; Traber, P.; Singer, M.V.; Rossol, S. Hepatitis B Core Antigen Is a Potent Inductor of Interleukin-18 in Peripheral Blood Mononuclear Cells of Healthy Controls and Patients with Hepatitis B Infection. J. Med. Virol. 2003, 71, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lan, P.; Hou, X.; Han, Q.; Lu, N.; Li, T.; Jiao, C.; Zhang, J.; Zhang, C.; Tian, Z. HBV Inhibits LPS-Induced NLRP3 Inflammasome Activation and IL-1β Production via Suppressing the NF-ΚB Pathway and ROS Production. J. Hepatol. 2017, 66, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, X.K.; Cheng, Y.J.; Zhang, Q.; Wu, J.; Lu, S.; Zhang, W.; Liu, Y.; Zhou, M.Y.; Wang, Y.; et al. Gasdermin D-Mediated Hepatocyte Pyroptosis Expands Inflammator Responses That Aggravate Acute Liver Failure by Upregulating Monocyte Chemotactic Protein 1/CC Chemokine Receptor-2 to Recruit Macrophages. World J. Gastroenterol. 2019, 25, 6527–6540. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Kanneganti, T.D.; van Damme, P.; vanden Berghe, T.; Vanoverberghe, I.; Vandekerckhove, J.; Vandenabeele, P.; Gevaert, K.; Núñe-az, G. Targeted Peptidecentric Proteomics Reveals Caspase-7 as a Substrate of the Caspase-1 Inflammasomes. Mol. Cell. Proteom. 2008, 7, 2350–2363. [Google Scholar] [CrossRef] [PubMed]
- Samir, P.; Malireddi, R.K.S.; Kanneganti, T.D. The PANoptosome: A Deadly Protein Complex Driving Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687.e13. [Google Scholar] [CrossRef]
- Zheng, M.; Kanneganti, T.D. The Regulation of the ZBP1-NLRP3 Inflammasome and Its Implications in Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Immunol. Rev. 2020, 297, 26–38. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Apoptosis | MPT-Mediated Necrosis | Necroptosis | Autophagy | Pyroptosis | Ferroptosis | |
|---|---|---|---|---|---|---|
| Alcoholic Liver Diseases (ALD) | -Extensively studied in human and murine models. -ROS production and CHOP-dependent apoptosis [61,62,63,64]. -FAS, FASL, and TNF well characterized [66,67]. -Casp8Δhepa mice had less steatosis and cell death [72]. -Pan-caspase inhibitor protects and no switch to necroptosis [72,73]. -BID KO mice protected from apoptosis but not inflammation [74] | -Higher RIPK3, but the source is not clear [30,76]. -RIPK3 global KO mice treated with alcohol had less steatosis, inflammation, and liver injury [76]. -Global RIPK3 KO less transaminases but not difference in inflammation and neutrophil infiltration [77]. -Daily 7-Nec1 reduced inflammation but did not prevent liver injury [77]. -No transcriptional induction of RIPK3 but proteasome inhibition resulted in increase [77]. -No induction of RIPK3 in Gao-Binge model. -Further studies needed to explore the role of pMLKL. Unconfirmed. | -Interplay of autophagy and apoptosis is highly likely in pathogenesis. -Ethanol induces autophagy through ROS and activation of AAMPK and inhibition of mTORC1 [79,82]. -Dose and duration of alcohol can influence autophagy with acute and lower dose models, promoting and higher doses inhibiting some components and preventing autophagy [79]. -Parkin-mediated mitophagy and CMA were shown to be protective [85,87]. | -More prominent in inflammatory cells than hepatocytes [43,89,90]. -NLRP3, CASP-1, and ASC upregulated with ethanol [91]. -Alcohol induces CASP-1, NLRP3, IL-1. CASP-1 KO, and IL-1 antagonist protection [92]. -Elevated CASP11/4 activate GSDMD, more prominent in macrophages [96]. | -Several studies suggest, however, that GPX4 may not be the initiator [57]. -SIRT1 deficiency may be protective [98]. -Lipin1 overexpression (lipid metabolism) worsens injury, increased iron accumulation, impaired ferroptotic gene expression, GPX4 not altered [57]. | |
| Non-Alcoholic Fatty Liver Diseases (NASH/NAFLD) | -Patients with NASH have positive detection of CASP3/7 and TUNEL [99]. -CASP3/8 KO mice on MCD diet protected from apoptosis [105,106]. -Emricasan protects mice from HFD [107]. Caspase inhibitors not successful in human clinical trials [111,112,113]. -Conflicting results of pan-caspase inhibition in different diets [108,109]. -CASP6 interplay with CASP3/7 leads to persistent apoptosis involving the AMPK pathway [114]. -Both intrinsic (via lipotoxocity and organelle stress) and extrinsic (via cell surface receptor) pathways contribute to injury [117,118,119,120]. | -CypD KO mice have lower mitochondrial stress, steatosis, and TG on HFD [16]. -Cyclophilin inhibitor reduced inflammation, steatosis, and ballooning in mice on HFD [121]. | -Increased RIPK3 in humans with NASH [27,122,123]. -RIPK3 induction with MCD diet further exacerbated with CASP8 deletion [123]. -Increased RIPK3 and pMLKL in HFD mice [124]. -RIPK3 KO mice shown to be glucose intolerant on chow diet [124,125] and had worse injury [124]. -RIPK3 deficiency leads to less inflammation, steatosis, and fibrosis, in CD-HFD and MCD diet [126]. -MLKL KO mice on HFD had increased MLKL, RIPK1, and pMLKL. [127]. -RIPK1 inhibitor prevented inflammation and decreased fibrosis in HFD mice [128]. -Autophagic markers were abrogated by MLKL KO, suggesting that MLKL may play a role in autophagic flux [129]. -Occurrence of necroptosis under basal conditions without pan-caspase inhibition is questionable. | -In NASH inflammasome activation is triggered by lipotoxicity, organelle stress, and hepatocyte death [133]. -Inflammasome-mediated dysbiosis has been shown to regulate NAFLD and obesity in MCD diet [134]. -CASP1 KO, ASC KO, or NLRP3 KO mice on MCD diet showed increased transaminases, worsening inflammation, and steatosis compared to WT controls. More injury was evident in IL-18 KO mice but not in IL-1R KO mice [134]. -NLRP3 inhibitor, in foz/foz mice fed HFD and WT mice with MCD diet improved inflammation, but there was no effect on steatosis [135]. -GSDMD expression was increased in db/db mice on MCD diet, and GSDMD KO mice on HFD had attenuated steatosis, improved ALT and TG levels, and less fibrosis [47]. | -Vit E and phlebotomy improves liver chemistry in NAFLD/NASH [138,139] -GPX4 induction has been observed following Western and MCD diets [140]. -GPX4 activator administration promoted cell survival and improved AST/ALT [140]. -Trolox (Vit E analogue and antioxidant) led to less cell death [141]. -Ferrostatin treatment reversed mitochondrial morphological changes observed with MCD diet [142]. -Distinguishing ROS leading to MPT and necrosis from ferroptosis is still difficult. | |
| Acetaminophen (APAP) toxicity | -APAP cell death is not morphologically apoptotic. -Translocation of tBID to mitochondria during APAP does not mediate MOMP [170,174]. -Caspase inhibitors do not protect from APAP toxicity [171,174]. -No evidence MOMP in APAP toxicity [155,161]. -TUNEL staining is not specific to apoptosis and the pattern of the TUNEL stain in APAP is cytoplasmic not nuclear [177]. -Lack of PUMA protects against hepatocyte death but through preventing necrosis not apoptosis [167]. RIPK1 and JNK inhibition and siRNA knockdown abrogates PUMA’s upregulation [167]. | -CypD-deficient mice are protected against APAP hepatotoxicity and cell death [15]. -Cyclosporin protects from APAP [163,164]. -Inhibition, knockdown or KO of MAPK proteins, including MLKL3, ASK1, MKK4, and JNK protects against necrotic cell death [155,156,157,158,159,160]. -AIF-deficient mice have less injury in APAP toxicity [162]. -Knockdown or inhibition of GSK3β and RIPK1 protects from APAP liver injury [30,166,167]. -RIPK1 participates upstream, of JNK and has a non-necroptotic function [30,167]. | -Nec-1 has been shown to protect from APAP; however, it has off-target effects [27,179,180]. -Controversial results with global RIPK3 KO mice treated with APAP have been reported [27,30,165,181]. -MLKL KO mice not protected from APAP [30,36]. -RIPK1 may have a role in APAP toxicity independent of its function in necroptosis and upstream of JNK signaling [30,167]. | -Activation of autophagy protective in APAP [182,183,184,185]. -Autophagy and mitophagy limit ROS generation and are thought to limit the expansion of necrotic foci [188]. -LC3 transgenic mice show an increase in autophagosomes surrounding necrotic foci after APAP administration [152,188]. -APAP increases autophagic flux and its inhibition increases APAP protein adducts [152]. | -CASP1, ASC, and NLRP3 KO mice have been shown to be less sensitive to APAP toxicity [189]. -No protection with anti-IL-1β antibodies or KO of IL-1β, CASP1 or NALP3 [190,191,192]. - TLR4 signaling in macrophages proposed to aggravate APAP injury by generating IL-1α [192]. However, this is controversial. -GSDMD KO mice have increased injury in APAP toxicity [194]. -Further investigation is needed to determine pyroptosis in APAP toxicity. | -Antioxidants such as Fer-1 and deferoxamine may alleviate APAP toxicity with less lipid peroxidation, GSH depletion, and iron accumulation [198,199,200]. -No cleaved caspase 3 and increased RIPK3 expression was detected with Fer-1 treatment [201]. -DFO protected against APAP by maintaining GSH levels [201]. -It is difficult to determine if antioxidants work to increase GSH and prevent APAP necrosis or if they are preventing “ferroptosis”. -No data on GPX4 KO mice are available; therefore, the contribution of ferroptosis is controversial. |
| Autoimmune Hepatitis (AIH) including mouse models of Concanavalin A (ConA) and α-galactoceramide (α-GalCer) models | -Councilman bodies that are apoptotic have been seen in liver biopsy of AIH patients [203]. -In the ConA model IFN-γ, IL-4, IL-6, FAS, and TRAIL participation has been reported, but TNF is known as the principal cytokine involved in toxicity [206]. -TNF induces FasL-dependent apoptosis with transcriptional induction of FAS via the NFκB pathway [27,207]. -Caspase inhibition in ConA model is largely ineffective [209]. | -Increased hepatic MLKL mRNA and protein expression in the liver biopsy of patients with AIH [36]. -Nec-1 has been shown to protect against ConA [201,202,203,204,205,206,207,208,209,210,211,212]. -MLKL global KO has protected from Con but MLKL-hepatocyte-specific KO is not protected [36,214]. -RIPK1 inhibition has shown to be effective upstream of MLKL in a ConA model [36]. -RIPK1-deficient hepatocytes die of TNF and caspase-dependent apoptosis with low dose ConA injection [215,216]. -RIPK1 knockdown promoted increased TNF-mediated apoptosis in αGalCer [205]. -RIPK3 deficiency in αGalCer controversial. One paper has shown a modest protective effect by limiting cytokine production and NK activation [217]. In other, RIPK3 global KO and MLKL global KO mice not protected from αGalCer [205]. | -NLRP3 and CASP1 KO mice were protected in a ConA model [219]. -CASP1 is needed for IL-1β production [219]. -IL-1 suppression reduces liver injury through reduced TNF and IL-17 secretion in a ConA model [219]. -NKT cells express OX40 and high levels of CASP1. The activation of CASP1 led to pro–IL-1β maturation and GSDMD cleavage and pyroptosis [221]. -Low-dose ConA induces TNF superfamily receptor, OX40, in the liver and blocking OX40 can prevent ConA hepatitis [221]. -Further studies on GSDMD are needed to study Pyroptosis in ConA and αGalCer. | -IDO-dependent ferroptosis has been reported in the ConA model [222]. -Downregulation of Cav-1 increases RNS production and injury is dampened with Fer-1 administration [223]. -No data with GPX4; therefore, results are inconclusive. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. https://doi.org/10.3390/ijms21249682
Shojaie L, Iorga A, Dara L. Cell Death in Liver Diseases: A Review. International Journal of Molecular Sciences. 2020; 21(24):9682. https://doi.org/10.3390/ijms21249682
Chicago/Turabian StyleShojaie, Layla, Andrea Iorga, and Lily Dara. 2020. "Cell Death in Liver Diseases: A Review" International Journal of Molecular Sciences 21, no. 24: 9682. https://doi.org/10.3390/ijms21249682
APA StyleShojaie, L., Iorga, A., & Dara, L. (2020). Cell Death in Liver Diseases: A Review. International Journal of Molecular Sciences, 21(24), 9682. https://doi.org/10.3390/ijms21249682