Skeletal Phenotypes Due to Abnormalities in Mitochondrial Protein Homeostasis and Import


 and
and
Abstract
1. Introduction
1.1. Mitochondrial Protein Homeostasis
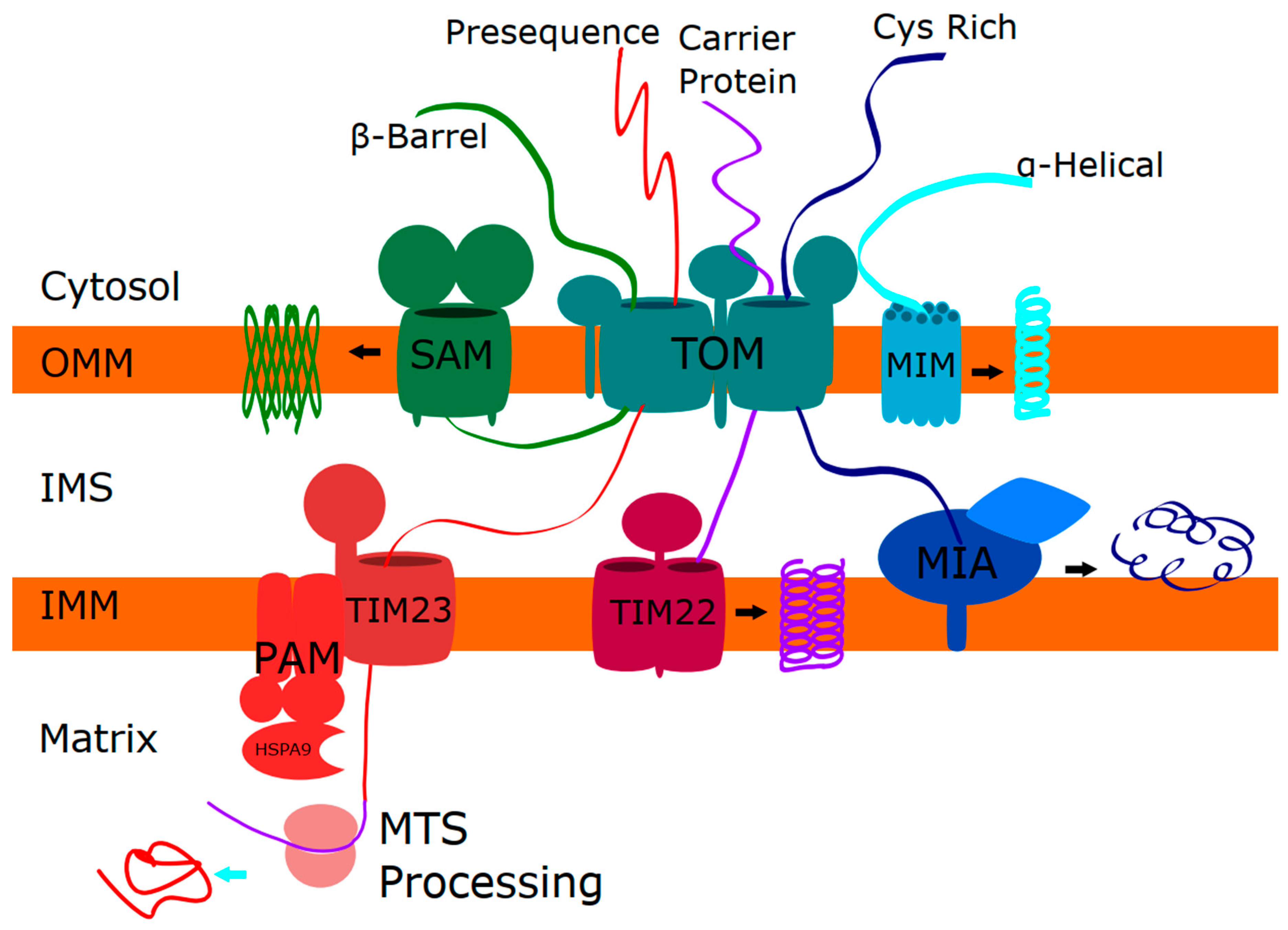
1.2. Mitochondrial Protein Import
2. Skeletal Phenotypes due to Disorders of Mitochondrial Protein Function
2.1. LONP1 and CODAS (Cerebral, Ocular, Dental, Auricular, and Skeletal Syndrome)
2.2. HSPA9 and EVEN-PLUS (Epiphyseal, Vertebral, and Ocular Changes Plus Associated Findings of Severe Microtia, Nasal Hypoplasia, and Other Malformations)
3. Expanding the List of Mitochondrial Skeletal Disorders
3.1. IARS2 and CAGSSS (Cataracts, Growth Hormone Deficiency, Sensory Neuropathy, Sensorineural Hearing Loss, Skeletal Dysplasia Syndrome)
3.2. PAM16 and SMDMDM (Spondylometaphyseal Dysplasia, Megarbane–Dagher–Melki Type)
3.3. AIFM1 and SEMD-HL (Spondyloepimetaphyseal Dysplasia, X-linked, with Hypomyelinating Leukodystrophy)
3.4. PISD
4. Integrating the Mitochondrial Functions Underlying Skeletal Phenotypes
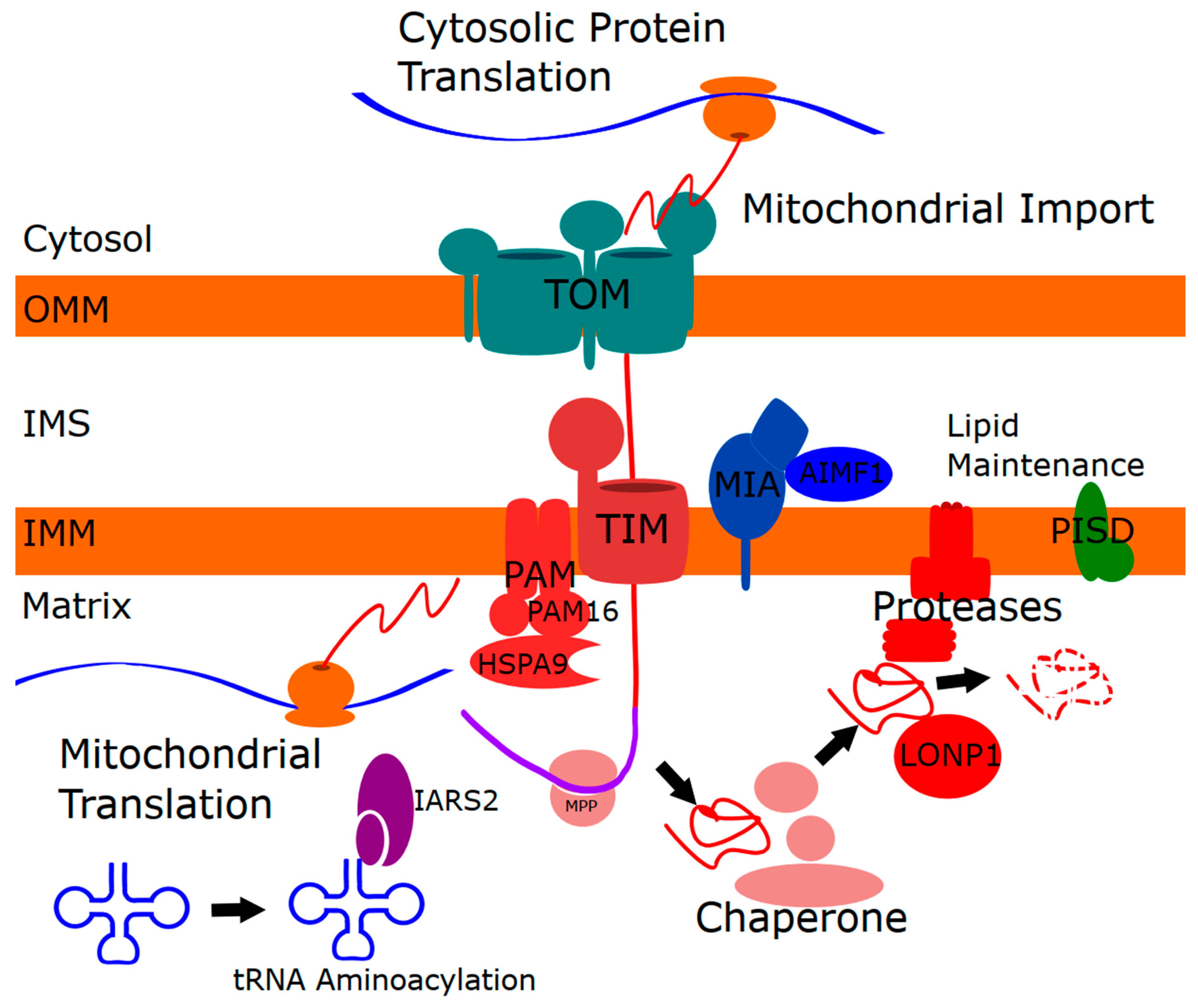
4.1. Linking Mitochondrial Protein Import to Mitochondrial Proteases and Chaperones
4.2. Mitochondrial Lipid Homeostasis
Connecting Mitochondrial Lipids to Mitochondrial Protein Homeostasis and Import
4.3. Mitochondrial tRNAs
4.3.1. Mitochondrial Aminoacyl-tRNA Synthetases
4.3.2. Connecting Mitochondrial tRNAs to Mitochondrial Protein Homeostasis
5. Additional Features in Mitochondrial Skeletal Disorders
6. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Schon, K.R.; Ratnaike, T.; van den Ameele, J.; Horvath, R.; Chinnery, P.F. Mitochondrial Diseases: A Diagnostic Revolution. Trends Genet. 2020, 36, 702–717. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Oláhová, M.; McFarland, R.; Taylor, R.W. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J. Inherit. Metab. Dis. 2020, 43, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Clark-Walker, G.D. The petite mutation in yeasts: 50 years on. Int. Rev. Cytol. 2000, 194, 197–238. [Google Scholar] [PubMed]
- Bernardi, G. Lessons from a small, dispensable genome: The mitochondrial genome of yeast. Gene 2005, 354, 189–200. [Google Scholar] [CrossRef]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A. 2019, 179, 2393–2419. [Google Scholar] [CrossRef]
- Ruan, L.; Wang, Y.; Zhang, X.; Tomaszewski, A.; McNamara, J.T.; Li, R. Mitochondria-Associated Proteostasis. Annu. Rev. Biophys. 2020, 49, 41–67. [Google Scholar] [CrossRef] [PubMed]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Høj, P.B.; Hoogenraad, N.J. Selective Induction of Mitochondrial Chaperones in Response to Loss of the Mitochondrial Genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Melber, A.; Haynes, C.M. UPRmt regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef]
- Hansen, K.G.; Herrmann, J.M. Transport of Proteins into Mitochondria. Protein J. 2019, 38, 330–342. [Google Scholar] [CrossRef]
- Poveda-Huertes, D.; Mulica, P.; Vögtle, F.N. The versatility of the mitochondrial presequence processing machinery: Cleavage, quality control and turnover. Cell Tissue Res. 2017, 367, 73–81. [Google Scholar] [CrossRef]
- Singh, R.; Jamdar, S.N.; Goyal, V.D.; Kumar, A.; Ghosh, B.; Makde, R.D. Structure of the human aminopeptidase XPNPEP3 and comparison of its in vitro activity with Icp55 orthologs: Insights into diverse cellular processes. J. Biol. Chem. 2017, 292, 10035–10047. [Google Scholar] [CrossRef] [PubMed]
- Jacques, S.; van der Sloot, A.M.; C. Huard, C.; Coulombe-Huntington, J.; Tsao, S.; Tollis, S.; Bertomeu, T.; Culp, E.J.; Pallant, D.; Cook, M.A.; et al. Imipridone Anticancer Compounds Ectopically Activate the ClpP Protease and Represent a New Scaffold for Antibiotic Development. Genetics 2020, 214, 1103–1120. [Google Scholar] [CrossRef]
- Mossmann, D.; Meisinger, C.; Vögtle, F.-N. Processing of mitochondrial presequences. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2012, 1819, 1098–1106. [Google Scholar] [CrossRef]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.H.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. A 2015, 167, 1501–1509. [Google Scholar] [CrossRef]
- Royer-Bertrand, B.; Castillo-Taucher, S.; Moreno-Salinas, R.; Cho, T.-J.; Chae, J.-H.; Choi, M.; Kim, O.-H.; Dikoglu, E.; Campos-Xavier, B.; Girardi, E.; et al. Mutations in the heat-shock protein A9 (HSPA9) gene cause the EVEN-PLUS syndrome of congenital malformations and skeletal dysplasia. Sci. Rep. 2015, 5, 17154. [Google Scholar] [CrossRef]
- Pinti, M.; Gibellini, L.; Nasi, M.; De Biasi, S.; Bortolotti, C.A.; Iannone, A.; Cossarizza, A. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim. Biophys. Acta 2016, 1857, 1300–1306. [Google Scholar] [CrossRef]
- Gibellini, L.; De Gaetano, A.; Mandrioli, M.; Van Tongeren, E.; Bortolotti, C.A.; Cossarizza, A.; Pinti, M. Chapter One—The biology of Lonp1: More than a mitochondrial protease. In International Review of Cell and Molecular Biology; Galluzzi, L., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 354, pp. 1–61. [Google Scholar]
- Zurita Rendón, O.; Shoubridge, E.A. LONP1 Is Required for Maturation of a Subset of Mitochondrial Proteins, and Its Loss Elicits an Integrated Stress Response. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef]
- Shebib, S.M.; Reed, M.H.; Shuckett, E.P.; Cross, H.G.; Perry, J.B.; Chudley, A.E. Newly recognized syndrome of cerebral, ocular, dental, auricular, skeletal anomalies: CODAS syndrome—A case report. Am. J. Med. Genet. 1991, 40, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Inui, T.; Anzai, M.; Takezawa, Y.; Endo, W.; Kakisaka, Y.; Kikuchi, A.; Onuma, A.; Kure, S.; Nishino, I.; Ohba, C.; et al. A novel mutation in the proteolytic domain of LONP1 causes atypical CODAS syndrome. J. Hum. Genet. 2017, 62, 653–655. [Google Scholar] [CrossRef]
- Peter, B.; Waddington, C.L.; Oláhová, M.; Sommerville, E.W.; Hopton, S.; Pyle, A.; Champion, M.; Ohlson, M.; Siibak, T.; Chrzanowska-Lightowlers, Z.M.A.; et al. Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum. Mol. Genet. 2018, 27, 1743–1753. [Google Scholar] [CrossRef]
- Khan, A.O.; AlBakri, A. Clinical features of LONP1-related infantile cataract. J. AAPOS Off. Publ. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2018, 22, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Hannah-Shmouni, F.; MacNeil, L.; Brady, L.; Nilsson, M.I.; Tarnopolsky, M. Expanding the Clinical Spectrum of LONP1-Related Mitochondrial Cytopathy. Front. Neurol. 2019, 10, 981. [Google Scholar] [CrossRef]
- Nimmo, G.A.M.; Venkatesh, S.; Pandey, A.K.; Marshall, C.R.; Hazrati, L.-N.; Blaser, S.; Ahmed, S.; Cameron, J.; Singh, K.; Ray, P.N.; et al. Bi-allelic mutations of LONP1 encoding the mitochondrial LonP1 protease cause pyruvate dehydrogenase deficiency and profound neurodegeneration with progressive cerebellar atrophy. Hum. Mol. Genet. 2019, 28, 290–306. [Google Scholar] [CrossRef]
- Besse, A.; Brezavar, D.; Hanson, J.; Larson, A.; Bonnen, P.E. LONP1 de novo dominant mutation causes mitochondrial encephalopathy with loss of LONP1 chaperone activity and excessive LONP1 proteolytic activity. Mitochondrion 2020, 51, 68–78. [Google Scholar] [CrossRef]
- Jo, D.S.; Park, S.J.; Kim, A.-K.; Park, N.Y.; Kim, J.B.; Bae, J.-E.; Park, H.J.; Shin, J.H.; Chang, J.W.; Kim, P.K.; et al. Loss of HSPA9 induces peroxisomal degradation by increasing pexophagy. Autophagy 2020, 1–15. [Google Scholar] [CrossRef]
- Wadhwa, R.; Taira, K.; Kaul, S.C. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: What, when, and where? Cell Stress Chaperones 2002, 7, 309–316. [Google Scholar] [CrossRef]
- Finka, A.; Sharma, S.K.; Goloubinoff, P. Multi-layered molecular mechanisms of polypeptide holding, unfolding and disaggregation by HSP70/HSP110 chaperones. Front. Mol. Biosci. 2015, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Voos, W. Chaperone-protease networks in mitochondrial protein homeostasis. Biochim. Biophys. Acta 2013, 1833, 388–399. [Google Scholar] [CrossRef]
- Katiyar, A.; Fujimoto, M.; Tan, K.; Kurashima, A.; Srivastava, P.; Okada, M.; Takii, R.; Nakai, A. HSF1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio 2020. [Google Scholar] [CrossRef] [PubMed]
- Moseng, M.A.; Nix, J.C.; Page, R.C. Biophysical Consequences of EVEN-PLUS Syndrome Mutations for the Function of Mortalin. J. Phys. Chem. B 2019, 123, 3383–3396. [Google Scholar] [CrossRef]
- Younger, G.; Vetrini, F.; Weaver, D.D.; Lynnes, T.C.; Treat, K.; Pratt, V.M.; Torres-Martinez, W. EVEN-PLUS syndrome: A case report with novel variants in HSPA9 and evidence of HSPA9 gene dysfunction. Am. J. Med. Genet. A. 2020. [Google Scholar] [CrossRef]
- Schmitz-Abe, K.; Ciesielski, S.J.; Schmidt, P.J.; Campagna, D.R.; Rahimov, F.; Schilke, B.A.; Cuijpers, M.; Rieneck, K.; Lausen, B.; Linenberger, M.L.; et al. Congenital sideroblastic anemia due to mutations in the mitochondrial HSP70 homologue HSPA9. Blood 2015, 126, 2734–2738. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Schelling, C.; Kato, H.; Rapaport, D.; Woitalla, D.; Schiesling, C.; Schulte, C.; Sharma, M.; Illig, T.; Bauer, P.; et al. Dissecting the role of the mitochondrial chaperone mortalin in Parkinson’s disease: Functional impact of disease-related variants on mitochondrial homeostasis. Hum. Mol. Genet. 2010, 19, 4437–4452. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Buhas, D.; Majewski, J.; Sasarman, F.; Papillon-Cavanagh, S.; Thiffault, I.; Thiffaut, I.; Sheldon, K.M.; Massicotte, C.; Patry, L.; et al. Mutation in the nuclear-encoded mitochondrial isoleucyl-tRNA synthetase IARS2 in patients with cataracts, growth hormone deficiency with short stature, partial sensorineural deafness, and peripheral neuropathy or with Leigh syndrome. Hum. Mutat. 2014, 35, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Mehawej, C.; Delahodde, A.; Legeai-Mallet, L.; Delague, V.; Kaci, N.; Desvignes, J.-P.; Kibar, Z.; Capo-Chichi, J.-M.; Chouery, E.; Munnich, A.; et al. The impairment of MAGMAS function in human is responsible for a severe skeletal dysplasia. PLoS Genet. 2014, 10, e1004311. [Google Scholar] [CrossRef]
- Mierzewska, H.; Rydzanicz, M.; Biegański, T.; Kosinska, J.; Mierzewska-Schmidt, M.; Ługowska, A.; Pollak, A.; Stawiński, P.; Walczak, A.; Kędra, A.; et al. Spondyloepimetaphyseal dysplasia with neurodegeneration associated with AIFM1 mutation—A novel phenotype of the mitochondrial disease. Clin. Genet. 2017, 91, 30–37. [Google Scholar] [CrossRef]
- Girisha, K.M.; von Elsner, L.; Neethukrishna, K.; Muranjan, M.; Shukla, A.; Bhavani, G.S.; Nishimura, G.; Kutsche, K.; Mortier, G. The homozygous variant c.797G>A/p.(Cys266Tyr) in PISD is associated with a Spondyloepimetaphyseal dysplasia with large epiphyses and disturbed mitochondrial function. Hum. Mutat. 2019, 40, 299–309. [Google Scholar] [CrossRef]
- Zhao, T.; Goedhart, C.M.; Sam, P.N.; Sabouny, R.; Lingrell, S.; Cornish, A.J.; Lamont, R.E.; Bernier, F.P.; Sinasac, D.; Parboosingh, J.S.; et al. PISD is a mitochondrial disease gene causing skeletal dysplasia, cataracts, and white matter changes. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Peter, V.G.; Quinodoz, M.; Pinto-Basto, J.; Sousa, S.B.; Di Gioia, S.A.; Soares, G.; Ferraz Leal, G.; Silva, E.D.; Pescini Gobert, R.; Miyake, N.; et al. The Liberfarb syndrome, a multisystem disorder affecting eye, ear, bone, and brain development, is caused by a founder pathogenic variant in thePISD gene. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Mordaunt, D.A.; Savaririyan, R. Does IARS2 deficiency cause an intrinsic disorder of bone development (skeletal dysplasia) or are the reported skeletal changes secondary to growth hormone deficiency and neuromuscular involvement? Hum. Mutat. 2015, 36, 388. [Google Scholar] [CrossRef] [PubMed]
- Samuels, M.E.; Alos, N.; Deal, C.L. Response to: Does IARS2 deficiency cause an intrinsic disorder of bone development (skeletal dysplasia) or are the reported skeletal changes secondary to growth hormone deficiency and neuromuscular involvement? Hum. Mutat. 2015, 36, 389. [Google Scholar] [CrossRef]
- Jabbour, S.; Harissi-Dagher, M. Recessive Mutation in a Nuclear-Encoded Mitochondrial tRNA Synthetase Associated With Infantile Cataract, Congenital Neurotrophic Keratitis, and Orbital Myopathy. Cornea 2016, 35, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Moosa, S.; Haagerup, A.; Gregersen, P.A.; Petersen, K.K.; Altmüller, J.; Thiele, H.; Nürnberg, P.; Cho, T.-J.; Kim, O.-H.; Nishimura, G.; et al. Confirmation of CAGSSS syndrome as a distinct entity in a Danish patient with a novel homozygous mutation in IARS2. Am. J. Med. Genet. A. 2017, 173, 1102–1108. [Google Scholar] [CrossRef]
- Vona, B.; Maroofian, R.; Bellacchio, E.; Najafi, M.; Thompson, K.; Alahmad, A.; He, L.; Ahangari, N.; Rad, A.; Shahrokhzadeh, S.; et al. Expanding the clinical phenotype of IARS2-related mitochondrial disease. BMC Med. Genet. 2018, 19, 196. [Google Scholar] [CrossRef]
- Takezawa, Y.; Fujie, H.; Kikuchi, A.; Niihori, T.; Funayama, R.; Shirota, M.; Nakayama, K.; Aoki, Y.; Sasaki, M.; Kure, S. Novel IARS2 mutations in Japanese siblings with CAGSSS, Leigh, and West syndrome. Brain Dev. 2018, 40, 934–938. [Google Scholar] [CrossRef]
- Frazier, A.E.; Dudek, J.; Guiard, B.; Voos, W.; Li, Y.; Lind, M.; Meisinger, C.; Geissler, A.; Sickmann, A.; Meyer, H.E.; et al. Pam16 has an essential role in the mitochondrial protein import motor. Nat. Struct. Mol. Biol. 2004, 11, 226–233. [Google Scholar] [CrossRef]
- Sinha, D.; Joshi, N.; Chittoor, B.; Samji, P.; D’Silva, P. Role of Magmas in protein transport and human mitochondria biogenesis. Hum. Mol. Genet. 2010, 19, 1248–1262. [Google Scholar] [CrossRef]
- Pais, J.E.; Schilke, B.; Craig, E.A. Reevaluation of the role of the Pam18:Pam16 interaction in translocation of proteins by the mitochondrial Hsp70-based import motor. Mol. Biol. Cell 2011, 22, 4740–4749. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Srivastava, S.; Krishna, L.; D’Silva, P. Unraveling the intricate organization of mammalian mitochondrial presequence translocases: Existence of multiple translocases for maintenance of mitochondrial function. Mol. Cell. Biol. 2014, 34, 1757–1775. [Google Scholar] [CrossRef]
- Mégarbané, A.; Dagher, R.; Melki, I. Sib pair with previously unreported skeletal dysplasia. Am. J. Med. Genet. A. 2008, 146A, 2916–2919. [Google Scholar] [CrossRef]
- Mégarbané, A.; Mehawej, C.; El Zahr, A.; Haddad, S.; Cormier-Daire, V. A second family with autosomal recessive spondylometaphyseal dysplasia and early death. Am. J. Med. Genet. A. 2014, 164A, 1010–1014. [Google Scholar] [CrossRef]
- Moosa, S.; Fano, V.; Obregon, M.G.; Altmüller, J.; Thiele, H.; Nürnberg, P.; Nishimura, G.; Wollnik, B. A novel homozygous PAM16 mutation in a patient with a milder phenotype and longer survival. Am. J. Med. Genet. A. 2016, 170, 2436–2439. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Miramar, M.D.; Costantini, P.; Ravagnan, L.; Saraiva, L.M.; Haouzi, D.; Brothers, G.; Penninger, J.M.; Peleato, M.L.; Kroemer, G.; Susin, S.A. NADH Oxidase Activity of Mitochondrial Apoptosis-inducing Factor. J. Biol. Chem. 2001, 276, 16391–16398. [Google Scholar] [CrossRef]
- Sevrioukova, I.F. Structure/Function Relations in AIFM1 Variants Associated with Neurodegenerative Disorders. J. Mol. Biol. 2016, 428, 3650–3665. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Prabhu, S.P.; Lidov, H.G.W.; Shi, J.; Anselm, I.; Brownstein, C.A.; Bainbridge, M.N.; Beggs, A.H.; Vargas, S.O.; Agrawal, P.B. AIFM1 mutation presenting with fatal encephalomyopathy and mitochondrial disease in an infant. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001560. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Dai, A.-Y.; Tao, K.; Xiao, Q.; Huang, Z.-L.; Gao, M.; Li, H.; Wang, X.; Cao, W.-X.; Feng, W.-L. Heat shock protein-70 neutralizes apoptosis inducing factor in Bcr/Abl expressing cells. Cell. Signal. 2015, 27, 1949–1955. [Google Scholar] [CrossRef]
- Hangen, E.; Féraud, O.; Lachkar, S.; Mou, H.; Doti, N.; Fimia, G.M.; Lam, N.-V.; Zhu, C.; Godin, I.; Muller, K.; et al. Interaction between AIF and CHCHD4 Regulates Respiratory Chain Biogenesis. Mol. Cell 2015, 58, 1001–1014. [Google Scholar] [CrossRef]
- Meyer, K.; Buettner, S.; Ghezzi, D.; Zeviani, M.; Bano, D.; Nicotera, P. Loss of apoptosis-inducing factor critically affects MIA40 function. Cell Death Dis. 2015, 6, e1814. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, C.; Arena, G.; Nedara, K.; Edwards, R.; Brenner, C.; Tokatlidis, K.; Modjtahedi, N. AIF meets the CHCHD4/Mia40-dependent mitochondrial import pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165746. [Google Scholar] [CrossRef]
- Miyake, N.; Wolf, N.I.; Cayami, F.K.; Crawford, J.; Bley, A.; Bulas, D.; Conant, A.; Bent, S.J.; Gripp, K.W.; Hahn, A.; et al. X-linked hypomyelination with spondylometaphyseal dysplasia (H-SMD) associated with mutations in AIFM1. Neurogenetics 2017, 18, 185–194. [Google Scholar] [CrossRef]
- Bano, D.; Prehn, J.H.M. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef]
- Rinaldi, C.; Grunseich, C.; Sevrioukova, I.F.; Schindler, A.; Horkayne-Szakaly, I.; Lamperti, C.; Landouré, G.; Kennerson, M.L.; Burnett, B.G.; Bönnemann, C.; et al. Cowchock syndrome is associated with a mutation in apoptosis-inducing factor. Am. J. Hum. Genet. 2012, 91, 1095–1102. [Google Scholar] [CrossRef]
- Sancho, P.; Sánchez-Monteagudo, A.; Collado, A.; Marco-Marín, C.; Domínguez-González, C.; Camacho, A.; Knecht, E.; Espinós, C.; Lupo, V. A newly distal hereditary motor neuropathy caused by a rare AIFM1 mutation. Neurogenetics 2017, 18, 245–250. [Google Scholar] [CrossRef]
- Zong, L.; Guan, J.; Ealy, M.; Zhang, Q.; Wang, D.; Wang, H.; Zhao, Y.; Shen, Z.; Campbell, C.A.; Wang, F.; et al. Mutations in apoptosis-inducing factor cause X-linked recessive auditory neuropathy spectrum disorder. J. Med. Genet. 2015, 52, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bing, D.; Li, J.; Xie, L.; Xiong, F.; Lan, L.; Wang, D.; Guan, J.; Wang, Q. High Frequency of AIFM1 Variants and Phenotype Progression of Auditory Neuropathy in a Chinese Population. Neural Plast. 2020, 2020, 5625768. [Google Scholar] [CrossRef]
- Heimer, G.; Eyal, E.; Zhu, X.; Ruzzo, E.K.; Marek-Yagel, D.; Sagiv, D.; Anikster, Y.; Reznik-Wolf, H.; Pras, E.; Oz Levi, D.; et al. Mutations in AIFM1 cause an X-linked childhood cerebellar ataxia partially responsive to riboflavin. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2018, 22, 93–101. [Google Scholar] [CrossRef]
- Pandolfo, M.; Rai, M.; Remiche, G.; Desmyter, L.; Vandernoot, I. Cerebellar ataxia, neuropathy, hearing loss, and intellectual disability due to AIFM1 mutation. Neurol. Genet. 2020, 6, e420. [Google Scholar] [CrossRef]
- Ardissone, A.; Piscosquito, G.; Legati, A.; Langella, T.; Lamantea, E.; Garavaglia, B.; Salsano, E.; Farina, L.; Moroni, I.; Pareyson, D.; et al. A slowly progressive mitochondrial encephalomyopathy widens the spectrum of AIFM1 disorders. Neurology 2015, 84, 2193–2195. [Google Scholar] [CrossRef]
- Kettwig, M.; Schubach, M.; Zimmermann, F.A.; Klinge, L.; Mayr, J.A.; Biskup, S.; Sperl, W.; Gärtner, J.; Huppke, P. From ventriculomegaly to severe muscular atrophy: Expansion of the clinical spectrum related to mutations in AIFM1. Mitochondrion 2015, 21, 12–18. [Google Scholar] [CrossRef]
- Ghezzi, D.; Sevrioukova, I.; Invernizzi, F.; Lamperti, C.; Mora, M.; D’Adamo, P.; Novara, F.; Zuffardi, O.; Uziel, G.; Zeviani, M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am. J. Hum. Genet. 2010, 86, 639–649. [Google Scholar] [CrossRef]
- Berger, I.; Ben-Neriah, Z.; Dor-Wolman, T.; Shaag, A.; Saada, A.; Zenvirt, S.; Raas-Rothschild, A.; Nadjari, M.; Kaestner, K.H.; Elpeleg, O. Early prenatal ventriculomegaly due to an AIFM1 mutation identified by linkage analysis and whole exome sequencing. Mol. Genet. Metab. 2011, 104, 517–520. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucińska-Więckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosińska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 174. [Google Scholar] [CrossRef]
- Diodato, D.; Tasca, G.; Verrigni, D.; D’Amico, A.; Rizza, T.; Tozzi, G.; Martinelli, D.; Verardo, M.; Invernizzi, F.; Nasca, A.; et al. A novel AIFM1 mutation expands the phenotype to an infantile motor neuron disease. Eur. J. Hum. Genet. EJHG 2016, 24, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, R.; Nanowski, T.S.; Beigneux, A.; Kulinski, A.; Young, S.G.; Vance, J.E. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 2005, 280, 40032–40040. [Google Scholar] [CrossRef]
- Tasseva, G.; Bai, H.D.; Davidescu, M.; Haromy, A.; Michelakis, E.; Vance, J.E. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 2013, 288, 4158–4173. [Google Scholar] [CrossRef]
- Heden, T.D.; Johnson, J.M.; Ferrara, P.J.; Eshima, H.; Verkerke, A.R.P.; Wentzler, E.J.; Siripoksup, P.; Narowski, T.M.; Coleman, C.B.; Lin, C.-T.; et al. Mitochondrial PE potentiates respiratory enzymes to amplify skeletal muscle aerobic capacity. Sci. Adv. 2019, 5, eaax8352. [Google Scholar] [CrossRef]
- MacVicar, T.; Ohba, Y.; Nolte, H.; Mayer, F.C.; Tatsuta, T.; Sprenger, H.-G.; Lindner, B.; Zhao, Y.; Li, J.; Bruns, C.; et al. Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 2019, 575, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Tricarico, R.; Savage, M.; Golemis, E.A.; Hall, M.J. Disease-Associated Genetic Variation in Human Mitochondrial Protein Import. Am. J. Hum. Genet. 2019, 104, 784–801. [Google Scholar] [CrossRef]
- Barth, P.G.; Scholte, H.R.; Berden, J.A.; Van der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.; Van ’t Veer-Korthof, E.T.; Van der Harten, J.J.; Sobotka-Plojhar, M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef]
- Haghighi, A.; Haack, T.B.; Atiq, M.; Mottaghi, H.; Haghighi-Kakhki, H.; Bashir, R.A.; Ahting, U.; Feichtinger, R.G.; Mayr, J.A.; Rötig, A.; et al. Sengers syndrome: Six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients. Orphanet J. Rare Dis. 2014, 9, 119. [Google Scholar] [CrossRef]
- Davey, K.M.; Parboosingh, J.S.; McLeod, D.R.; Chan, A.; Casey, R.; Ferreira, P.; Snyder, F.F.; Bridge, P.J.; Bernier, F.P. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J. Med. Genet. 2006, 43, 385–393. [Google Scholar] [CrossRef]
- Lu, Y.-W.; Claypool, S.M. Disorders of phospholipid metabolism: An emerging class of mitochondrial disease due to defects in nuclear genes. Front. Genet. 2015, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Srivastava, S.; D’Silva, P. Functional Diversity of Human Mitochondrial J-proteins Is Independent of Their Association with the Inner Membrane Presequence Translocase. J. Biol. Chem. 2016, 291, 17345–17359. [Google Scholar] [CrossRef]
- Kang, Y.; Stroud, D.A.; Baker, M.J.; De Souza, D.P.; Frazier, A.E.; Liem, M.; Tull, D.; Mathivanan, S.; McConville, M.J.; Thorburn, D.R.; et al. Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 2017, 67, 457–470.e5. [Google Scholar] [CrossRef]
- Vukotic, M.; Nolte, H.; König, T.; Saita, S.; Ananjew, M.; Krüger, M.; Tatsuta, T.; Langer, T. Acylglycerol Kinase Mutated in Sengers Syndrome Is a Subunit of the TIM22 Protein Translocase in Mitochondria. Mol. Cell 2017, 67, 471–483.e7. [Google Scholar] [CrossRef]
- Mårtensson, C.U.; Becker, T. Acylglycerol Kinase: Mitochondrial Protein Transport Meets Lipid Biosynthesis. Trends Cell Biol. 2017, 27, 700–702. [Google Scholar] [CrossRef]
- Quiros, P.M.; Langer, T.; Lopez-Otin, C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol 2015, 16, 345–359. [Google Scholar] [CrossRef]
- Levytskyy, R.M.; Bohovych, I.; Khalimonchuk, O. Metalloproteases of the Inner Mitochondrial Membrane. Biochemistry 2017, 56, 4737–4746. [Google Scholar] [CrossRef]
- Gebert, N.; Ryan, M.T.; Pfanner, N.; Wiedemann, N.; Stojanovski, D. Mitochondrial protein import machineries and lipids: A functional connection. Biochim. Biophys. Acta 2011, 1808, 1002–1011. [Google Scholar] [CrossRef]
- Malhotra, K.; Modak, A.; Nangia, S.; Daman, T.H.; Gunsel, U.; Robinson, V.L.; Mokranjac, D.; May, E.R.; Alder, N.N. Cardiolipin mediates membrane and channel interactions of the mitochondrial TIM23 protein import complex receptor Tim50. Sci. Adv. 2017, 3, e1700532. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Sissler, M.; González-Serrano, L.E.; Westhof, E. Recent Advances in Mitochondrial Aminoacyl-tRNA Synthetases and Disease. Trends Mol. Med. 2017, 23, 693–708. [Google Scholar] [CrossRef]
- Shutt, T.E.; Shadel, G.S. A compendium of human mitochondrial gene expression machinery with links to disease. Env. Mol Mutagen 2010, 51, 360–379. [Google Scholar] [CrossRef]
- González-Serrano, L.E.; Chihade, J.W.; Sissler, M. When a common biological role does not imply common disease outcomes: Disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J. Biol. Chem. 2019, 294, 5309–5320. [Google Scholar] [CrossRef]
- van der Knaap, M.S.; Bugiani, M.; Mendes, M.I.; Riley, L.G.; Smith, D.E.C.; Rudinger-Thirion, J.; Frugier, M.; Breur, M.; Crawford, J.; van Gaalen, J.; et al. Biallelic variants in LARS2 and KARS cause deafness and (ovario)leukodystrophy. Neurology 2019, 92, e1225–e1237. [Google Scholar] [CrossRef]
- Guo, M.; Schimmel, P. Essential Non-Translational Functions of tRNA Synthetases. Nat. Chem. Biol. 2013, 9, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.A.R.; Ibba, M. Aminoacyl-tRNA Synthetases. RNA 2020. [Google Scholar] [CrossRef]
- Wang, M.; Sips, P.; Khin, E.; Rotival, M.; Sun, X.; Ahmed, R.; Widjaja, A.A.; Schafer, S.; Yusoff, P.; Choksi, P.K.; et al. Wars2 is a determinant of angiogenesis. Nat. Commun. 2016, 7, 12061. [Google Scholar] [CrossRef]
- Ferreira, N.; Perks, K.L.; Rossetti, G.; Rudler, D.L.; Hughes, L.A.; Ermer, J.A.; Scott, L.H.; Kuznetsova, I.; Richman, T.R.; Narayana, V.K.; et al. Stress signaling and cellular proliferation reverse the effects of mitochondrial mistranslation. EMBO J. 2019, 38, e102155. [Google Scholar] [CrossRef]
- Coyne, L.P.; Chen, X.J. Consequences of inner mitochondrial membrane protein misfolding. Mitochondrion 2019, 49, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Seiferling, D.; Szczepanowska, K.; Becker, C.; Senft, K.; Hermans, S.; Maiti, P.; König, T.; Kukat, A.; Trifunovic, A. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep. 2016, 17, 953–964. [Google Scholar] [CrossRef]
- Picchioni, D.; Antolin-Fontes, A.; Camacho, N.; Schmitz, C.; Pons-Pons, A.; Rodríguez-Escribà, M.; Machallekidou, A.; Güler, M.N.; Siatra, P.; Carretero-Junquera, M.; et al. Mitochondrial Protein Synthesis and mtDNA Levels Coordinated through an Aminoacyl-tRNA Synthetase Subunit. Cell Rep. 2019, 27, 40–47.e5. [Google Scholar] [CrossRef]
- Liu, T.; Lu, B.; Lee, I.; Ondrovicová, G.; Kutejová, E.; Suzuki, C.K. DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J. Biol. Chem. 2004, 279, 13902–13910. [Google Scholar] [CrossRef]
- Ali, A.T.; Idaghdour, Y.; Hodgkinson, A. Analysis of mitochondrial m1A/G RNA modification reveals links to nuclear genetic variants and associated disease processes. Commun. Biol. 2020, 3, 147. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 2013, 9, 1750–1757. [Google Scholar] [CrossRef]
- Miller, W.L.; Bose, H.S. Early steps in steroidogenesis: Intracellular cholesterol trafficking. J. Lipid Res. 2011, 52, 2111–2135. [Google Scholar] [CrossRef]
- Bose, H.S.; Gebrail, F.; Marshall, B.; Perry, E.W.; Whittal, R.M. Inner Mitochondrial Translocase Tim50 Is Central in Adrenal and Testicular Steroid Synthesis. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef]
- Elustondo, P.; Martin, L.A.; Karten, B. Mitochondrial cholesterol import. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Steroid hormone synthesis in mitochondria. Mol. Cell. Endocrinol. 2013, 379, 62–73. [Google Scholar] [CrossRef]
- Zhang, L.; Prietsch, S.O.; Ducharme, F.M. Inhaled corticosteroids in children with persistent asthma: Effects on growth. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Tack, L.J.W.; Tatsi, C.; Stratakis, C.A.; Lodish, M.B. Effects of Glucocorticoids on Bone: What we can Learn from Pediatric Endogenous Cushing’s Syndrome. Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Metab. 2016, 48, 764–770. [Google Scholar] [CrossRef]
- Minnetti, M.; Caiulo, S.; Ferrigno, R.; Baldini-Ferroli, B.; Bottaro, G.; Gianfrilli, D.; Sbardella, E.; De Martino, M.C.; Savage, M.O. Abnormal linear growth in paediatric adrenal diseases: Pathogenesis, prevalence and management. Clin. Endocrinol. (Oxf.) 2020, 92, 98–108. [Google Scholar] [CrossRef]
- Urban, R.C.; Cotlier, E. Corticosteroid-induced cataracts. Surv. Ophthalmol. 1986, 31, 102–110. [Google Scholar] [CrossRef]
- De, P.; Roy, S.G.; Kar, D.; Bandyopadhyay, A. Excess of glucocorticoid induces myocardial remodeling and alteration of calcium signaling in cardiomyocytes. J. Endocrinol. 2011, 209, 105–114. [Google Scholar] [CrossRef]
- Chow, J.; Rahman, J.; Achermann, J.C.; Dattani, M.T.; Rahman, S. Mitochondrial disease and endocrine dysfunction. Nat. Rev. Endocrinol. 2017, 13, 92–104. [Google Scholar] [CrossRef]
- Chatzispyrou, I.A.; Held, N.M.; Mouchiroud, L.; Auwerx, J.; Houtkooper, R.H. Tetracycline antibiotics impair mitochondrial function and its experimental use confounds research. Cancer Res. 2015, 75, 4446–4449. [Google Scholar] [CrossRef]
- Villanueva, A.R.; Frost, H.M. Bone formation in human osteogenesis imperfecta, measured by tetracycline bone labeling. Acta Orthop. Scand. 1970, 41, 531–538. [Google Scholar] [CrossRef]
- Manolagas, S.C.; O’Brien, C.A.; Almeida, M. The role of estrogen and androgen receptors in bone health and disease. Nat. Rev. Endocrinol. 2013, 9, 699–712. [Google Scholar] [CrossRef]
- Dobson, P.F.; Dennis, E.P.; Hipps, D.; Reeve, A.; Laude, A.; Bradshaw, C.; Stamp, C.; Smith, A.; Deehan, D.J.; Turnbull, D.M.; et al. Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss. Sci. Rep. 2020, 10, 11643. [Google Scholar] [CrossRef]
- Gandhi, S.S.; Muraresku, C.; McCormick, E.M.; Falk, M.J.; McCormack, S.E. Risk factors for poor bone health in primary mitochondrial disease. J. Inherit. Metab. Dis. 2017, 40, 673–683. [Google Scholar] [CrossRef]
- Gottschalk, W.K.; Lutz, M.W.; He, Y.T.; Saunders, A.M.; Burns, D.K.; Roses, A.D.; Chiba-Falek, O. The Broad Impact of TOM40 on Neurodegenerative Diseases in Aging. J. Park. Dis. Alzheimers Dis. 2014, 1. [Google Scholar] [CrossRef]
- Lautenschäger, J.; Kaminski Schierle, G.S. Mitochondrial degradation of amyloidogenic proteins—A new perspective for neurodegenerative diseases. Prog. Neurobiol. 2019, 181, 101660. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Disorder | Gene Name/Protein Function | Skeletal Phenotypes and Anomalies | Notable Reported Characteristics |
|---|---|---|---|---|
| LONP1 | CODAS (cerebral, ocular, dental, auricular and skeletal): OMIM 600373 | Lon peptidase 1, mitochondrial. Matrix ATP-dependent protease. | Spondylo-epi-(meta)-physeal dysplasia, short stature, facial dysmorphism, hip dysplasia | Cataracts, developmental delay, dental, hearing loss |
| HSPA9 | EVEN-PLUS (epiphyseal, vertebral, ear, nose, plus associated findings): OMIM 616854 | Heat shock protein family A (Hsp70) member 9 (aka Mortalin, mtHsp70, GRP75)/Mitochondrial chaperone. | Spondylo-epi-(meta)-physeal dysplasia, short stature, facial dysmorphism, scoliosis, hip dysplasia | Cataracts, cardiac malformations, dental, genital anomalies, developmental delay |
| IARS2 | CAGSSS (cataracts, growth hormone deficiency, sensory neuropathy, sensorineural hearing loss, and skeletal dysplasia): OMIM 616007 | Isoleucyl-tRNA synthetase 2, mitochondrial/tRNA synthetase | Spondylo-epi-(meta)-physeal dysplasia, short stature | Cataracts, neurodevelopmental delay, seizures, peripheral neuropathy hearing loss, growth hormone deficiency |
| PAM16 | SMDMDM Spondylometaphyseal dysplasia, Megarbane-Dagher-Melki-type: OMIM 613320 | Presequence translocase associated motor 16. ( aka MAGMAS, TIMM16)/Involved in mitochondrial protein import. | Severe spondylodysplastic dysplasia, short stature, facial dysmorphism | Cardiomyopathy, developmental delay |
| AIFM1 | SEMD-HL (Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy): OMIM 300232 | Apoptosis inducing factor mitochondria associated 1/Involved in mitochondrial protein import, apoptosis and assembly of mitochondrial oxidative. phosphorylation complexes. | Spondylo-epi-(meta)-physeal dysplasias, short stature, midface hypoplasia | Myelination, progressive neurodegeneration of the central and peripheral nervous system |
| PISD | SEMD (Spondylometaphyseal dysplasia). Liberfarb syndrome: OMIM 618889 | Phosphatidylserine decarboxylase/Converts phosphatidylserine to phosphatidylethanolamine in the IMM. | Spondylo-epi-(meta)-physeal dysplasias, short stature, mid-face hypoplasia | Cataracts, white matter changes |
| Gene | Disorder | Gene Name/Protein Function | Reported Skeletal Anomalies | Notable Reported Characteristics |
|---|---|---|---|---|
| CLPB | MEGCANN (3-methylglutaconic aciduria, type VII, with cataracts, neurologic involvement and neutropenia): OMIM 616271 | Caseinolytic mitochondrial matrix peptidase chaperone subunit B/Protein disaggregase associated with IMM (inner mitochondrial membrane. | Extremity rhizomelia, impaired growth, facial dysmorphism | Cataracts, neurologic deterioration, 3-methylglutaconic aciduria, neutropenia |
| HSPD1 | SPG13 (Spastic paraplegia 13, autosomal dominant: OMIM 605280. Leukodystrophy, hypomyelinating, 4: OMIM 612233 | Heat shock protein family D (Hsp60) member/Matrix chaperone. | N/A | Dilated cardiomyopathy, leukodystrophy, hypotonia, psychomotor developmental delay, spastic paraplegia |
| CLPP | PRLTS3 (Perrault syndrome 3): OMIM 614129 | Caseinolytic mitochondrial matrix peptidase proteolytic subunit/Mitochondrial protease associated with IMM. | Short stature, facial dysmorphism | Premature ovarian failure, ataxia, microcephaly, learning difficulties, sensorineural hearing loss |
| SPATA5 | EHLMRS (Epilepsy, Hearing Loss and Mental Retardation Syndrome): OMIM 613940 | Spermatogenesis associated 5. AAA family of ATPases/Unclear molecular function. Role in maintaining mitochondrial function. | Short stature, scoliosis, hip dysplasia | Cataracts, epilepsy, hearing loss and intellectual disability |
| HTRA2 | MGCA8 (3-methylglutaconic aciduria type VII): OMIM 617248 | HtrA serine peptidase 2/IMS (inner membrane space) protease associated with IMM. | N/A | Cataracts, 3-methylglutaconic aciduria, seizures, hypotonia, abnormal movements, neutropenia |
| AFG3L2 | OPA12 (Optic atrophy 12): OMIM 618977. SPAX5 (Spastic ataxia 5, autosomal recessive): OMIM 614487. SCA28 (Spinocerebellar ataxia 28): OMIM 610246 | AFG3 like matrix AAA peptidase subunit 2/IMM protease. | N/A | Optic atrophy, spinocerebellar ataxia, spastic ataxia, chronic progressive external ophthalmoplegia |
| SPG7 | SPG7 (Spastic paraplegia 7, autosomal recessive): OMIM 607259 | SPG7 matrix AAA peptidase subunit, paraplegin/IMM protease. | N/A | Spastic paraplegia, ataxia, optic atrophy, cortical atrophy, cerebellar atrophy, chronic progressive external ophthalmoplegia |
| Gene | Disorder | Gene Name/Protein Function | Reported Skeletal Anomalies | Notable Reported Characteristics |
|---|---|---|---|---|
| TOMM70 | Multi-OXPHOS deficiencies: PMID 31907385 | Translocase of outer mitochondrial membrane 70/Mitochondrial import. | Short stature | Developmental delay, microchephaly, severe anemia, lactic acidosis |
| TIMM50 | MGCA9 (3-methylglutaconic aciduria, type IX): OMIM 617698 | Translocase of inner mitochondrial membrane 50/Mitochondrial import. | Short stature, dysmorphic facial features, hip dysplasia, scoliosis and osteoarticular issues | Cardiomyopathy, left ventricle dilation, cardiorespiratory arrest, 3-methylglutaconic aciduria, early-onset seizures, severely delayed psychomotor development, intellectual disability, hypotonia or spasticity |
| AGK | Senger syndrome, MTDPS10 (cardiomyopathic mitochondrial DNA depletion syndrome-10): OMIM 212350. CTRCT38 (Cataract 38, autosomal recessive): OMIM 614691 | Acylglycerol kinase/Roles in mitochondrial lipid metabolism and mitochondrial import. | N/A | Cataracts, hypertrophic cardiomyopathy, skeletal myopathy, exercise intolerance |
| DNAJC19 | DCMA (Dilated Cardiomyopathy with Ataxia), 3-methylglutaconic aciduria type V (MGCA5): OMIM 610198 | DNAJ heat shock protein family (Hsp40) member C19/Roles in mitochondrial import and cardiolipin metabolism. | Growth retardation | Cataracts, dilated cardiomyopathy, ataxia, 3-methylglutaconic aciduria, genital anomalies |
| GFER | MPMCD (Myopathy, mitochondrial progressive, with congenital cataract and developmental delay): OMIM 613076 | Growth factor, augmenter of liver regeneration. (aka ERV1)/Mitochondrial import MIA (mitochondrial import and assembly) component. | Short stature, facial dysmorphology, scoliosis, hip dysplasia | Cataracts, intellectual disability, hearing loss, hypotonia, developmental delay |
| PMPCA | SCAR2 (Autosomal Recessive Spinocerebellar Ataxia-2): OMIM 213200 | Peptidase, mitochondrial processing subunit alpha/MTS (mitochondrial targeting sequence) cleavage. | N/A | Cataracts, spinocerebellar ataxia, intellectual disability |
| PMPCB | MMDS6 (Multiple mitochondrial dysfunctions syndrome 6): OMIM 617954 | Peptidase, mitochondrial processing subunit beta/MTS cleavage. | N/A | Early onset severe neurodegeneration, hypotonia, intellectual disability, seizures, microcephaly, motor abnormalities |
| MIPEP | COXPD31 (Combined oxidative phosphorylation deficiency 31): OMIM 617228 | Mitochondrial intermediate peptidase/MTS cleavage. | Short stature, facial dysmorphology | Cataracts, left ventricular contraction, dilated cardiomyopathy, global developmental delay, severe hypotonia, epilepsy, microcephaly |
| XPNPEP3 | NPHPL1 (Nephronophthisis-like nephropathy 1): OMIM 613159 | X-prolyl aminopeptidase 3/MTS cleavage. Role in cilia. | N/A | Hypertrophic dilated cardiomyopathy, renal failure, ciliopathy, essential tremor, hearing loss, muscle fatigue, seizures, and developmental delay |
| IMMP2L | Neurodevelopmental disorders: PMID 25478008 | Inner mitochondrial membrane peptidase subunit 2/MTS cleavage. | N/A | Autism spectrum disorder, attention-deficit hyperactivity disorder, and schizophrenia |
| Gene | Disorder | Gene Name/Protein Function | Reported Skeletal Anomalies | Notable Reported Characteristics |
|---|---|---|---|---|
| TAZ | Barth syndrome: OMIM 302060 | Tafazzin/Remodeling of cardiolipin acyl side chains. | Pre-pubertal growth delay, facial dysmorphism | Dilated cardiomyopathy, 3-methylglutaconic aciduria, neutropenia, motor delay |
| SERAC1 | MEGDEL (3-methylglutaconic aciduria with deafness, encephalopathy, and Leigh-like syndrome):OMIM 614739 | Serine active site containing 1/Mediates phospholipid exchange. | Short stature, scoliosis, dysmorphology | Impaired psychomotor function, encephalopathy, deafness, 3-methylglutaconic aciduria, spasticity |
| OPA3 | Costeff Syndrome (3-methylgutaconic aciduria, type III; MGCA3): OMIM 258501. ADOAC (Autosomal Dominant Optic Atrophy and Cataract): OMIM 165300 | Outer mitochondrial membrane lipid metabolism regulator OPA3/Implicated in lipid metabolism | Growth retardation | Cataracts, optic atrophy, early-onset extrapyramidal movement disorder, and cognitive deficits, 3-methylglutaconic aciduria, lipodystrophy |
| AGK | Senger syndrome, MTDPS10 (cardiomyopathic mitochondrial DNA depletion syndrome-10): OMIM 212350. CTRCT38 (Cataract 38, autosomal recessive): OMIM 614691 | Acylglycerol kinase/Roles in mitochondrial lipid metabolism and mitochondrial import. | N/A | Cataracts, hypertrophic cardiomyopathy, skeletal myopathy, exercise intolerance |
| DNAJC19 | DCMA (Dilated Cardiomyopathy with Ataxia), 3-methylglutaconic aciduria type V (MGCA5): OMIM 610198 | DNAJ heat shock protein family (Hsp40) member C19/Roles in mitochondrial import and cardiolipin metabolism. | Growth retardation | Cataracts, dilated cardiomyopathy, ataxia, 3-methylglutaconic aciduria, genital anomalies |
| Gene | Disorder | Gene Name | Reported Skeletal Anomalies | Notable Reported Characteristics |
|---|---|---|---|---|
| LARS2 | PRLTS4 (Perrault syndrome 4): OMIM 615300 | Leucyl-tRNA synthetase 2, mitochondrial. | Dysmorphic facial features, scoliosis, Marfan habitus | Premature ovarian failure, sensorineural hearing loss |
| HARS2 | PRLTS 2 (Perrault syndrome 2): OMIM 614926 | Histidyl-tRNA synthetase 2, mitochondrial. | N/A | Premature ovarian failure, sensorineural hearing loss |
| VARS2 | COXPD20 (Combined oxidative phosphorylation deficiency 20): OMIM 615917 | Valyl-tRNA synthetase 2, mitochondrial. | Growth deficiency, facial dymorphisms, hip dysplasia | Hypertrophic cardiomyopathy, muscle weakness, hypotonia, central neurologic disease, epilepsy |
| WARS2 | NEMMLAS (Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures): OMIM 617710 | Tryptophanyl tRNA synthetase 2, mitochondrial. | Short stature/growth retardation, dysmorphic features | Delayed psychomotor development, intellectual disability, abnormal motor function, seizures |
| GARS | CMT2D (Charcot–Marie–Tooth disease, type 2D): OMIM 601472. HMN5A (Neuronopathy, distal hereditary motor, type VA): OMIM 600794 | glycyl-tRNA synthetase 1. Mitochondrial and cytosolic. | Growth retardation, facial features, scoliosis | Axonal neuropathy, distal motor neuronopathy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, T.; Goedhart, C.; Pfeffer, G.; Greenway, S.C.; Lines, M.; Khan, A.; Innes, A.M.; Shutt, T.E. Skeletal Phenotypes Due to Abnormalities in Mitochondrial Protein Homeostasis and Import. Int. J. Mol. Sci. 2020, 21, 8327. https://doi.org/10.3390/ijms21218327
Zhao T, Goedhart C, Pfeffer G, Greenway SC, Lines M, Khan A, Innes AM, Shutt TE. Skeletal Phenotypes Due to Abnormalities in Mitochondrial Protein Homeostasis and Import. International Journal of Molecular Sciences. 2020; 21(21):8327. https://doi.org/10.3390/ijms21218327
Chicago/Turabian StyleZhao, Tian, Caitlin Goedhart, Gerald Pfeffer, Steven C Greenway, Matthew Lines, Aneal Khan, A Micheil Innes, and Timothy E Shutt. 2020. "Skeletal Phenotypes Due to Abnormalities in Mitochondrial Protein Homeostasis and Import" International Journal of Molecular Sciences 21, no. 21: 8327. https://doi.org/10.3390/ijms21218327
APA StyleZhao, T., Goedhart, C., Pfeffer, G., Greenway, S. C., Lines, M., Khan, A., Innes, A. M., & Shutt, T. E. (2020). Skeletal Phenotypes Due to Abnormalities in Mitochondrial Protein Homeostasis and Import. International Journal of Molecular Sciences, 21(21), 8327. https://doi.org/10.3390/ijms21218327