Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease

 and
and 
Abstract
1. Introduction
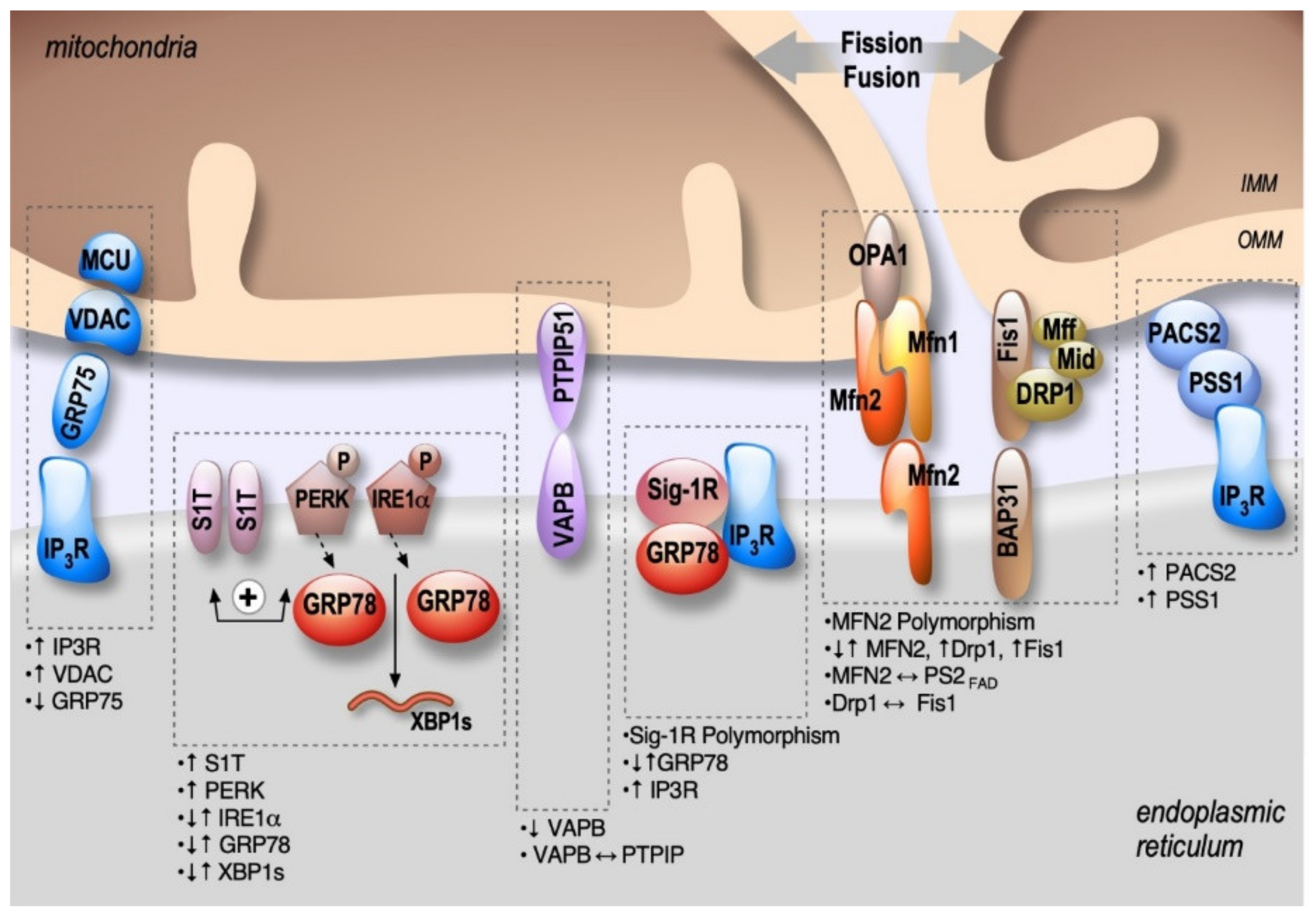
2. Structural and Functional Partners of MAMs Are Involved in AD
2.1. Mitofusins: MFNs

2.2. IP3R-Grp75-VDAC
2.3. PACS2-PSS1
2.4. BAP31-FIS1-DRP1
2.5. VAPB-PTPIP51
2.6. Sig-1R-Grp78
2.7. Protein Kinase-Like Endoplasmic Reticulum Kinase: PERK

{kind=link}
{kind=link}
{kind=link}
| Proteins | Alterations | Study Models | References |
|---|---|---|---|
| MFNs | Polymorphism | AD patients | [40] |
| ↓MFN2 & MFN1 (proteins and mRNA) | AD brains (hippocampus & frontal cortex) & N2a “OE” APPswe | [41,42,43] | |
| ↑MFN2 | MCI cytoplasmic hybrid (cybrid) cells | [44] | |
| ↑MFN2⟷PS2 (FAD), ↑MAMs | MFN2 KO MEFs FAD PS2 (N141I) Mice | [46] | |
| IP3R-Grp75-VDAC | ↑VDAC Aβ⟷VDAC pTau⟷VDAC | Tg2576 and J20 mice APP, APP/PS1, 3xTgAD mice & AD brains (cortical tissues) | [53] [55] |
| ↑VDAC, ↑IP3R activity, ↑MAMs | Aβ-treated hippocampal neurons | [54] | |
| VDAC dephosphorylation | AD brains | [56] | |
| ↓Grp75 | AD brains (temporal and parietal cortex) | [52] | |
| FAD PS1/2⟷IP3R | Sf9 cells | [62] | |
| PACS2-PSS1 | ↑PACS2, ↑PSS1 | APPswe/LDN mice AD brains cortex | [54] |
| BAP31-FIS1-DRP1 | ↑Fis1, ↑Drp1 | AD brains (frontal cortex) | [42] |
| ↑Drp1⟷Fis | Aβ42-treated neurons, SAD/FAD fibroblasts, N2a “OE” APPswe, & 5xTgAD mice | [80] | |
| pTau⟷Drp1 ↓Drp1 mito localization | AD brains, APP, APP/PS1 & 3xTgAD mice Drosophila “OE” Tau R406W | [55] [133] | |
| Aβ⟷Drp1 | AD brains (frontal cortex) | [42] | |
| VAPB-PTPIP51 | ↓VAPB, ↓PTPIP51 ↓VAPB⟷PTPIP51 | AD brains (subicular with GVB neurons) AD brains (cortex) | [90] [92] |
| Sig-1R-Grp78 | ↓Sig-1R Sig-1R polymorphism | AD brains AD patients | [109] [110,111] |
| ↑Grp78 | Streptozotocin model, APP/PS1 mice & Aβ25-35-treated rats | [134,135,136] | |
| ↓Grp78 mRNA | AD brains (FAD PS1) | [101] | |
| =Grp78 expression | 5xFAD mice | [102] | |
| PERK | ↑PERK pathway | AD brains 5xFAD mice APP/PS1, APP(SL)/PS1 KI & rTg4510 mice APOE4 mice | [121,122,123,124,125,126] [123] [124,125,126,127] [128,129] |
| IRE1α | ↑↓IRE1α pathway, ↑↓XBP1s | AD brains & 5xFAD mice Aβ42o-treated SH-SY5Y & CHO “OE” APPLDN | [137] [138] |
| S1T | ↑S1T | AD brains, SHSY-5Y “OE”APPswe or treated with Aβo | [139] |
2.8. Active Inositol-Requiring Transmembrane Kinase and Endonuclease Alpha: IRE1α
2.9. The Truncated Variant of the Sarco-Endoplasmic Reticulum Ca2+-ATPase 1: S1T
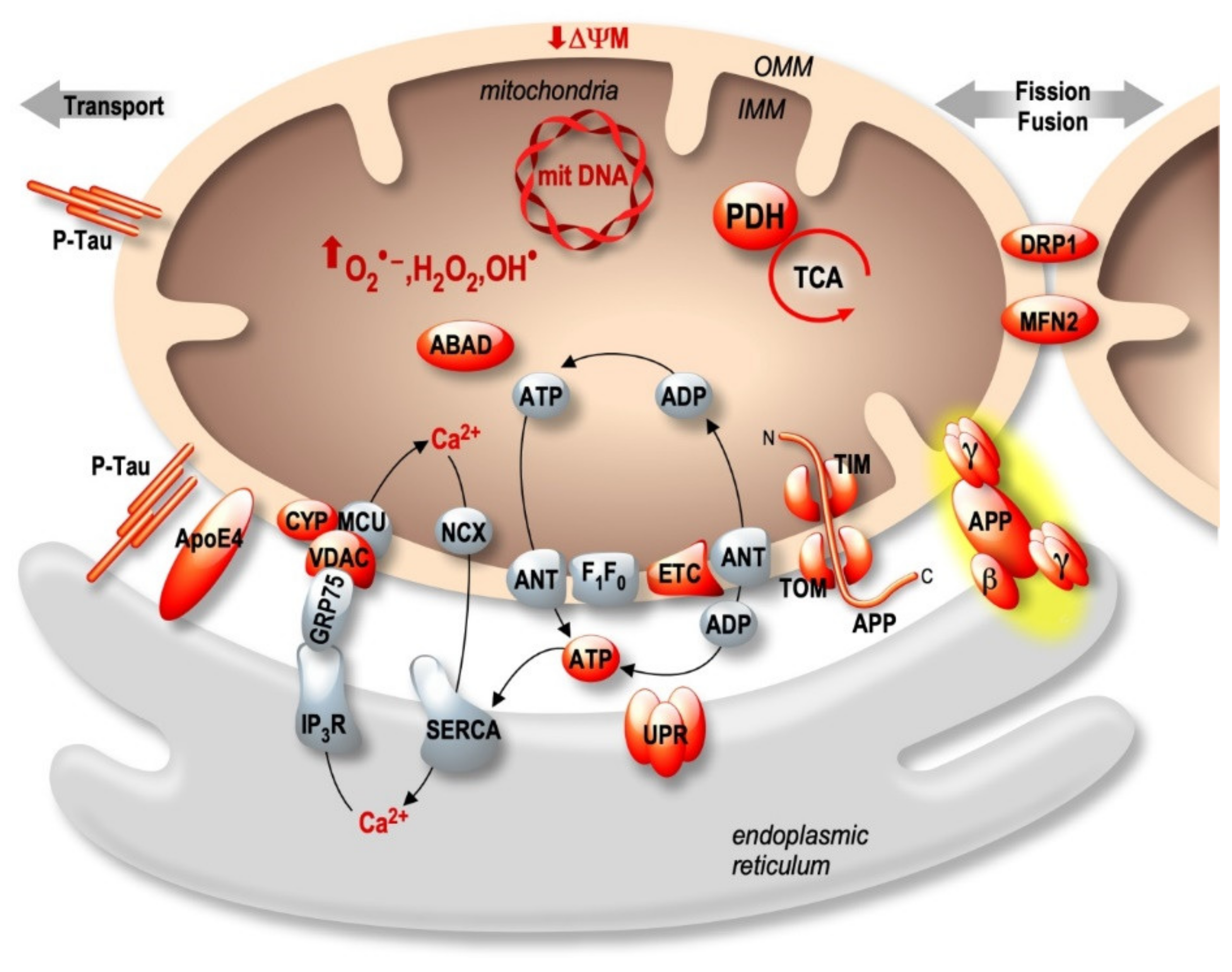
3. The MAMs Hypothesis in the Pathophysiological Process of Alzheimer’s Disease
3.1. Apolipoprotein E: APOE
3.2. Presenilins
3.3. APP-Derived C-Terminal Fragments (APP-CTFs) and Aβ
3.4. Tau

| Proteins | Alterations | Study Models | Ref |
|---|---|---|---|
| APOEε4 | ↓DRP1, ↑MFN1 Mito fusion (↑COX1, ↑TOMM40) Mitophagy failure (↓PINK1, ↑Parkin, ↑p62) | AD brains APOEε4 KI mice | [160] |
| ↑VDAC1, ↑IP3R, ↑BAP31, ↑Fis1, ↑MFN2, ↓Grp75 | N2acells “OE” APOE4 | [161] | |
| ↑PS, ↑CO, ↑CE, ↑Lipid droplets | APOE4 KI mice astrocytes CM treatment | [163] | |
| PS1 & PS2 | ↑LC3-II and ↑autophagosomes | MEFs (PS1KO, PS2KO, & PSDKO) HepG2 cells (PS1“SI”) | [165,166] |
| PS2⟷Mfn2 ↑MAMs | MEFs (MFN2 KO), Human FAD fibroblasts, SHSY-5Y & primary neurons “OE” MFN2 | [46,172] | |
| ↑ER-mitochondria coupling Autophagy blockade (↑LC3-II, ↑p62) | FAD PS2 (T122R, N141I) &. SHSY-5Y “OE” FAD PS2 | [174] | |
| ↓IP3R1/3, ↓MIRO1, ↓KIF5C ↑MAMs | Purkinje & SHSY-5Y “OE” FAD PS1(E280A) | [176] | |
| Autophagy impairment | FAD PS1 (A246E, M233T, H163Y, M146L, L392V) fibroblasts | [177] | |
| ↑MAMs ↑ MAMs function (↑CE, ↑PS, ↑PE, ↑lipid droplets) | PS1 KO, PS2 KO, PSDKO FAD and SAD patient fibroblasts | [190] | |
| APP, Aβ & APP-CTFs | APP in mitochondria | HCN-1A, COS cells “OE” APPWT or APP chimeric mutants & APPswe mice | [169,180,181] |
| APP, APP-CTFs, Aβ in MAMs ↑MAMs ↑ceramide, ↓sphingomyelin,↑lipid droplets (Aβ dependant) | SHSY-5Y “OE” APPswe, APPLDN Mice brains (WT,APP23 & APP23/PS45) PSDKO +/− DAPT | [185,186,187,190,194] | |
| ↑mito ROS, ↓complex I &IV activities (Aβ-dependant) | AD brains; Aβ-treated CGCs | [195,196] | |
| ↑mito ROS, ↓complex I activity Mitophagy failure (APP-CTFs dependant) | 3xTgAD, 2xTgAD, AAV-C99 mice, SHSY-5Y cells “OE”APPswe or C99 +/− γ-secretase inhibitor AD brains | [202] | |
| Tau | ↓complex V, ↓ATP, ↑mitROS | TauP301L mice | [206] |
| Mitophagy failure (↓Parkin mitochondrial translocation) | N2a “OE” TauP301L | [207] | |
| ↑OPA1, ↑MFN1/2, Mitochondrial fragmentation, ↓complex I activity, ↓ATP | HEK293 & rat primary hippocampal neurons “OE” Tau | [208] | |
| Mitochondria & ER distributions | CHO and N2a cells “OE” Tau | [210] | |
| ↓Mito axonal movement | Hippocampal neurons “OE” Tau45-230 | [211] | |
| ↑MAMs | Motor neurons “OE” TauP301L | [209] | |
| ↓MAMs | TauP301L mice | [212] |
4. MAMs as a Potential Therapeutic Strategy
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Checler, F. Processing of the beta-amyloid precursor protein and its regulation in Alzheimer’s disease. J. Neurochem. 1995, 65, 1431–1444. [Google Scholar] [CrossRef] [PubMed]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 2020, 61, 40–48. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Flammang, B.; Pardossi-Piquard, R.; Sevalle, J.; Debayle, D.; Dabert-Gay, A.S.; Thévenet, A.; Lauritzen, I.; Checler, F. Evidence that the amyloid-β protein precursor intracellular domain, AICD, derives from β-secretase-generated C-terminal fragment. J. Alzheimer’s Dis. 2012, 30, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.B.; Moore, S.; Daria, A.; et al. n-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Song, M.; Liu, X.; Su Kang, S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping Yu, S.; et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 2015, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.; Carrete, A.; Paumier, J.M.; Py, N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer’s disease. Cell. Mol. Life Sci. 2016, 73, 217–236. [Google Scholar] [CrossRef]
- Bourgeois, A.; Lauritzen, I.; Lorivel, T.; Bauer, C.; Checler, F.; Pardossi-Piquard, R. Intraneuronal accumulation of C99 contributes to synaptic alterations, apathy-like behavior, and spatial learning deficits in 3 × TgAD and 2 × TgAD mice. Neurobiol. Aging 2018, 71, 21–31. [Google Scholar] [CrossRef]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255. [Google Scholar] [CrossRef]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. Erratum: A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ. Neuron 2019, 104, 256–270. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria—ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Hajnóczky, G. Mitochondria and endoplasmic reticulum: The lethal interorganelle cross-talk. J. Bioenerg. Biomembr. 2005, 37, 191–206. [Google Scholar] [CrossRef]
- Veeresh, P.; Kaur, H.; Sarmah, D.; Mounica, L.; Verma, G.; Kotian, V.; Kesharwani, R.; Kalia, K.; Borah, A.; Wang, X.; et al. Endoplasmic reticulum–mitochondria crosstalk: From junction to function across neurological disorders. Ann. N. Y. Acad. Sci. 2019, 1457, 41–60. [Google Scholar] [CrossRef]
- Van Vliet, A.R.; Agostinis, P. Mitochondria-Associated membranes and ER stress. In Current Topics in Microbiology and Immunology; Springer Nature Switzerland AG: Cham, Switzerland, 2018; Volume 414, pp. 73–102. [Google Scholar]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef]
- Hashimoto, S.; Saido, T.C. Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 1–22. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef]
- Cipolat, S.; De Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacín, M.; Zorzano, A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Martorell-Riera, A.; Segarra-Mondejar, M.; Muñoz, J.P.; Ginet, V.; Olloquequi, J.; Pérez-Clausell, J.; Palacín, M.; Reina, M.; Puyal, J.; Zorzano, A.; et al. Mfn2 downregulation in excitotoxicity causes mitochondrial dysfunction and delayed neuronal death. EMBO J. 2014, 33, 2388–2407. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Kim, Y.J.; Park, J.K.; Kang, W.S.; Kim, S.K.; Han, C.; Na, H.R.; Park, H.J.; Kim, J.W.; Kim, Y.Y.; Park, M.H.; et al. Association between mitofusin 2 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population. Psychiatry Investig. 2017, 14, 81–85. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Choi, H.; Min, J.S.; Kim, B.; Lee, S.R.; Yun, J.W.; Choi, M.S.; Chang, K.T.; Lee, D.S. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J. Neurochem. 2015, 132, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Wu, L.; Huang, S.; Zhong, C.; Li, G.; Yu, H.; Swerdlow, R.H.; Chen, J.X.; Yan, S.S. Oxidative stress-mediated activation of extracellualr signal-regulated kinase contributes to mild cognitive impairment-related mitochondrial dysfuntion. Free Radic. Biol. Med. 2014, 75, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Leal, N.S.; Schreiner, B.; Pinho, C.M.; Filadi, R.; Wiehager, B.; Karlström, H.; Pizzo, P.; Ankarcrona, M. Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production. J. Cell. Mol. Med. 2016, 20, 1686–1695. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol triphosphate and calcium signalling. Nature 1993, 361, 315–325. [Google Scholar] [CrossRef]
- Joseph, S.K.; Hajnóczky, G. IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 2007, 12, 951–968. [Google Scholar] [CrossRef]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Yoo, B.C.; Kim, S.H.; Cairns, N.; Fountoulakis, M.; Lubec, G. Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2001, 280, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Vilariño, M.; Cabodevilla, F.; Del Río, J.; Frechilla, D.; Pérez-Mediavilla, A. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer’s disease transgenic mice: An insight into the pathogenic effects of amyloid-β. J. Alzheimers Dis. 2011, 23, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Echevarria, C.; Díaz, M.; Ferrer, I.; Canerina-Amaro, A.; Marin, R. Aβ promotes VDAC1 channel dephosphorylation in neuronal lipid rafts. Relevance to the mechanisms of neurotoxicity in Alzheimer’s disease. Neuroscience 2014, 278, 354–366. [Google Scholar] [CrossRef]
- Manczak, M.; Sheiko, T.; Craigen, W.J.; Reddy, P.H. Reduced VDAC1 protects against Alzheimer’s disease, mitochondria, and synaptic deficiencies. J. Alzheimer’s Dis. 2013, 37, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef]
- Cheung, K.; Mei, L.; Mak, D.D.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-function enhancement of InsP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef]
- Demuro, A.; Parker, I. Cytotoxicity of intracellular Aβ42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 2013, 33, 3824–3833. [Google Scholar] [CrossRef]
- Shilling, D.; Müller, M.; Takano, H.; Mak, D.O.D.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. J. Neurosci. 2014, 34, 6910–6923. [Google Scholar] [CrossRef]
- Cheung, K.H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.Y.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef]
- Del Prete, D.; Checler, F.; Chami, M. Ryanodine receptors: Physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 2014, 9, 21. [Google Scholar] [CrossRef]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-Translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef] [PubMed]
- Bussiere, R.; Lacampagne, A.; Reiken, S.; Liu, X.; Scheuerman, V.; Zalk, R.; Martin, C.; Checler, F.; Marks, A.R.; Chami, M. Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J. Biol. Chem. 2017, 292, 10153–10168. [Google Scholar] [CrossRef] [PubMed]
- Oules, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Brechot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; LaFerla, F.M.; Parker, I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Parker, I.; Laferla, F. Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer’s mouse models. Ann. N. Y. Acad. Sci. 2007, 1097, 265–277. [Google Scholar] [CrossRef]
- Li, C.; Li, L.; Yang, M.; Zeng, L.; Sun, L. PACS-2: A key regulator of mitochondria-associated membranes (MAMs). Pharmacol. Res. 2020, 160, 105080. [Google Scholar] [CrossRef]
- Werneburg, N.W.; Bronk, S.F.; Guicciardi, M.E.; Thomas, L.; Dikeakos, J.D.; Thomas, G.; Gores, G.J. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein-induced lysosomal translocation of proapoptotic effectors is mediated by phosphofurin acidic cluster sorting protein-2 (PACS-2). J. Biol. Chem. 2012, 287, 24427–24437. [Google Scholar] [CrossRef]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.-H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef]
- Youker, R.T.; Shinde, U.; Day, R.; Thomas, G. At the crossroads of homoeostasis and disease: Roles of the PACS proteins in membrane traffic and apoptosis. Biochem. J. 2009, 421, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.J.; Vance, J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 2000, 275, 34534–34540. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Pagliuso, A.; Cossart, P.; Stavru, F. The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 2018, 75, 355–374. [Google Scholar] [CrossRef]
- Shen, Q.; Yamano, K.; Head, B.P.; Kawajiri, S.; Cheung, J.T.M.; Wang, C.; Cho, J.-H.; Hattori, N.; Youle, R.J.; van der Bliek, A.M. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol. Biol. Cell 2014, 25, 145–159. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Rojansky, R.; Cha, M.-Y.; Chan, D.C. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 2016, 5. [Google Scholar] [CrossRef]
- Namba, T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2017, 9, 6128–6143. [Google Scholar] [CrossRef]
- Wang, T.; Chen, J.; Hou, Y.; Yu, Y.; Wang, B. BAP31 deficiency contributes to the formation of amyloid-β plaques in Alzheimer’s disease by reducing the stability of RTN3. FASEB J. 2019, 33, 4936–4946. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.W.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef]
- Galmes, R.; Houcine, A.; Vliet, A.R.; Agostinis, P.; Jackson, C.L.; Giordano, F. ORP5/ORP8 localize to endoplasmic reticulum–mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 2016, 17, 800–810. [Google Scholar] [CrossRef]
- Tokutake, Y.; Yamada, K.; Ohata, M.; Obayashi, Y.; Tsuchiya, M.; Yonekura, S. ALS-Linked P56S-VAPB mutation impairs the formation of multinuclear myotube in C2C12 Cells. Int. J. Mol. Sci. 2015, 16, 18628–18641. [Google Scholar] [CrossRef]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER-Mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Yamoah, A.; Tripathi, P.; Sechi, A.; Köhler, C.; Guo, H.; Chandrasekar, A.; Nolte, K.W.; Wruck, C.J.; Katona, I.; Anink, J.; et al. Aggregates of RNA binding proteins and ER chaperones linked to exosomes in granulovacuolar degeneration of the Alzheimer’s disease brain. J. Alzheimer’s Dis. JAD 2020, 139–156. [Google Scholar] [CrossRef]
- Llorens-Marítin, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7. [Google Scholar] [CrossRef]
- Lau, D.H.W.; Paillusson, S.; Hartopp, N.; Rupawala, H.; Mórotz, G.M.; Gomez-Suaga, P.; Greig, J.; Troakes, C.; Noble, W.; Miller, C.C.J. Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 2020, 143, 105020. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. σ-1 receptors (σ1 binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: Roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 2003, 306, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR 1- and SOD 1 -linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. The potential role of sigma-1 receptors in lipid transport and lipid raft reconstitution in the brain: Implication for drug abuse. Life Sci. 2005, 77, 1612–1624. [Google Scholar] [CrossRef]
- Gorbatyuk, M.; Gorbatyuk, O. The Molecular Chaperone GRP78/BiP as a Therapeutic Target for Neurodegenerative Disorders: A Mini Review. J. Genet. Syndr. Gene Ther. 2013, 4. [Google Scholar] [CrossRef]
- Prasad, M.; Pawlak, K.J.; Burak, W.E.; Perry, E.E.; Marshall, B.; Whittal, R.M.; Bose, H.S. Mitochondrial metabolic regulation by GRP78. Sci. Adv. 2017, 3. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Scheper, W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener. Dis. 2012, 10, 212–215. [Google Scholar] [CrossRef]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular mechanisms of ER stress and UPR in the pathogenesis of Alzheimer’s disease. Mol. Neurobiol. 2020, 57, 2902–2919. [Google Scholar] [CrossRef]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485. [Google Scholar] [CrossRef]
- Sadleir, K.R.; Popovic, J.; Vassar, R. ER stress is not elevated in the 5XFAD mouse model of Alzheimer’s disease. J. Biol. Chem. 2018, 293, 18434–18443. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Morin-Surun, M.P.; Collin, T.; Denavit-Saubie, M.; Baulieu, E.E.; Monnet, F.P. Intracellular σ1 receptor modulates phospholipase C and protein kinase C activities in the brainstem. Proc. Natl. Acad. Sci. USA 1999, 96, 8196–8199. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Hayashi, T.; Harvey, B.K.; Wang, Y.; Wu, W.W.; Shen, R.F.; Zhang, Y.; Becker, K.G.; Hoffer, B.J.; Su, T.P. Sigma-1 receptors regulate hippocampal dendritic spine formation via a free radical-sensitive mechanism involving Rac1·GTP pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 22468–22473. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal sigma-1 receptors: Signaling functions and protective roles in neurodegenerative diseases. Front. Neurosci. 2019, 13, 1–20. [Google Scholar] [CrossRef]
- Delprat, B.; Crouzier, L.; Su, T.-P.; Maurice, T. At the crossing of ER stress and MAMs: A key role of Sigma-1 receptor? Adv. Exp. Med. Biol. 2020, 1131, 699–718. [Google Scholar] [CrossRef]
- Jansen, K.L.R.; Faull, R.L.M.; Storey, P.; Leslie, R.A. Loss of sigma binding sites in the CA1 area of the anterior hippocampus in Alzheimer’s disease correlates with CA1 pyramidal cell loss. Brain Res. 1993, 623, 299–302. [Google Scholar] [CrossRef]
- Fehér, Á.; Juhász, A.; László, A.; Kálmán, J.; Pákáski, M.; Kálmán, J.; Janka, Z. Association between a variant of the sigma-1 receptor gene and Alzheimer’s disease. Neurosci. Lett. 2012, 517, 136–139. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, L.; Halliday, G.; Dobson-Stone, C.; Wang, Y.; Tang, H.-D.; Cao, L.; Deng, Y.-L.; Wang, G.; Zhang, Y.-M.; et al. Genetic polymorphisms in Sigma-1 receptor and apolipoprotein E interact to influence the severity of Alzheimers disease. Curr. Alzheimer Res. 2011, 8, 765–770. [Google Scholar] [CrossRef]
- Fisher, A.; Bezprozvanny, I.; Wu, L.; Ryskamp, D.A.; Bar-Ner, N.; Natan, N.; Brandeis, R.; Elkon, H.; Nahum, V.; Gershonov, E.; et al. AF710B, a Novel M1/sigma1 agonist with therapeutic efficacy in animal models of Alzheimer’s disease. Neurodegener. Dis. 2016, 16, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Hall, H.; Iulita, M.F.; Gubert, P.; Flores Aguilar, L.; Ducatenzeiler, A.; Fisher, A.; Cuello, A.C. AF710B, an M1/sigma-1 receptor agonist with long-lasting disease-modifying properties in a transgenic rat model of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Strehaiano, M.; Duhr, F.; Chevallier, N. Amyloid toxicity is enhanced after pharmacological or genetic invalidation of the σ1 receptor. Behav. Brain Res. 2018, 339, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lahmy, V.; Meunier, J.; Malmström, S.; Naert, G.; Givalois, L.; Kim, S.H.; Villard, V.; Vamvakides, A.; Maurice, T. Blockade of tau hyperphosphorylation and aβ 1-42 generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and σ 1 receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2013, 38, 1706–1723. [Google Scholar] [CrossRef]
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25–35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell. Neurosci. 2015, 8, 1–11. [Google Scholar] [CrossRef]
- Tsai, S.Y.A.; Pokrass, M.J.; Klauer, N.R.; Nohara, H.; Su, T.P. Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc. Natl. Acad. Sci. USA 2015, 112, 6742–6747. [Google Scholar] [CrossRef]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Munoz, J.P.; Ivanova, S.; Sanchez-Wandelmer, J.; Martinez-Cristobal, P.; Noguera, E.; Sancho, A.; Diaz-Ramos, A.; Hernandez-Alvarez, M.I.; Sebastian, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef]
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK2 regulates endoplasmic reticulum-mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. 2020, 39, e100875. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. PERK mediates eIF2alpha phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Page, G.; Rioux Bilan, A.; Ingrand, S.; Lafay-Chebassier, C.; Pain, S.; Perault Pochat, M.C.; Bouras, C.; Bayer, T.; Hugon, J. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience 2006, 139, 1343–1354. [Google Scholar] [CrossRef]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C., 3rd; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef]
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015, 130, 633–642. [Google Scholar] [CrossRef]
- Segev, Y.; Barrera, I.; Ounallah-Saad, H.; Wibrand, K.; Sporild, I.; Livne, A.; Rosenberg, T.; David, O.; Mints, M.; Bramham, C.R.; et al. PKR inhibition rescues memory deficit and ATF4 overexpression in ApoE epsilon4 human replacement mice. J. Neurosci. 2015, 35, 12986–12993. [Google Scholar] [CrossRef]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef]
- Yang, W.; Zhou, X.; Zimmermann, H.R.; Cavener, D.R.; Klann, E.; Ma, T. Repression of the eIF2alpha kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2016, 41, 19–24. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Gobert, D.; Stern, E.; Gamache, K.; Colina, R.; Cuello, C.; Sossin, W.; Kaufman, R.; Pelletier, J.; Rosenblum, K.; et al. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 2007, 129, 195–206. [Google Scholar] [CrossRef]
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 2014, 158, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization In Vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Biswas, J.; Gupta, S.; Verma, D.K.; Gupta, P.; Singh, A.; Tiwari, S.; Goswami, P.; Sharma, S.; Singh, S. Involvement of glucose related energy crisis and endoplasmic reticulum stress: Insinuation of streptozotocin induced Alzheimer’s like pathology. Cell. Signal. 2018, 42, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Wang, S.; Wang, Z.; Wang, Z.; Sun, C.; Zhang, Y. Inhibition of PTEN attenuates endoplasmic reticulum stress and apoptosis via activation of PI3K/AKT pathway in Alzheimer’s disease. Neurochem. Res. 2017, 42, 3052–3060. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Liu, G.; Yang, L. Crocin improves cognitive behavior in rats with Alzheimer’s disease by regulating endoplasmic reticulum stress and apoptosis. Biomed. Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, S.; Schuck, F.; Grosgen, S.; Riemenschneider, M.; Hartmann, T.; Postina, R.; Grimm, M.; Endres, K. Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer’s disease. FASEB J. 2014, 28, 978–997. [Google Scholar] [CrossRef]
- Gerakis, Y.; Dunys, J.; Bauer, C.; Checler, F. Aβ42 oligomers modulate β-secretase through an XBP-1s-dependent pathway involving HRD1. Sci. Rep. 2016, 6, 37436. [Google Scholar] [CrossRef]
- Bussiere, R.; Oules, B.; Mary, A.; Vaillant-Beuchot, L.; Martin, C.; El Manaa, W.; Vallee, D.; Duplan, E.; Paterlini-Brechot, P.; Alves Da Costa, C.; et al. Upregulation of the sarco-endoplasmic reticulum calcium ATPase 1 truncated isoform plays a pathogenic role in Alzheimer’s disease. Cells 2019, 8, 1539. [Google Scholar] [CrossRef]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.P. Sigma-1 receptor chaperone at the ER-Mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Takeda, K.; Nagashima, S.; Shiiba, I.; Uda, A.; Tokuyama, T.; Ito, N.; Fukuda, T.; Matsushita, N.; Ishido, S.; Iwawaki, T.; et al. MITOL prevents ER stress-induced apoptosis by IRE1alpha ubiquitylation at ER-mitochondria contact sites. EMBO J. 2019, 38, e100999. [Google Scholar] [CrossRef]
- Son, S.M.; Byun, J.; Roh, S.E.; Kim, S.J.; Mook-Jung, I. Reduced IRE1alpha mediates apoptotic cell death by disrupting calcium homeostasis via the InsP3 receptor. Cell Death Dis. 2014, 5, e1188. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef]
- Cissé, M.; Duplan, E.; Lorivel, T.; Dunys, J.; Bauer, C.; Meckler, X.; Gerakis, Y.; Lauritzen, I.; Checler, F. The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol. Psychiatry 2017, 22, 1562–1575. [Google Scholar] [CrossRef]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef]
- Waldherr, S.M.; Strovas, T.J.; Vadset, T.A.; Liachko, N.F.; Kraemer, B.C. Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat. Commun. 2019, 10, 4443. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Chami, M.; Gozuacik, D.; Saigo, K.; Capiod, T.; Falson, P.; Lecoeur, H.; Urashima, T.; Beckmann, J.; Gougeon, M.L.; Claret, M.; et al. Hepatitis B virus-related insertional mutagenesis implicates SERCA1 gene in the control of apoptosis. Oncogene 2000, 19, 2877–2886. [Google Scholar] [CrossRef]
- Chami, M.; Gozuacik, D.; Lagorce, D.; Brini, M.; Falson, P.; Peaucellier, G.; Pinton, P.; Lecoeur, H.; Gougeon, M.L.; le Maire, M.; et al. SERCA1 truncated proteins unable to pump calcium reduce the endoplasmic reticulum calcium concentration and induce apoptosis. J. Cell Biol. 2001, 153, 1301–1314. [Google Scholar] [CrossRef]
- Chami, M.; Oules, B.; Szabadkai, G.; Tacine, R.; Rizzuto, R.; Paterlini-Brechot, P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 2008, 32, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 2006, 26, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Lane-Donovan, C.; Wong, W.M.; Durakoglugil, M.S.; Wasser, C.R.; Jiang, S.; Xian, X.; Herz, J. Genetic restoration of plasma apoe improves cognition and partially restores synaptic defects in ApoE-deficient mice. J. Neurosci. 2016, 36, 10141–10150. [Google Scholar] [CrossRef] [PubMed]
- Neu, S.C.; Pa, J.; Kukull, W.; Beekly, D.; Kuzma, A.; Gangadharan, P.; Wang, L.-S.; Romero, K.; Arneric, S.P.; Redolfi, A.; et al. Apolipoprotein E genotype and sex risk factors for Alzheimer disease: A meta-analysis. JAMA Neurol. 2017, 74, 1178–1189. [Google Scholar] [CrossRef]
- Mahley, R.W.; Huang, Y. Apolipoprotein E sets the stage: Response to injury triggers neuropathology. Neuron 2012, 76, 871–885. [Google Scholar] [CrossRef]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Masliah, E.; Pinkas-Kramarski, R.; Michaelson, D.M. The Effects of APOE4 on mitochondrial dynamics and proteins in vivo. J. Alzheimer’s Dis. 2019, 70, 861–875. [Google Scholar] [CrossRef]
- Orr, A.L.; Kim, C.; Jimenez-Morales, D.; Newton, B.W.; Johnson, J.R.; Krogan, N.J.; Swaney, D.L.; Mahley, R.W. Neuronal apolipoprotein E4 expression results in proteome-wide alterations and compromises bioenergetic capacity by disrupting mitochondrial function. J. Alzheimer’s Dis. 2019, 68, 991–1011. [Google Scholar] [CrossRef]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.-P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, N.; Walter, J. Presenilins and γ-secretase in membrane proteostasis. Cells 2019, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Neely, K.M.; Green, K.N.; LaFerla, F.M. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a γ-secretase-independent manner. J. Neurosci. 2011, 31, 2781–2791. [Google Scholar] [CrossRef]
- Száraz, P.; Bánhegyi, G.; Marcolongo, P.; Benedetti, A. Transient knockdown of presenilin-1 provokes endoplasmic reticulum stress related formation of autophagosomes in HepG2 cells. Arch. Biochem. Biophys. 2013, 538, 57–63. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in Presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef]
- Area-Gomez, E.; De Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca 2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy 2019, 15, 2044–2062. [Google Scholar] [CrossRef] [PubMed]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; De Strooper, B.; Bezprozvanny, I. Familial Alzheimer disease-linked mutations specifically disrupt Ca 2+ leak function of presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Falla, D.; Barrera-Ocampo, A.; Hagel, C.; Korwitz, A.; Vinueza-Veloz, M.F.; Zhou, K.; Schonewille, M.; Zhou, H.; Velazquez-Perez, L.; Rodriguez-Labrada, R.; et al. Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J. Clin. Investig. 2014, 124, 1552–1567. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.C.; Zheng, H. Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2012, 2, 1–17. [Google Scholar] [CrossRef]
- Anandatheerthavarada, H.K.; Devi, L. Mitochondrial translocation of amyloid precursor protein and its cleaved products: Relevance to mitochondrial dysfunction in Alzheimer’s disease. Rev. Neurosci. 2007, 18, 343–354. [Google Scholar] [CrossRef]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef]
- Pagani, L.; Eckert, A. Amyloid-beta interaction with mitochondria. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, P.F.; Wiehager, B.; Sakai, J.; Frykman, S.; Behbahani, H.; Winblad, B.; Ankarcrona, M. Mitochondrial γ-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein. FASEB J. 2011, 25, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol. Dis. 2012, 45, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Hedskog, L.; Wiehager, B.; Ankarcrona, M. Amyloid-β peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimer’s Dis. 2015, 43, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP -C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Suski, J.M.; Oulès, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Bréchot, P.; et al. Localization and processing of the amyloid-β protein precursor in mitochondria-associated membranes. J. Alzheimer’s Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef]
- Hansson, C.A.; Frykman, S.; Farmery, M.R.; Tjernberg, L.O.; Nilsberth, C.; Pursglove, S.E.; Ito, A.; Winblad, B.; Cowburn, R.F.; Thyberg, J.; et al. Nicastrin, presenilin, APH-1, and PEN-2 form active γ-secretase complexes in mitochondria. J. Biol. Chem. 2004, 279, 51654–51660. [Google Scholar] [CrossRef]
- Ankarcrona, M.; Hultenby, K. Presenilin-1 is located in rat mitochondria. Biochem. Biophys. Res. Commun. 2002, 295, 766–770. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef]
- Marquer, C.; Devauges, V.; Cossec, J.C.; Liot, G.; Lécart, S.; Saudou, F.; Duyckaerts, C.; Lévêque-Fort, S.; Potier, M.C. Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J. 2011, 25, 1295–1305. [Google Scholar] [CrossRef]
- Cordy, J.M.; Hussain, I.; Dingwall, C.; Hooper, N.M.; Turner, A.J. Exclusively targeting β-secretase to lipid rafts by GPI-anchor addition up-regulates β-site processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2003, 100, 11735–11740. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of γ-secretase with lipid rafts in post-golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Vargas, T.; Ugalde, C.; Spuch, C.; Antequera, D.; Morán, M.J.; Martín, M.A.; Ferrer, I.; Bermejo-Pareja, F.; Carro, E. Aβ accumulation in choroid plexus is associated with mitochondrial-induced apoptosis. Neurobiol. Aging 2010, 31, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain Complexes I and IV are impaired by β-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Alzheimer’s APP mangles mitochondria. Nat. Med. 2006, 12, 1241–1243. [Google Scholar] [CrossRef]
- Pavlov, P.F.; Petersen, C.H.; Glaser, E.; Ankarcrona, M. Mitochondrial accumulation of APP and Aβ: Significance for Alzheimer disease pathogenesis. J. Cell. Mol. Med. 2009, 13, 4137–4145. [Google Scholar] [CrossRef]
- Agrawal, R.R.; Montesinos, J.; Larrea, D.; Area-Gomez, E.; Pera, M. The silence of the fats: A MAM’s story about Alzheimer. Neurobiol. Dis. 2020, 145, 105062. [Google Scholar] [CrossRef]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef]
- Montesinos, J.; Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Agrawal, R.R.; Velasco, K.R.; Yun, T.D.; Stavrovskaya, I.G.; Xu, Y.; Koo, S.Y.; et al. The Alzheimer’s disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. EMBO J. 2020, 39. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2020. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-β toxicity in alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Zhang, B.; Lee, V.M.Y.; Trojanowski, J.Q. Axonal transport defects: A common theme in neurodegenerative diseases. Acta Neuropathol. 2005, 109, 5–13. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Dröse, S.; Brandt, U.; Müller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Perreault, S.; Bousquet, O.; Lauzon, M.; Paiement, J.; Leclerc, N. Increased association between rough endoplasmic reticulum membranes and mitochondria in transgenic mice that express P301L Tau. J. Neuropathol. Exp. Neurol. 2009, 68, 503–514. [Google Scholar] [CrossRef]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E.-M.; Mandelkow, E. Overexpression of Tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef]
- Afreen, S.; Riherd Methner, D.N.; Ferreira, A. Tau45-230 association with the cytoskeleton and membrane-bound organelles: Functional implications in neurodegeneration. Neuroscience 2017, 362, 104–117. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Wang, Q.; Lin, Z.; Chen, M.L.; Sun, G.Z. Endoplasmic reticulum (ER) stress inhibitor salubrinal protects against ceramide-induced SH-SY5Y cell death. Biochem. Biophys. Res. Commun. 2012, 427, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, K.S.; Lee, H.J.; Kim, D.H.; Noh, Y.H.; Yu, K.; Jung, H.Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; et al. Activation of PERK signaling attenuates Aβ-mediated ER stress. PLoS ONE 2010, 5, e10489. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, Y.; Zhang, H.; Ma, Q.; Zhang, Y.; Xu, H. Salubrinal attenuates β-amyloid-induced neuronal death and microglial activation by inhibition of the NF-κB pathway. Neurobiol. Aging 2012, 33, 1007.e9–1007.e17. [Google Scholar] [CrossRef] [PubMed]
- Barreda-Manso, M.A.; Yanguas-Casás, N.; Nieto-Sampedro, M.; Romero-Ramírez, L. Salubrinal inhibits the expression of proteoglycans and favors neurite outgrowth from cortical neurons in vitro. Exp. Cell Res. 2015, 335, 82–90. [Google Scholar] [CrossRef]
- Paris, D.; Ganey, N.J.; Laporte, V.; Patel, N.S.; Beaulieu-Abdelahad, D.; Bachmeier, C.; March, A.; Ait-Ghezala, G.; Mullan, M.J. Reduction of β-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflamm. 2010, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.W.; Tsui, Y.T.C.; Wong, T.T.; Cheung, S.K.K.; Goggins, W.B.; Yi, L.M.; Cheng, K.K.; Baum, L. Effects of 17-allylamino-17-demethoxygeldanamycin (17-AAG) in transgenic mouse models of frontotemporal lobar degeneration and Alzheimer’s disease. Transl. Neurodegener. 2013, 2, 1–9. [Google Scholar] [CrossRef]
- Twaroski, Y.; Zaja, C.; Bosnjak, B. Altered Mitochondrial dynamics contributes to propofol-induced cell death in human stem cell-derived neurons. Physiol. Behav. 2015, 123, 1067–1083. [Google Scholar] [CrossRef]
- Xie, N.; Wang, C.; Lian, Y.; Wu, C.; Zhang, H.; Zhang, Q. Inhibition of mitochondrial fission attenuates Aβ-induced microglia apoptosis. Neuroscience 2014, 256, 36–42. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division inhibitor 1 protects against amyloid-β induced mitochondrial fragmentation and synaptic damage in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef]
- Yeon, J.Y.; Min, S.H.; Park, H.J.; Kim, J.W.; Lee, Y.H.; Park, S.Y.; Jeong, P.S.; Park, H.; Lee, D.S.; Kim, S.U.; et al. Mdivi-1, mitochondrial fission inhibitor, impairs developmental competence and mitochondrial function of embryos and cells in pigs. J. Reprod. Dev. 2015, 61, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Disatnik, M.; Monbureau, M.; Shamloo, M.; Mochly-rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388. [Google Scholar] [CrossRef] [PubMed]
- Arismendi-Morillo, G.; Castellano-Ramírez, A.; Seyfried, T.N. Ultrastructural characterization of the Mitochondria-associated membranes abnormalities in human astrocytomas: Functional and therapeutics implications. Ultrastruct. Pathol. 2017, 41, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Lynes, E.M.; Bui, M.; Yap, M.C.; Benson, M.D.; Schneider, B.; Ellgaard, L.; Berthiaume, L.G.; Simmen, T. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 2012, 31, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Maraver, J.; Loh, S.H.Y.; Martins, L.M. Forcing contacts between mitochondria and the endoplasmic reticulum extends lifespan in a Drosophila model of Alzheimer’s disease. Biol. Open 2020, 9. [Google Scholar] [CrossRef]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Yoon, J.H.; Cho, J.H.; Lee, S.J.; et al. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2020. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9521. https://doi.org/10.3390/ijms21249521
Eysert F, Kinoshita PF, Mary A, Vaillant-Beuchot L, Checler F, Chami M. Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(24):9521. https://doi.org/10.3390/ijms21249521
Chicago/Turabian StyleEysert, Fanny, Paula Fernanda Kinoshita, Arnaud Mary, Loan Vaillant-Beuchot, Frédéric Checler, and Mounia Chami. 2020. "Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 24: 9521. https://doi.org/10.3390/ijms21249521
APA StyleEysert, F., Kinoshita, P. F., Mary, A., Vaillant-Beuchot, L., Checler, F., & Chami, M. (2020). Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease. International Journal of Molecular Sciences, 21(24), 9521. https://doi.org/10.3390/ijms21249521