6. Materials and Methods
6.1. General
All reagents and solvents were purchased from Sigma Aldrich (Toluca, Mexico) and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) on Merck F253 silica gel aluminum sheets, and spots were revealed with ultraviolet (UV) light (254 nm). NMR experiments were carried out in Varian NMR System (500 MHz and 125 MHz), Varian Mercury (300 MHz and 75 MHz) and Bruker ASCEND (600 MHz and 150 MHz). 1H NMR and 13C spectra were assigned with the help of 2-D experiments (gHSQC and gHMBC). The chemical shifts (δ) are given in ppm. Mass spectra (MS) were recorded on a Bruker Amazon Speed (ESI). Infrared (IR) spectra were obtained on a Perkin Elmer FT-IR Spectrum 2000 spectrometer from the ENCB-IPN spectroscopy instrumentation center. Melting points were determined on an Electrothermal MEL-TEMP apparatus and are uncorrected. Microwave reactions were accomplished on a CEM Discovery SP apparatus.
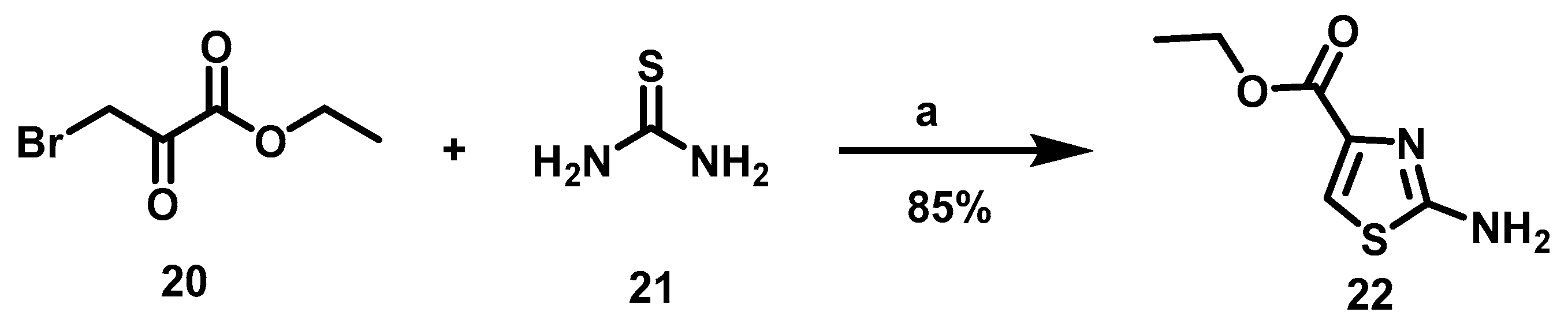
6.2. Procedure for the Synthesis of Ethyl 2-Aminothiazol-4-carboxylate, 22
Ethyl bromine pyruvate (7.69 mmol, 1 eq), thiourea (7.69 mmol, 1 eq) and 15 mL of EtOH were added to a flask equipped with a magnetic stirring bar and left to reaction mixture at rt for 16 h. At the end of this period, the solvent was evaporated under reduced pressure, followed by the addition of 40 mL of 20% aqueous potassium carbonate to the residue. The suspension formed was maintained under constant stirring for 30 min before subjecting it to vacuum filtration. The solid retained on the filter paper was washed with 40 mL of distilled water in two 20 mL portions and oven-dried at 40 °C.
Ethyl 2-Aminothiazol-4-Carboxylate, 22
Yield: 85%. White solid. 1H NMR (300 MHz, DMSO-d6) δ: 7.44 (s, 1H, H-5), 7.24 (s, 2H, NH), 4.17 (q, J = 7.10 Hz, 2H, H-8), 1.22 (t, J = 7.10 Hz, 3H, H-9). 13C NMR (75 MHz, DMSO-d6) δ 168.6 (C-2), 161.5 (C-6), 142.6 (C-4), 117.4 (C-5), 60.6 (C-8), 14.6 (C-9). DIP-MS (ESI; M + Na) m/z (calculated): 195.01, m/z (measured): 195.00.
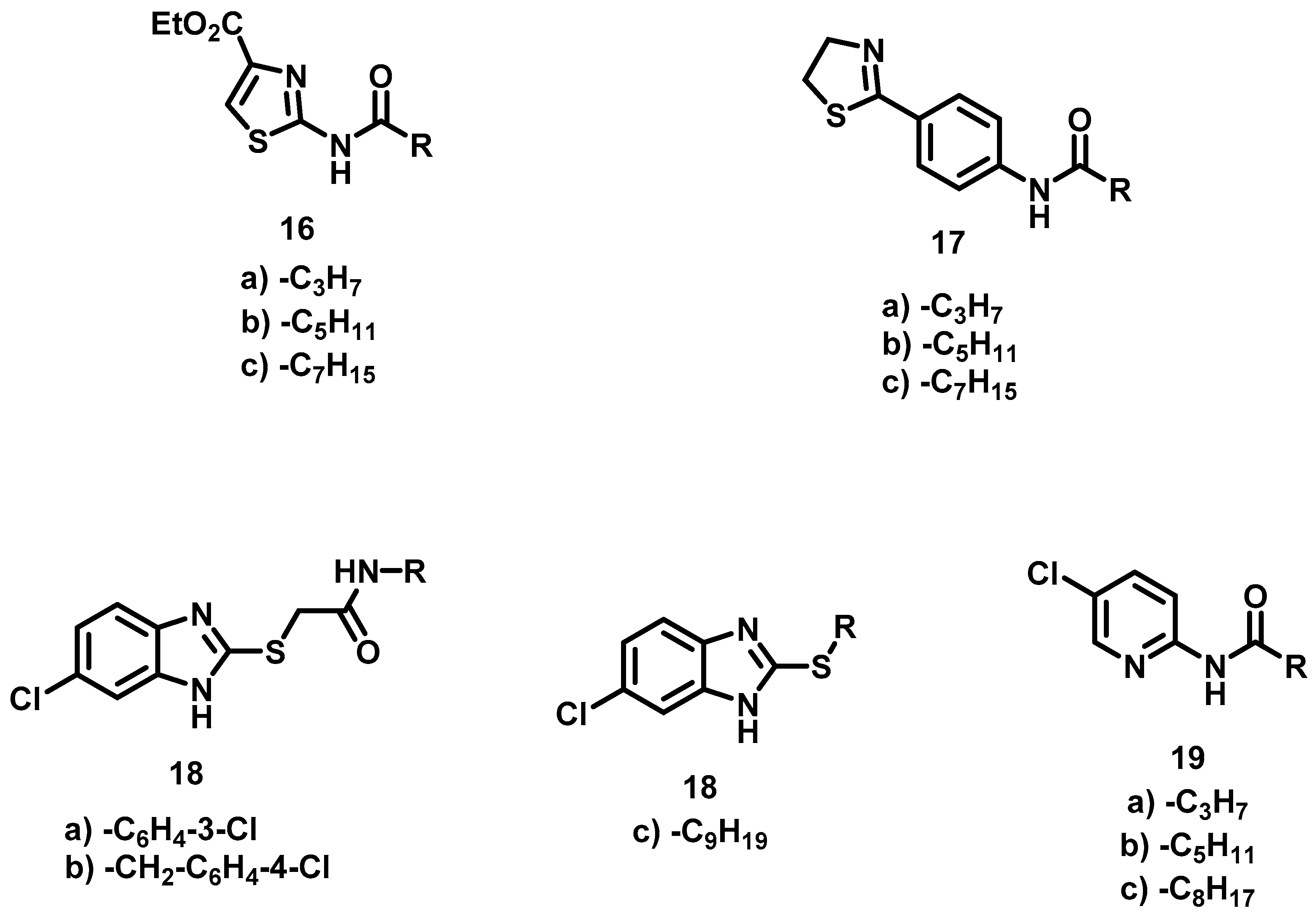
6.3. Synthesis of Ethyl 2-Acylamidothiazol-4-carboxylate, 16
2-Amino thiazole (1.74 mmol, 1 eq) and 2 mL of carboxylic acid (butanoic acid at 150 °C, hexanoic acid at 150 °C, octanoic acid at 180 °C) were added to a reaction vial equipped with magnetic stirring, the reaction mixture was warmed for 90 min. It was then transferred to an aqueous solution of 20% potassium carbonate (40 mL) and allowed the mixture to stir for 30 min. Subsequently, the suspension was vacuum filtered and the solid obtained washed with distilled water (2 × 20 mL) and oven-dried at 40 °C.
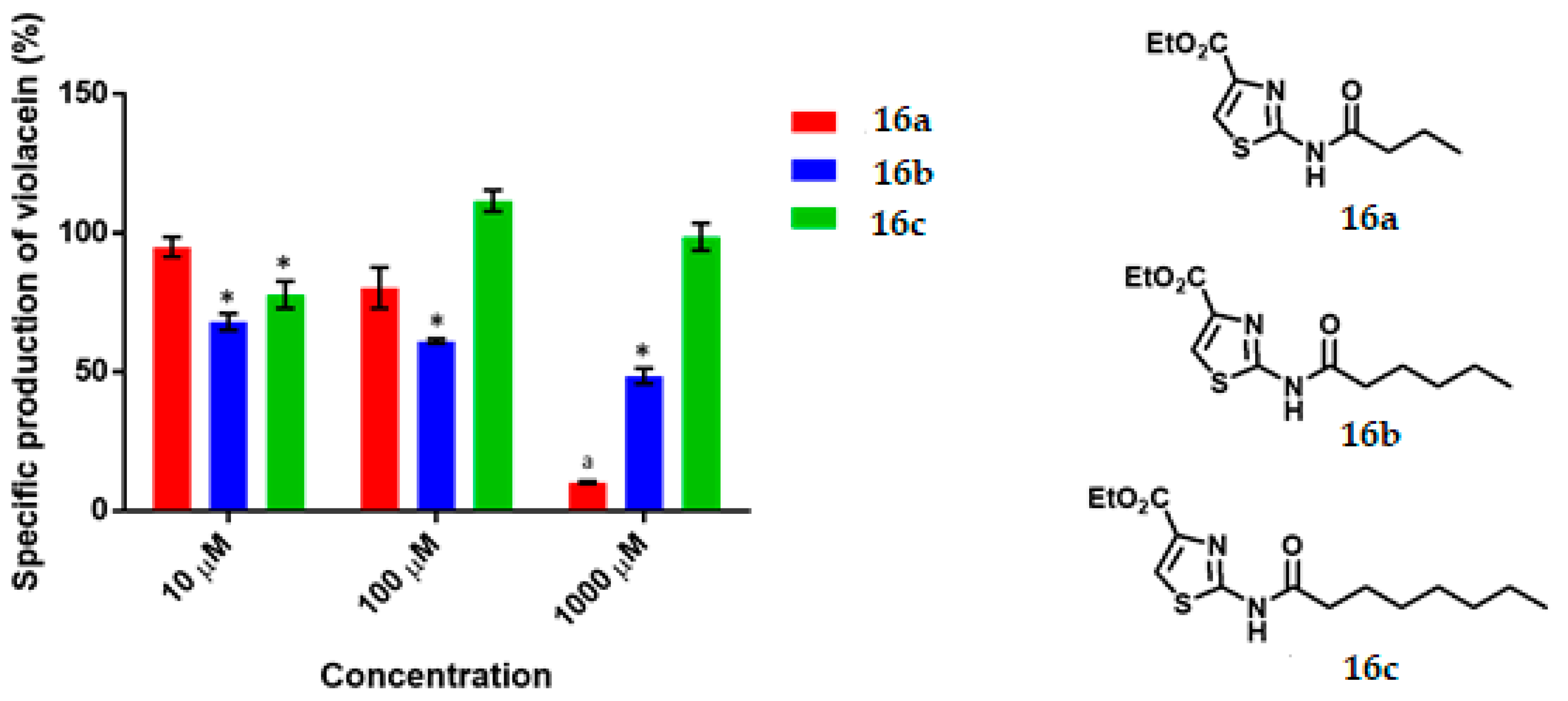
Ethyl 2-Butyramidothiazol-4-Carboxylate, 16a
Yield: 82%. Brownish solid; m.p. 145–148 °C. Lit. [
30] 150–151 °C
Rf = 0.6 (AcOEt). IR (CH
2Cl
2)
= 3256.8, 1720.1, 1690.4, 1546.0, 1215.1 cm
−1.
1H NMR (300 MHz, CDCl
3) δ: 7.81 (s, 1H, H-5′), 4.35 (q,
J = 7.12 Hz, 2H, H-3″), 2.43 (t,
J = 7.47 Hz, 2H, H-2), 1.70 (m, 2H, H-3), 1.35 (t,
J = 7.12 Hz, 3H, H-4″), 0.94 (t,
J = 7.47 Hz, 3H, H-4).
13C NMR (75 MHz, CDCl
3) δ 171.9 (C-1), 161.4 (C-1″), 159.0 (C-2′), 141.1 (C-4′), 122.2 (C-5′), 61.4 (C-3″), 37.9 (C-2), 18.3 (C-3), 14.3 (C-4″), 13.6 (C-4). DIP-MS (ESI; M + Na
+)
m/
z (calculated): 265.06,
m/
z (measured): 265.10.
Ethyl 2-Hexanamidothiazol-4-Carboxylate, 16b
Yield: 85%. Brownish solid; m.p. 129–132 °C. Lit. [
30] 135–136 °C
Rf = 0.66 (AcOEt). IR (CH
2Cl
2)
= 3256.8, 1723.4, 1680.7, 1547.6, 1211.1 cm
−1.
1H NMR (300 MHz, CDCl
3) δ: 7.82 (s, 1H, H-5′), 4.35 (q,
J = 7.13 Hz, 2H, H-3″), 2.44 (t,
J = 7.59 Hz, 2H, H-2), 1.68 (m, 2H, H-3), 1.36 (t,
J = 7.13 Hz, 3H, H-4″), 1.28 (m, 4H, H-4,5), 0.86 (m, 3H, H-6).
13C NMR (75 MHz, CDCl
3) δ 171.9 (C-1), 161.4 (C-1″), 158.8 (C-2), 141.19 (C-4′), 122.18 (C-5′), 61.37 (C-3″), 36.05 (C-2), 31.21 (C-4), 24.6 (C-3), 22.3 (C-5), 14.3 (C-4″), 13.8 (C-6). DIP-MS (ESI; M + Na)
m/
z (calculated): 293.09,
m/
z (measured): 293.16.
Ethyl 2-Octanamidothiazol-4-Carboxylate, 16c
Yield: 80%. Brownish solid; m.p. 102–104 °C. Lit. [
30] 109–111 °C.
Rf = 0.68 (AcOEt). IR (CH
2Cl
2)
= 3245.9, 1725.1, 1689.5, 1544.2, 1214.2 cm
−1.
1H NMR (300 MHz, CDCl
3) δ: 10.63 (s, 1H, NH), 7.82 (s, 1H, H-5′), 4.35 (q,
J = 7.11 Hz, 2H, H-3″), 2.44 (t,
J = 4.54 Hz, 2H, H-2), 1.65 (m, 2H, H-3), 1.36 (t,
J = 7.11 Hz, 3H, H-4″), 1.24 (m, 8H, H-4,5,6,7), 0.84 (t,
J = 6.19 Hz, 3H, H-8).
13C NMR (75 MHz, CDCl
3) δ 172.0 (C-1), 161.3 (C-1″), 158.9 (C-2′), 141.1 (C-4′), 122.2 (C-5′), 61.4 (C-3″), 36.1 (C-2), 31.6 (C-6), 29.0 (C-5), 28.9 (C-4), 24.9 (C-3), 22.5 (C-7), 14.31 (C-4″), 14.0 (C-8). DIP-MS (ESI; M + Na)
m/
z (calculated): 321.12,
m/
z (measured): 321.22.
6.4. Procedure for the Synthesis of 4-(Thiazolin-2-yl)–aniline, 25
4-aminobenzonitrile (4.23 mmol, 1 eq), cysteamine hydrochloride (6.34 mmol, 1.5 eq), potassium carbonate (21.16 mmol, 5 eq) and 5 mL of a EtOH/H2O (1:1) mixture were added to an ACE pressure tube equipped with a magnetic stirring bar. The reaction mixture was subjected to heating in a sand bath at 110 °C for 16 h. At the end of this period, it was transferred to a ball flask and the solvent was evaporated under reduced pressure. The residue was suspended in distilled water (20 mL) and liquid-liquid extractions were made with CH2Cl2 (3 × 15 mL), the combined organic phases were dried with anhydrous Na2SO4 and evaporated in a ball flask under reduced pressure. The residue was purified by chromatographic column, using silica gel as the stationary phase and an 8:2 hexane/AcOEt mixture as the mobile phase.
4-(Thiazolin-2-yl) Aniline, 25
Yield: 82%. Pink solid. 1H NMR (300 MHz, CDCl3) δ: 7.64 (d, J = 8.57 Hz, 2H, H-3), 6.63 (d, J = 8.57 Hz, 2H, H-2), 4.38 (t, J = 8.2 Hz, 2H, H-4′), 3.96 (s, 2H, NH), 3.34 (t, J = 8.2 Hz, 2H, H-5′). 13C NMR (75 MHz, CDCl3) δ 167.9 (C-2′), 149.3 (C-1), 130.0 (C-3), 123.5 (C-4), 114.2 (C-2), 64.9 (C-4′), 33.5 (C-5′). DIP-MS (ESI; M + H) m/z (calculated): 179.06, m/z (measured): 179.06.
6.5. Synthesis of N-[4-(Thiazolin-2-yl)-phenyl]-carboxamides, 17
4-(Thiazolin-2-yl)-Aniline (1.96 mmol, 1 eq) and 2.5 mL of carboxylic acid (butanoic, hexanoic, octanoic acids) were added to a reaction vial equipped with a magnetic stirring bar. The vial was closed and the reaction mixture warmed (butanoic acid at 150 °C, hexanoic acid at 150 °C, octanoic acid at 180 ° C) for 90 min. At the end of that period, the reaction mixture was transferred to an aqueous solution of 20% potassium carbonate (40 mL) and left to stand under constant stirring for 30 min. The suspension was subjected to vacuum filtration and the solid retained on the filter paper was washed with 40 mL of distilled water in two 20 mL portions, then oven-dried at 40 °C.
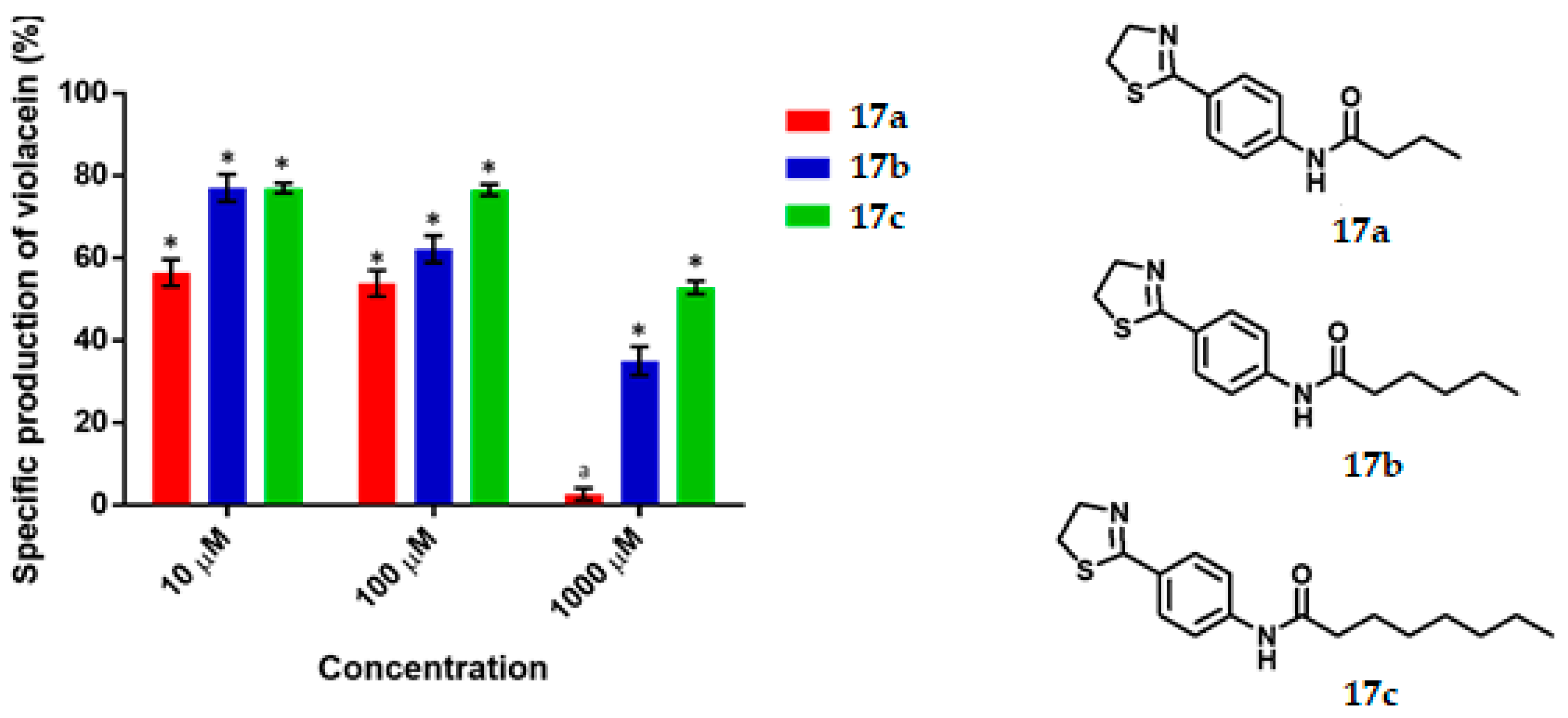
N-(4-(Thiazolin-2-yl) Phenyl) Butiramide, 17a
Yield: 85%. Yellow solid; m.p. 138–140 °C. Rf = 0.3 (AcOEt). IR (CH2Cl2) = 3296.6, 1667.6, 1598.3, 1527.5, 845.5 cm−1. 1H NMR (300 MHz, CDCl3) δ: 7.80 (s, 1H, NH), 7.76 (d, J = 8.58 Hz, 2H, H-3′), 7.58 (d, J = 8.58 Hz, 2H, H-2′), 4.42 (t, J = 8.28 Hz, 2H, H-4″), 3.39 (t, J = 8.28 Hz, 2H, H-5″), 2.32 (t, J = 7.42 Hz, 2H, H-2), 1.73 (m, 2H, H-3), 0.97 (t, J = 7.35 Hz, 3H, H-4). 13C NMR (75 MHz, CDCl3) δ 171.8 (C-1), 168.0 (C-2″), 140.7 (C-1′), 129.3 (C-3′), 128.7 (C-4′), 119.1 (C-2′), 65.0 (C-4″), 39.6 (C-2), 33.7 (C-5″), 19.0 (C-3), 13.7 (C-4). DIP-MS (ESI; M+H) m/z (calculated): 249.10, m/z (measured): 249.15.
N-(4-(Thiazolin-2-yl) Phenyl) Hexanamide, 17b
Yield: 88%. Brownish solid; m.p. 129–131 °C. Rf = 0.46 (AcOEt). IR (CH2Cl2) = 3301.0, 1669.4, 1598.7, 1529.8, 843.9 cm−1. 1H NMR (300 MHz, CDCl3) δ: 7.76 (d, J = 8.59 Hz, 2H, H-3′), 7.65 (s, 1H, NH), 7.58 (d, J = 8.59 Hz, 2H, H-2′), 4.42 (t, J = 8.3 Hz, 2H, H-4″), 3.39 (t, J = 8.3 Hz, 2H, H-5″), 2.34 (t, J = 7.55 Hz, 2H, H-2), 1.70 (m, 2H, H-3), 1.33 (m, 4H, H-4,5), 0.88 (t, J = 6.44 Hz, 3H, H-6). 13C NMR (75 MHz, CDCl3) δ 171.7 (C-1), 167.9 (C-2″), 140.6 (C-1′), 129.3 (C-3′), 128.7 (C-4′), 119.0 (C-2′), 65.0 (C-4″), 37.8 (C-2), 33.677 (C-5″), 31.4 (C-4), 25.2 (C-3), 22.4 (C-5), 13.9 (C-6). DIP-MS (ESI; M + Na) m/z (calculated): 299.11, m/z (measured): 299.20.
N-(4-(Thiazolin-2-yl) Phenyl) Octanamide, 17c
Yield: 79%. Brownish solid; m.p. 68–70 °C. Rf = 0.51 (AcOEt). IR (CH2Cl2) = 3301.9, 1667.5, 1597.6, 1530.8, 844.1 cm−1. 1H NMR (300 MHz, CDCl3) δ: 7.90 (1H, NH), 7.76 (2H, H-3′), 7.59 (2H, H-2′), 4.42 (2H, H-4″), 3.39 (2H, H-5″), 2.35 (2H, H-2), 1.69 (2H, H-3), 1.26 (8H, H-4,5,6,7), 0.86 (3H, H-8). 13C NMR (75 MHz, CDCl3) δ 171.7 (C-1), 167.9 C-2″), 140.7 (C-1′), 129.3 (C-3′), 118.9 (C-4′), 64.9 (C-4″), 33.7 (C-2), 31.6 (C-5″), 29.2, 29.0 28.9, 25.5, 22.6, 14.1 (C-8). DIP-MS (ESI; M + H) m/z (calculated): 305.16, m/z (measured): 305.27.
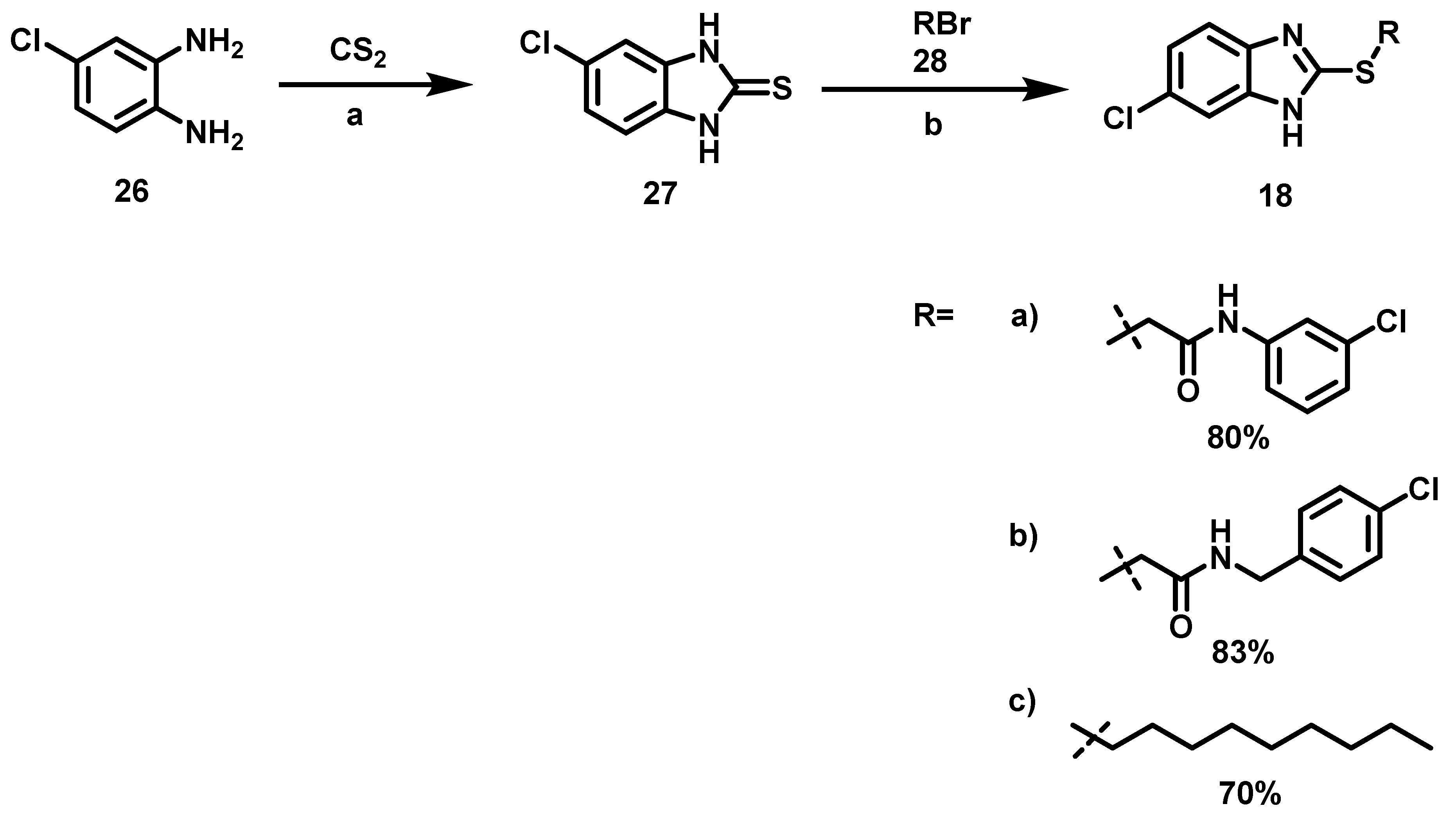
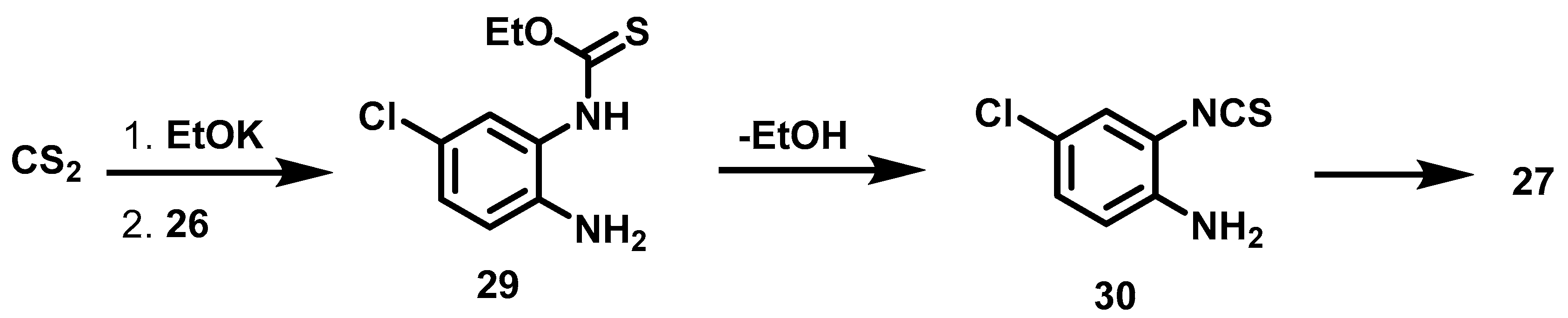
6.6. Procedure for the Synthesis of 5-Chloro-benzimidazole-2-thione, 27
4-Chloro-phenylenediamine (5.61 mmol, 1 Eq), carbon disulfide (7.23 mmol, 1.3 Eq), potassium hydroxide (16.83 mmol, 3 eq) and 5 mL of an ethanol/water mixture (7:3) were placed in an ACE pressure tube charged with magnetic stirring. The reaction mixture was heated at 80 °C for 5 h, then the solvent was evaporated under reduced pressure and 15 mL of a 5% aqueous HCl solution were added to the residue. The suspension was left with stirring for 15 min and subsequently it was vacuum filtered, the solid obtained was washed with 2 × 20 mL of distilled water, the solid was dried at 40 °C in an oven.
5-chloro-1,3-dihydro-2H-Benzimidazol-2-Thione, 27
Yield: 88%. Brownish solid. 1H NMR (300 MHz, Acetone-d6) δ: 7.17 (dd, J = 1.86, 0.86 Hz, 1H, H-6), 7.15 (d, J = 0.86 Hz, 1H, H-4), 7.13 (d, J = 1.86 Hz, 1H, H-7). 13C NMR (75 MHz, Acetone-d6) δ 170.4 (C-2), 133.768 (C-4a), 131.7 (C-7a), 127.1 (C-5), 122.1 (C-6), 110.5 (C-7), 109.4 (C-4). DIP-MS (ESI; M − H) m/z (calculated): 182.97, m/z (measured): 182.88.
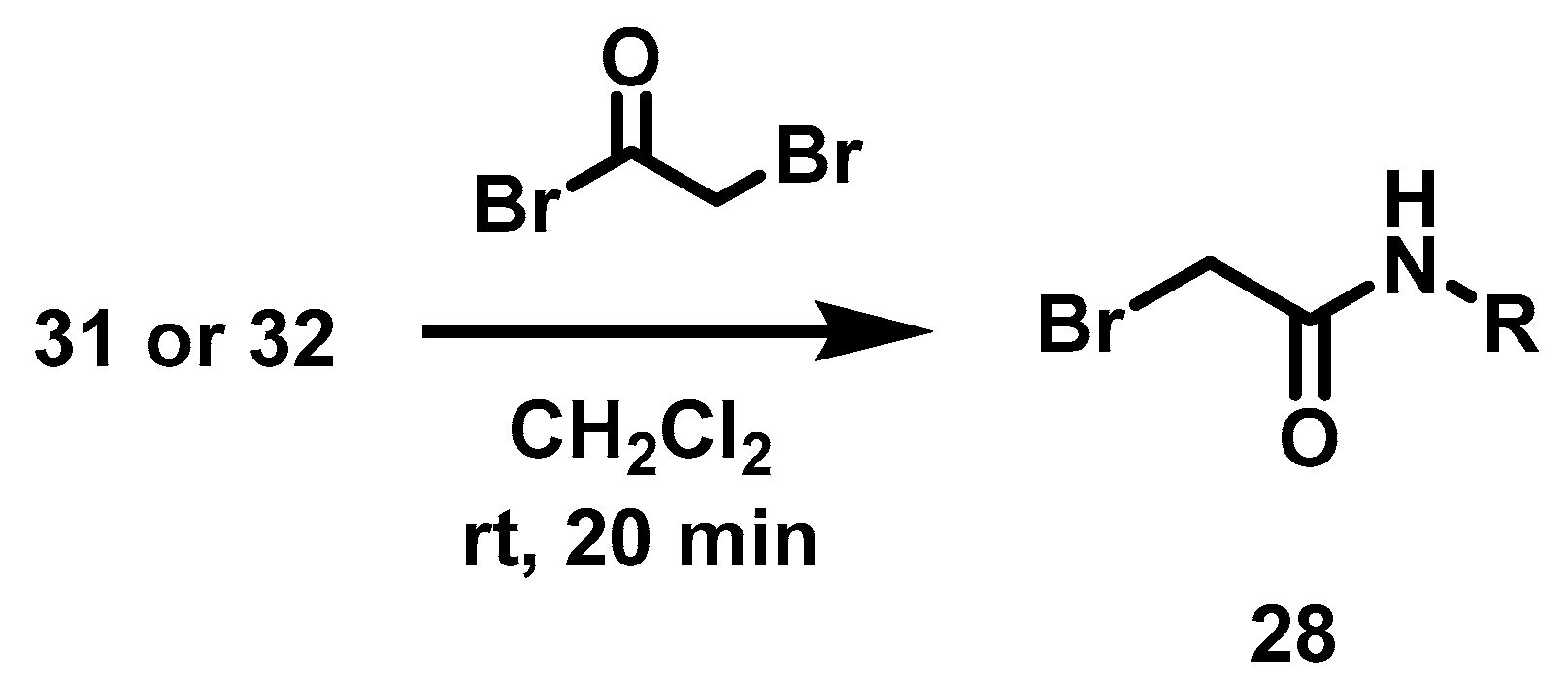
6.7. Alkylation of 5-Chloro-benzimidazole-2-thione, 18
Benzimidazol-2-thione (1.35 mmol, 1 eq), alkyl halide (1.35 mmol, 1 eq) and 15 mL of ethanol were placed in a 100 mL ball flask equipped with a magnetic stirring bar and the reaction mixture was left to stand for 16 h at 60 °C before evaporating the solvent. The solid was suspended in an aqueous solution of 20% potassium carbonate (30 mL) and vacuum filtered. This solid was washed with 2 × 20 mL of distilled water and oven-dried at 40 °C.
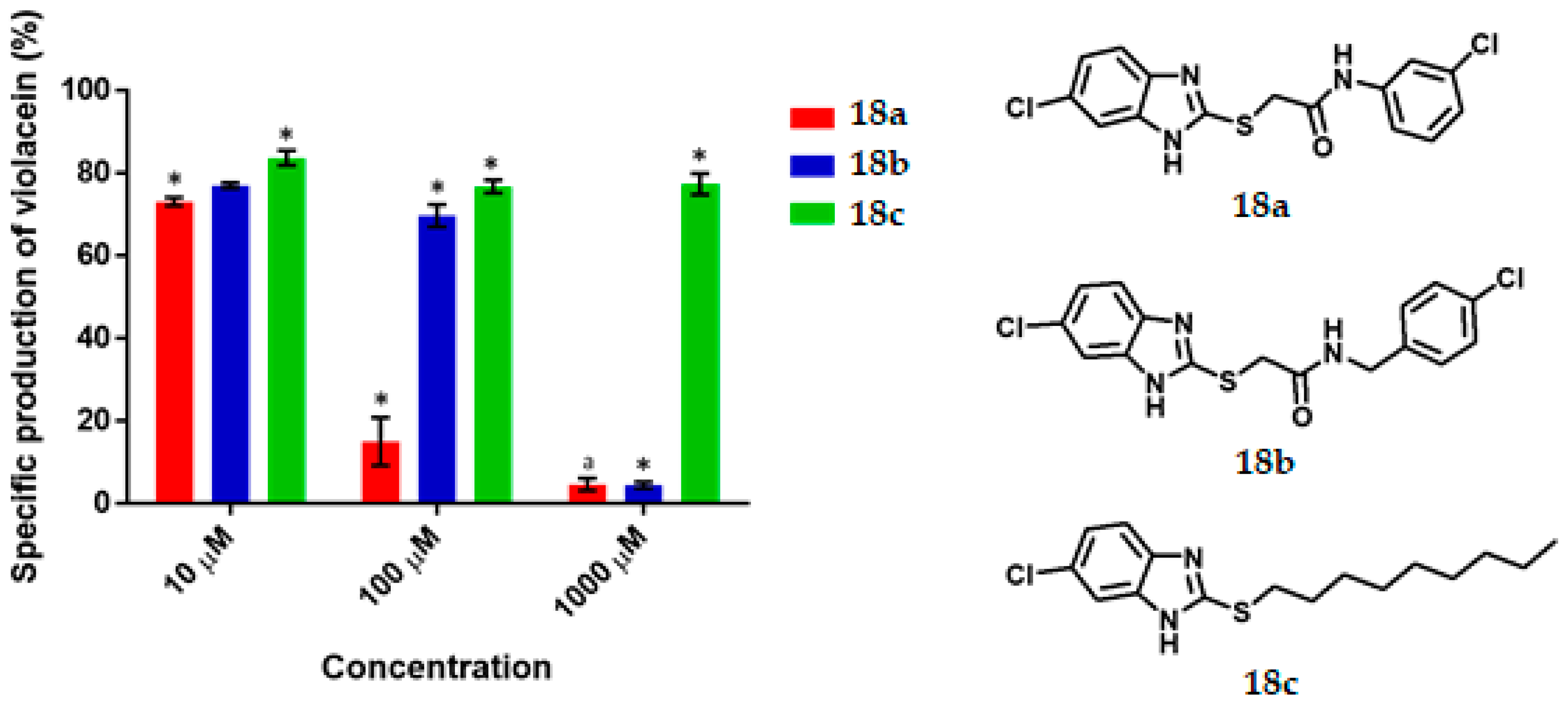
2-[(6-chloro-1H-Benzoimidazol-2-yl) thio)-N-(3-Chlorophenyl)] Acetamide, 18a
Yield: 80%. Purple solid; m.p. 228–231 °C. Rf = 0.28 (hexane/AcOEt 7:3). IR (CH2Cl2) = 1652.2, 1586.4, 1423.7 cm−1. 1H NMR (300 MHz, DMSO-d6) δ: 10.77 (s, 1H, NH), 7.77 (s, 1H, H-2′), 7.64 (d, J = 1.64 Hz, 1H, H-7″), 7.57 (d, J = 8.63 Hz, 1H, H-4″), 7.43 (d, J = 8.36 Hz, 1H, H-6′), 7.32 (m, 2H, H-5′,5″), 7.10 (d, J = 7.84 Hz, 1H, H-4′), 4.41 (s, 2H, H-2). 13C NMR (75 MHz, DMSO-d6) δ 166.1 (C-1), 152.3 (C-2″), 140.5 (C-1′), 137.2 (C-7a″), 134.9 (C-4a″), 133.5 (C-3′), 131.0 (C-5′), 128.3 (C-6′), 124.1 (C-5″), 123.8 (C-4′), 119.0 (C-2′), 118.0 (C-6′), 115.1 (C-4″), 113.8 (C-7″), 36.9 (C-2). DIP-MS (ESI; M-H) m/z (calculated): 349.99, m/z (measured): 349.93.
2-[(6-chloro-1H-Benzimidazol-2-yl) thio)-N-(4-chlorobenzyl] Acetamide, 18b
Yield: 83%. Blue solid; m.p. = 115–118 °C. Rf = 0.15 (hexane/AcOEt 7:3); IR (CH2Cl2) = 3735.7, 3649.3, 3283.2, 1640.0, 1557.9, 1393.9 cm−1. 1H NMR (300 MHz, DMSO-d6) δ: 8.87 (t, J = 5.95 Hz, 1H, NH), 7.54 (d, J = 1.98 Hz, 1H, H-7″), 7.48 (d, J = 8.64 Hz, 1H, H-4″), 7.23 (m, 5H, H-3′,4′,5″), 4.25 (d, J = 5.95 Hz, 2H, H-1′), 4.11 (s, 2H, H-2). 13C NMR (75 MHz, DMSO-d6) δ 167.5 (C-1), 152.1 (C-2″), 139.4 (C-7a″), 138.4 (C-2′), 136.9 (C-4a″), 131.7 (C-5′), 129.4 (C-3′), 128.5 (C-4′), 127.2 (C-6″), 123.0 (C-5″), 115.2 (C-4″), 114.0 (C-7″), 42.3 (C-1′), 35.5 (C-2). DIP-MS (ESI; M-H) m/z (calculated): 364.00, m/z (measured): 363.95.
6-chloro-2- (Nonylthio) -1H-Benzimidazole, 18c
Yield: 70%. Pink solid; m.p. 73–76 °C. Rf = 0.66 (hexane/AcOEt 7:3). IR (CH2Cl2) = 3032.6, 1389.6 cm−1. 1H NMR (300 MHz, CDCl3) δ: 9.891 (s, 1H, NH), 7.51 (d, J = 1.97 Hz, 1H, H-7), 7.42 (d, J = 8.56 Hz, 1H, H-4), 7.16 (dd, J = 8.56, 1.97 Hz, 1H, H-5), 3.28 (m, 2H, H-1′), 1.72 (m, 2H, H-2′), 1.28 (m, 12H, H-3′,4′,5′,6′,7′,8′), 0.85 (t, J = 6.83 Hz, 3H, H-9′). 13C NMR (75 MHz, CDCl3) δ 152.6 (C-2), 139.9 (C-7a), 137.9 (C-4a), 127.9 (C-6), 122.7 (C-5), 114.6 (C-4), 113.9 (C-7), 32.7 (C-1′), 31.8, 29.6 (C-2′), 29.4, 29.2, 29.1 (C-3′), 28.7 (C-4′), 22.7, 14.1 (C-9′). DIP-MS (ESI; M-H) m/z (calculated): 309.11, m/z (measured): 309.05.
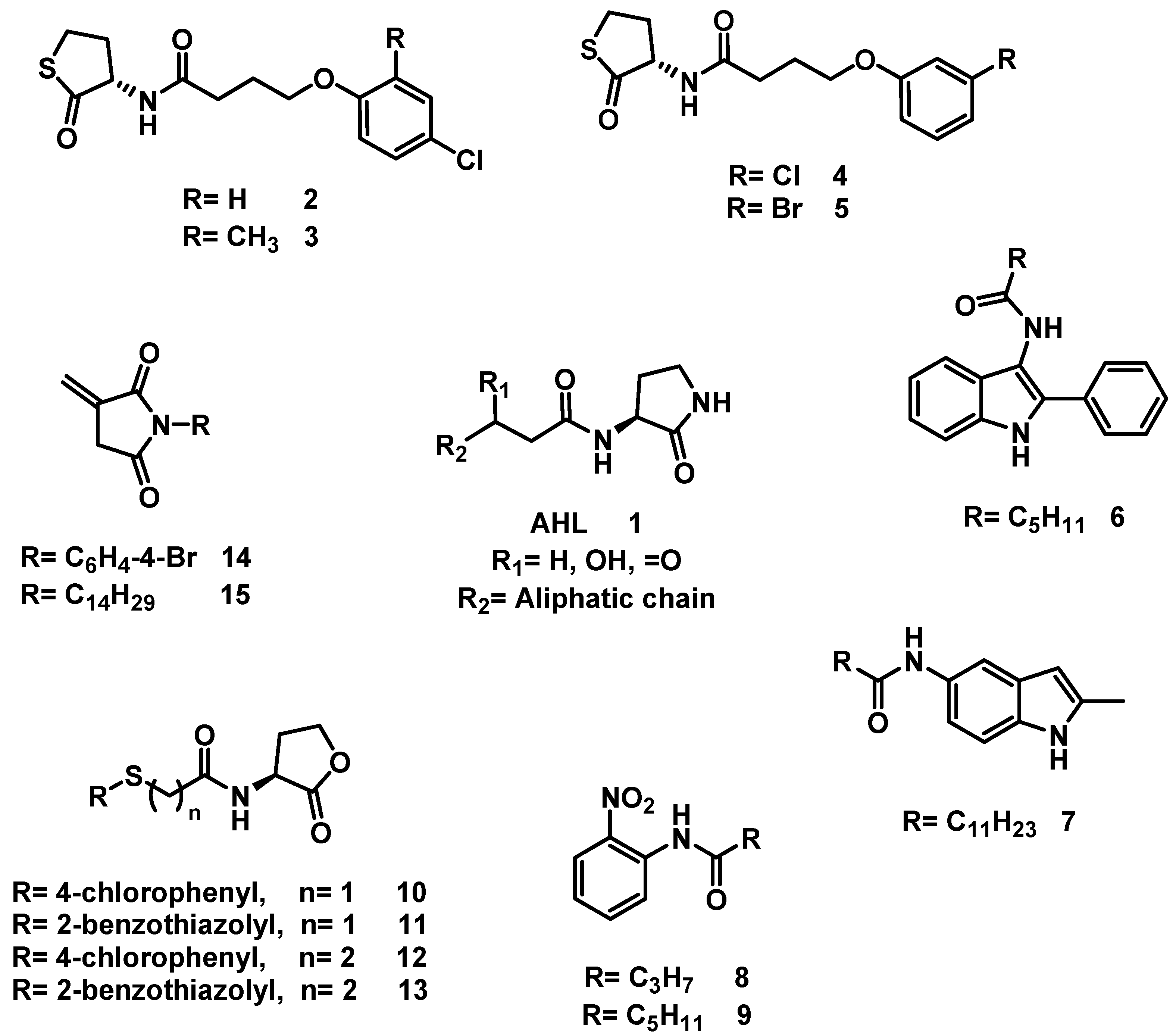
6.8. Procedure for the Synthesis of Compounds, 19
In a vial for microwave reaction equipped with a magnetic stirring bar, a mixture was made of 5-chloro-2-aminopyridine (3.11 mmol, 1 eq) and 2.5 mL of carboxylic acid (butanoic, hexanoic, nonanoic acids). The vial was closed, and the reaction mixture heated for 90 min (butanoic acid at 150 °C, hexanoic acid at 150 °C, nonanoic acid at 180 °C). Upon completion of that period, the reaction mixture was transferred to a 20% potassium carbonate solution (40 mL) and left to stir for 30 min before being vacuum filtered. The solid was washed with 40 mL of distilled water in two portions of 20 mL and oven-dried at 40 °C.
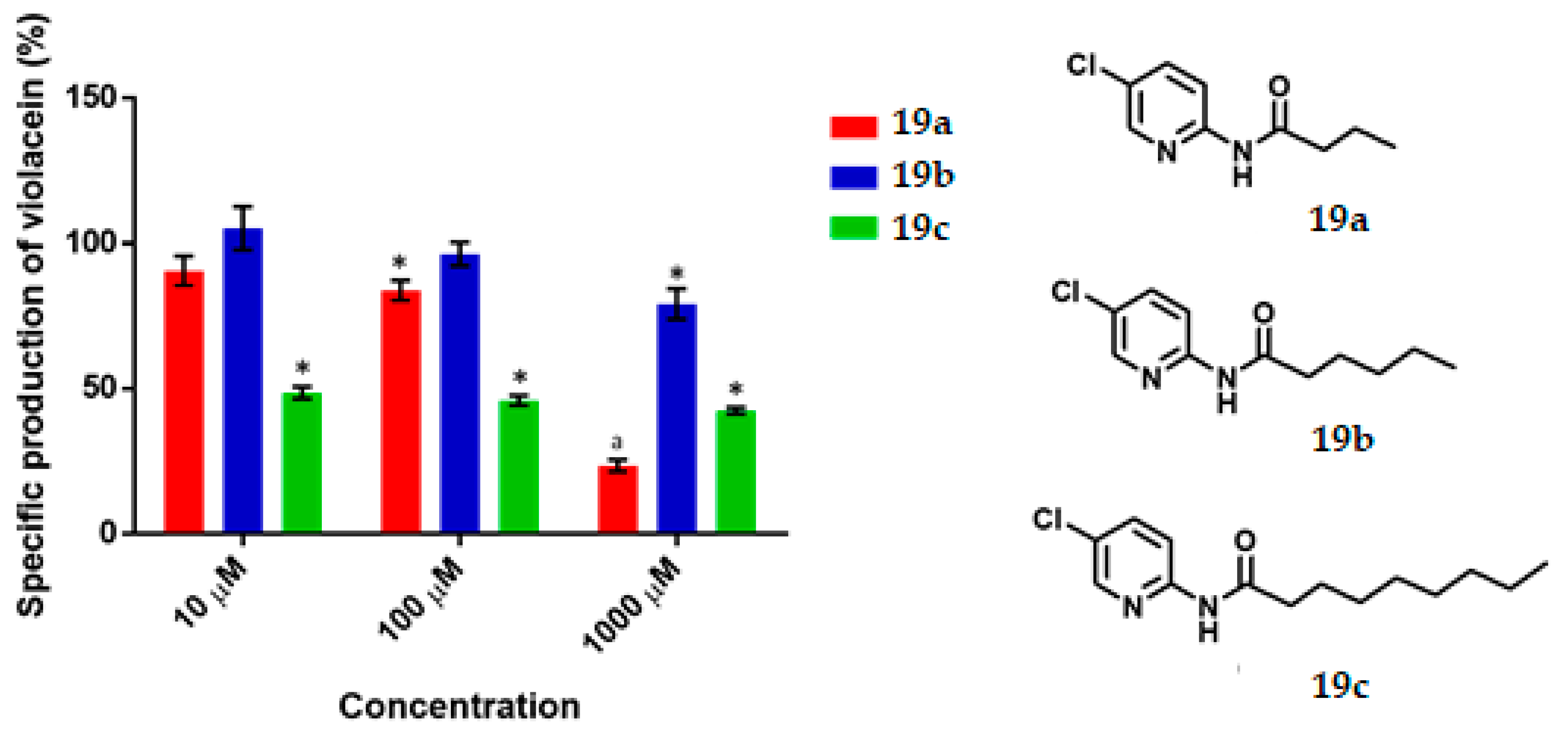
N-(5-Chloropyridin-2-yl) Butamide, 19a
Yield: 91%. White solid; m.p. 102–104 °C. Rf = 0.64 (hexane/AcOEt 7:3). IR (CH2Cl2) = 3300.9, 1697.8, 1582.4 cm−1. 1H NMR (300 MHz, CDCl3) δ: 8.41 (s, 1H, NH), 8.22 (d, J = 8.95 Hz, 1H, H-3′), 8.19 (d, J = 2.54 Hz, 1H, H-6′), 7.65 (dd, J = 8.95, 2.54 Hz, 1H, H-4′), 2.36 (t, J = 7.43 Hz, 2H, H-2), 1.74 (m, 2H, H-3), 0.98 (t, J = 7.38 Hz, 3H, H-4). DIP-MS (ESI; M + Na) m/z (calculated): 221.04, m/z (measured): 221.07.
N-(5-Chloropyridin-2-yl) Hexanamide, 19b
Yield: 87%. White solid; m.p. 103–106 °C. Rf = 0.73 (hexane/AcOEt 7:3). IR (CH2Cl2) = 3275.3, 1690.5, 1582.3 cm-1. NMR 1H (600 MHz, CDCl3) δ: 8.37 (s, 1H, NH), 8.23 (d, J = 8.9 Hz, 1H, H-3′), 8.21 (d, J = 2.5 Hz, 1H, H-6′), 7.66 (dd, J = 8.9, 2.5 Hz, 1H, H-4′), 2.49 (t, J = 7.58 Hz, 2H, H-2), 1.73 (m, 2H, H-3), 1.35 (m, 4H, H-4,5), 0.90 (t, J = 7.13 Hz, 3H, H-6). NMR 13C (125 MHz, CDCl3) δ 171.9 (C-1), 149.9 (C-2′), 146.3 (C-6′), 138.0 (C-4′), 126.5 (C-5′), 114.8 (C-3′), 37.7 (C-2), 31.3 (C-4), 25.0 (C-3), 22.4 (C-5), 13.9 (C-6). DIP-MS (ESI; M+Na) m/z (calculated): 249.07, m/z (measured): 249.11.
N-(5-Chloropyridin-2-yl) Nonamide, 19c
Yield: 90%. White solid; m.p. 64–66 °C. Rf = 0.8 (hexane/AcOEt 7:3). IR (CH2Cl2) = 3271.7, 1687.1, 1582.7 cm−1. 1H NMR (600 MHz, CDCl3) δ: 8.874 (s, 1H, NH), 8.25 (d, J = 8.88 Hz, 1H, H-3′), 8.19 (d, J = 2Hz, 1H, H-6′), 7.64 (dd, J = 8.88, 2 Hz, 1H, H-4′), 2.38 (t, J = 7.59 Hz, 2H, H-2), 1.70 (m, 2H, H-3), 1.28 (m, 10H, H-4,5,6,7,8), 0.85 (t, J = 6.88 Hz, 3H, H-9). 13C NMR (125 MHz, CDCl3) δ 172.1 (C-1), 150.1 (C-2′), 146.1 (C-6′), 138.7 (C-4′), 126.5 (C-5′), 115.0 (C-3′), 37.6 (C-2), 31.8, 29.3, 29.2, 29.1, 25.3 (C-3), 22.6, 14.0 (C-9). DIP-MS (ESI; M+Na) m/z (calculated): 291.12, m/z (measured): 291.19.
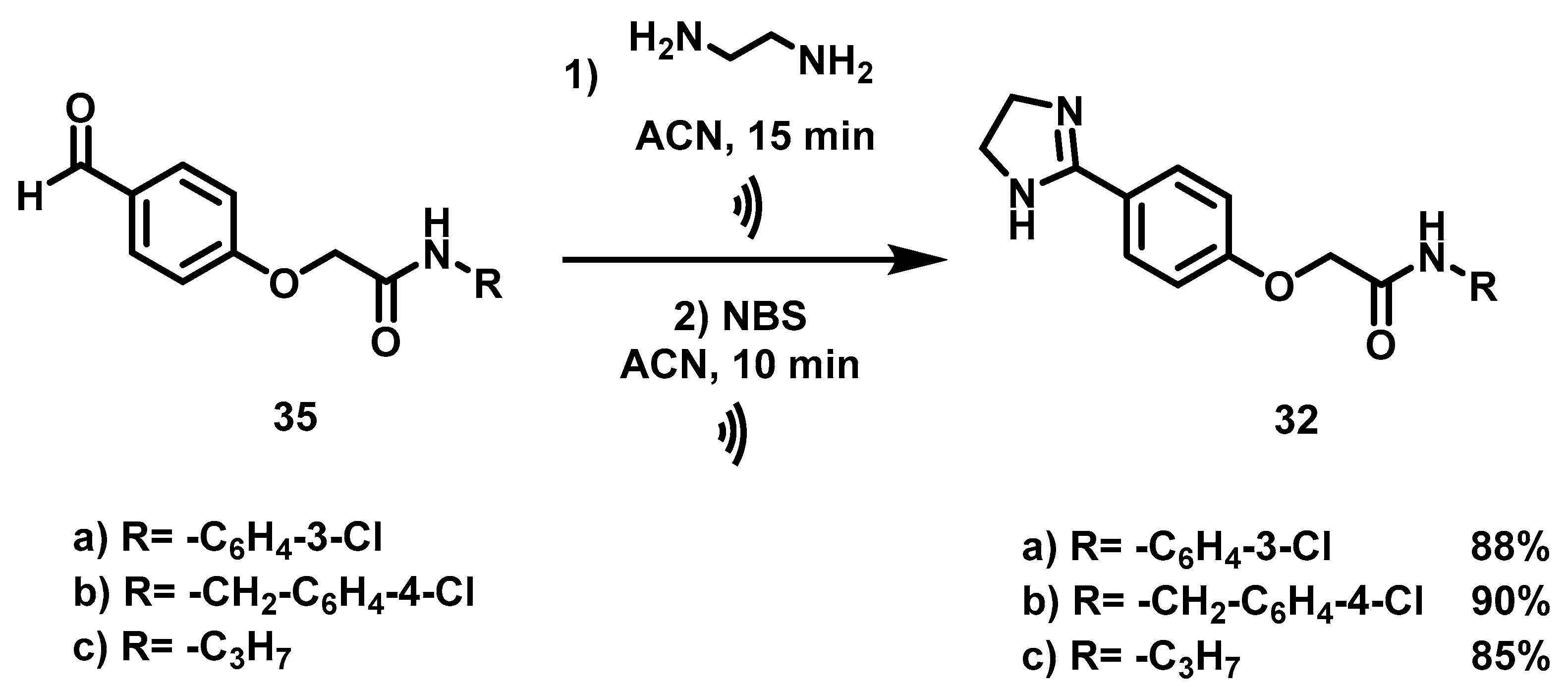
6.9. Procedure for the Synthesis of N-Substituted 2-(4-formylphenoxy) Acetamides, 35
In a 50 mL ball flask loaded with magnetic stirring bar, 15 mL of CH2Cl2, the corresponding amine (3.91 mmol, 1 eq) and bromine acetyl bromide (4.70 mmol, 1.2 eq) were added. The reaction mixture was stirred at rt for 20 min. Subsequently, 40 mL of a 20% aqueous sodium carbonate solution was added and separated by liquid-liquid extraction with CH2Cl2 (2 × 10 mL). The combined organic phases were dried with anhydrous Na2SO4 and filtered, then evaporated under vacuum. Once the residue was dry, it was transferred to a 100 mL ball flask equipped with a magnetic stirring bar and reacted with 4-hydroxybenzaldehyde (4.31 mmol, 1.1 eq) and DBU (3.91 mmol, 1 eq) at 70 °C for 6 h, employing acetonitrile (20 mL) as solvent. Upon completion of that period, the solvent was evaporated under reduced pressure, and 30 mL of a 5% HCl solution was added to the residue. Afterwards liquid-liquid extractions were carried out with AcOEt (3 × 10 mL) and the organic phases were dried with anhydrous Na2SO4 and filtered. The filtrate was evaporated under reduced pressure and the residue was purified by column chromatography, with silica gel as a stationary phase and a hexane/AcOEt mixture 7: 3 as the mobile phase.
N-(3-Chlorophenyl)-2-(4-Formylphenoxy) Acetamide, 35a
Yield: 65%. White solid. 1H NMR (600 MHz, DMSO-d6) δ: 10.364 (s, 1H, NH), 9.89 (s, 1H, CHO), 7.90 (d, J = 8.77 Hz, 2H, H-3″), 7.85 (t, J = 1.81 Hz, 1H, H-2′), 7.55 (d, J = 8.11 Hz, 1H, H-6′), 7.35 (t, J = 8.11 Hz, 1H, H-5′), 7.20 (d, J = 8.77 Hz, 2H, H-2″), 7.14 (d, J = 8.11 Hz, 1H, H-4′), 4.89 (s, 2H, H-2). 13C NMR (150 MHz, DMSO-d6) δ 191,7 (CHO), 166.8 (C-1), 163.1 (C-1″), 140.2 (C-1′), 133.6 (C-3′), 132.2 (C-3″), 130.9 (C-5′), 130.6 (C-4″), 123.9 (C-4′), 119.7 (C-2′), 118.5 (C-6′), 115.6 (C-2″), 67.5 (C-2). DIP-MS (ESI; M-H) m/z (calculated): 288.04, m/z (measured): 288.05.
N-(4-Chlorobenzyl)-2-(4-Formylphenoxy) Acetamide, 35b
Yield: 73%. White solid. 1H NMR (600 MHz, DMSO-d6) δ: 9.98 (s, 1H, CHO), 8.77 (t, J = 6 Hz, 1H, NH), 7.89 (d, J = 8.7 Hz, 2H, H-3″), 7.37 (d, J = 8.4 Hz, 2H, H-4′), 7.29 (d, J = 8.4 Hz, 2H, H-3′), 7.16 (d, J = 8.7 Hz, 2H, H-2″), 4.71 (s, 2H, H-2), 4.34 (d, J = 6 Hz, 2H, H-1′). 13C NMR (150 MHz, DMSO-d6) δ 191.3 (CHO), 167.2 (C-1), 162.5 (C-1″), 138.2 (C-2′), 131.6 (C-3″), 131.3 (C-5′), 130.1 (C-4″), 129.0 (C-3′), 128.1 (C-4′), 115.2 (C-2″), 66.9 (C-2), 41.1 (C-1′). DIP-MS (ESI; M-H) m/z (calculated): 302.05, m/z (measured): 302.09.
2-(4-Formylphenoxy)-N-propyl Acetamide, 35c
Yield: 79%. White solid. 1H NMR (500 MHz, DMSO-d6) δ: 9.87 (s, 1H, CHO), 8.15 (s, 1H, NH), 7.87 (d, J = 8.7 Hz, 2H, H-3″), 7.13 (d, J = 8.7 Hz, 2H, H-2″), 4.60 (s, 2H, H-2), 3.08 (dd, J = 13.15, 6.84, 2H, H-1′), 1.44 (m, 2H, H-2′), 0.82 (t, J = 7.42 Hz, 3H, H-3′). 13C NMR (125 MHz, DMSO-d6) δ 191.786 (CHO), 167.3 (C-1), 163.1 (C-1″), 132.1 (C-3″), 130.5 (C-4″), 115.6 (C-2″), 67.5 (C-2), 40.6 (C-1′), 22.8 (C-2′), 11.7 (C-3′). DIP-MS (ESI; M + Na) m/z (calculated): 244.09, m/z (measured): 244.10.
6.10. Synthesis of N-Substituted 2-(4-(imidazolin-2-yl)-phenoxy) Acetamides, 32
Once the aldehydes (1.20 mmol, 1 eq) were obtained, they were placed in a 100 mL ball flask containing 50 mL of acetonitrile. Afterwards, they were subjected to a reaction with ethylenediamine (1.20 mmol, 1 eq) with ultrasound for 15 min. Upon completion of this time, NBS (1.45 mmol, 1.2 Eq) was added to the reaction mixture and it was reacted by ultrasound for 10 min. Then the solvent was evaporated under reduced pressure, followed by the addition of 30 mL of an aqueous solution of 20% NaOH. The resulting suspension was stirred for 20 min and vacuum filtered. The solid obtained was washed with 40 mL of distilled water (2 × 20 mL) and oven-dried at 40 °C.
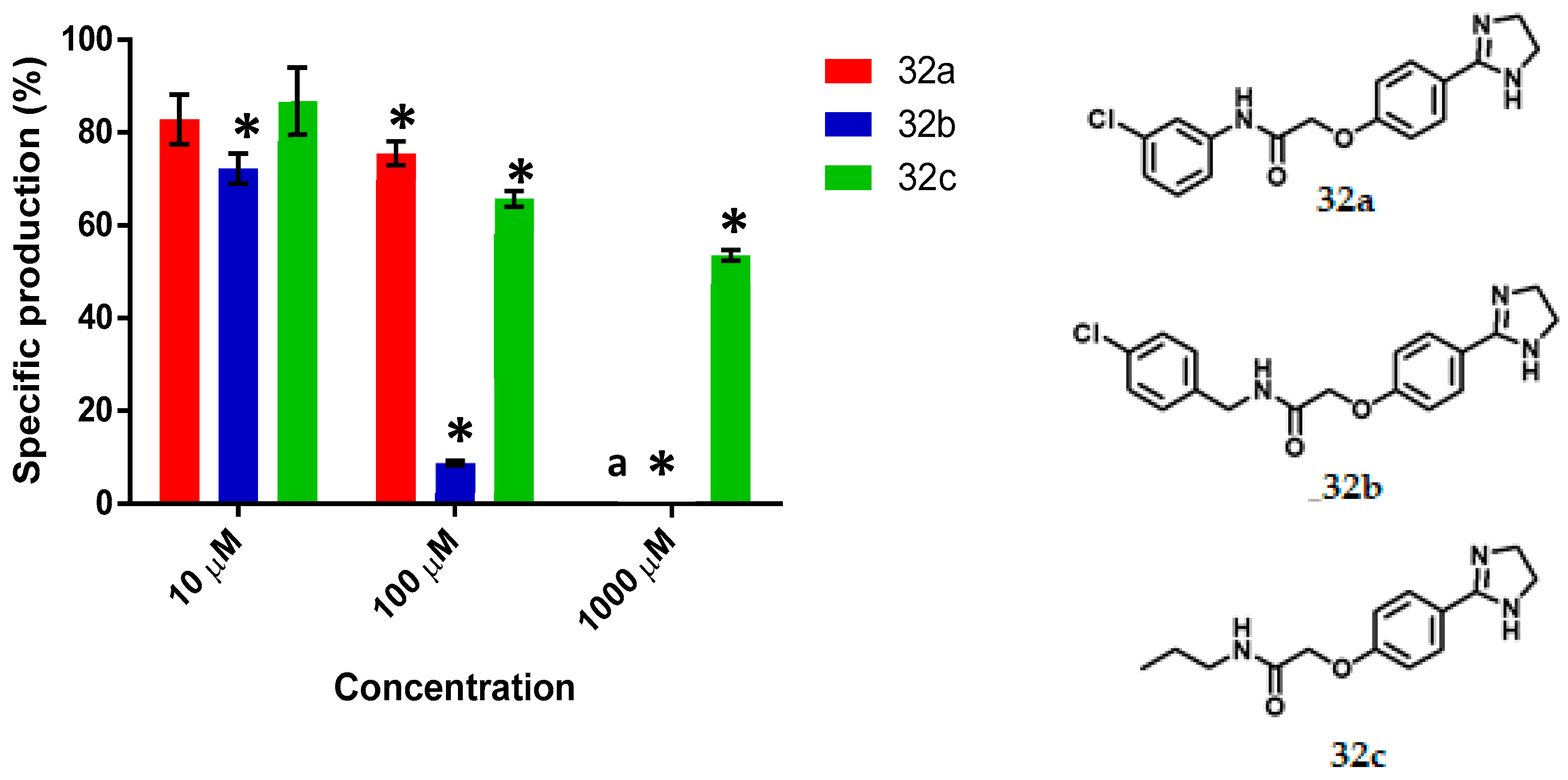
N-(3-Chlorophenyl)-2-(4-(Imidazolin-2-yl) Phenoxy) Acetamide, 32a
Yield: 88%. Orange solid; m.p. 192–195 °C. Rf = 0.17 (MeOH/NH4OH 20:1). IR (CH2Cl2) = 3584.6, 3203, 1655.9, 1532.8 cm–1. 1H NMR (500 MHz, DMSO-d6) δ: 10.47 (s, 1H, NH), 7.84 (t, J = 1.99 Hz, 1H, H-2′), 7.77 (d, J = 8.7 Hz, 2H, H-3″), 7.54 (m, 1H, H-6′), 7.33 (t, J = 8.12 Hz, 1H, H-5′), 7.13 (m, 1H, H-4′), 7.03 (d, J = 8.7 Hz, 2H, H-2″), 4.77 (s, 2H, H-2), 3.56 (s, 4H, H-7″). 13C NMR (125 MHz, DMSO-d6) δ 167.2 (C-1), 163.6 (C-5″), 159.7 (C-1″), 140.3 (C-1′), 133.5 (C-3′), 130.9 (C-5′), 129.1 (C-3″), 124.3 (C-4″), 123.8 (C-4′), 119.6 (C-2′), 118.5 (C-6′), 114.7 (C-2″), 67.4 (C-2), 49.9 (C-7″). DIP-MS (ESI; M-H2O + MeOH) m/z (calculated): 344.11, m/z (measured): 344.20.
N-(4-Chlorobenzyl)-2-(4-(Imidazolin-2-yl) Phenoxy) Acetamide, 32b
Yield: 90%. Yellow solid; m.p. 149–155 °C. Rf = 0.2 (MeOH/NH4OH 20:1). IR (CH2Cl2) = 3250.1, 1594.5, 1545.1, 1516.9, 839.1 cm−1. 1H NMR (500 MHz, DMSO-d6) δ: 8.74 (t, J = 6.14 Hz, 1H, NH), 7.79 (d, J = 8.78 Hz, 2H, H-3″), 7.38 (d, J = 8.4 Hz, 2H, H-4′), 7.29 (d, J = 8.4 Hz, 2H, H-3′), 7.02 (d, J = 8.78 Hz, 2H, H-2″), 4.62 (s, 2H, H-2), 4.35 (d, J = 6.14 Hz, 2H, H-1′), 3.59 (s, 4H, H-7″). 13C NMR (125 MHz, DMSO-d6) δ 168.1 (C-1), 163.6 (C-5″), 159.6 (C-1″), 138.8 (C-2′), 131. (C-5′), 129.6 (C-3′), 129.1 (C-3″), 128.6 (C-4′), 124.3 (C-4″), 114.8 (C-2″), 67.4 (C-2), 49.8 (C-7″), 41.7 (C-1′). DIP-MS (ESI; M+H) m/z (calculated): 344.11, m/z (measured): 344.02.
2-(4-(Imidazolin-2-yl) Phenoxy)-N-Propyl Acetamide, 32c
Yield: 85%. White solid); m.p. 175–178 °C. Rf = 0.15 (MeOH/NH4OH 20:1). IR (CH2Cl2) = 3376.1, 3202.8, 1659.4, 1615.4, 1537.3, 1519.5 cm-1. 1H NMR (500 MHz, DMSO-d6) δ: 8.10 (t, J = 5.36 Hz, 1H, NH), 7.75 (m, 2H, H-3″), 6.91 (m, 2H, H-2″), 4.50 (s, 2H, H-2), 3.56 (s, 4H, H-7″), 3.08 (m, 2H, H-1′), 1.43 (m, 2H, H-2′), 0.81 (t, J = 7.42 Hz, 3H, H-3′). 13C NMR (125 MHz, DMSO-d6) δ 167.7 (C-1), 163.6 (C-5″), 159.6 (C-1″), 129.0 (C-3″), 124.1 (C-4″), 114.7 (C-2″), 67.4 (C-2), 49.7 (C-7″), 40.5 (C-1′), 22.8 (C-2′), 11.7 (C-3′). DIP-MS (ESI; M+H) m/z (calculated): 262.15, m/z (measured): 262.04.
6.11. Preparation of Culture Media, Test Compounds, and Inoculum
The Luria Bertani (LB) broth was prepared in one liter of distilled water by adding 10 g peptone, 5 g yeast extract, and 5 g NaCl and then sterilized in an autoclave at 15 psi and 121 °C for 15 min. For the LB solid medium, 15 g of bacteriological agar was added and the solution was made up to a liter with distilled water. Thioglycolate medium BBL was prepared according to the fabricant specifications with distilled water and then sterilized in an autoclave at 15 psi and 121 °C for 15 min. C. violaceum CV026 was always grown in the presence of 30 μg/mL of kanamycin.
The amount of each test compound required for a concentration of 100 mM Pleasewas weighed. With the resulting solution, serial 1:10 dilutions were prepared to obtain concentrations 1000 μM, 100 μM and 10 μM.
6.12. Preparation of CV026 Suspensions
C. violaceum CV026 from a cryovial was streaked onto L-agar plates containing 30 μg/mL kanamycin. Bacteria were incubated at 29 °C for 24 h. The next day an isolated colony was inoculated into 5 mL LB medium with 30 μg/mL kanamycin, followed by incubation at 29 °C and 200 rpm for 15 h.
6.13. Evaluation of Compounds as Quorum Sensing Inhibitors in Chromobacterium violaceum CV026
C. violaceum CV026 was cultured in 50 mL of thioglycolate medium until reaching an optical density of 0.12 to 600 nm, then 430 μL of an 80 μM C6-AHL solution were added (680 nM final concentration) to the broth. Subsequently 1980 μL of the suspension was transferred to a 5 mL tube and 20 μL of the dilutions of the test compounds were added until reaching the final concentration of 1000 μM, 100 μM, 10 μM. Tubes were incubated at 29 °C and 800 rpm for 16 h. Upon completion of the incubation time, cell density was determined by absorbance at 720 nm by using the thioglycolate medium as the blank. Finally, the absorbance of violacein was measured. 500 μL of the bacterial culture was placed in a 2 mL tube and then 500 μL of acetone were added. The tubes were vortexed and centrifuged at 15,000 rpm for 1 min. The specific production of violacein was calculated by dividing the value of the reading at 577 nm by that at 720 nm. Experiments with additional concentrations were performed in order to determinate the IC50 values. Each experiment was performed 6 times/compound, and the results were graphed. Statistical significance was analyzed by Student’s t-test.
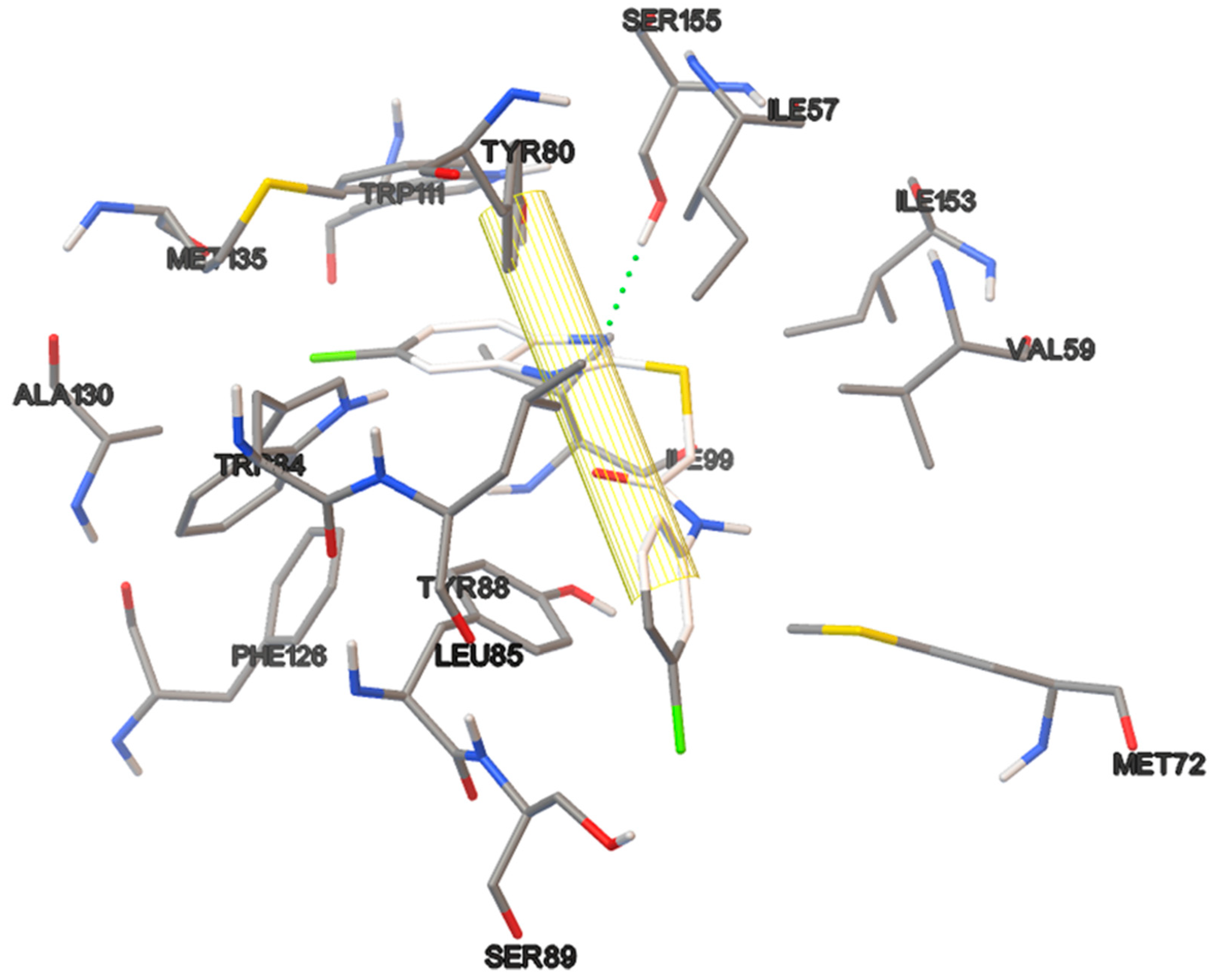
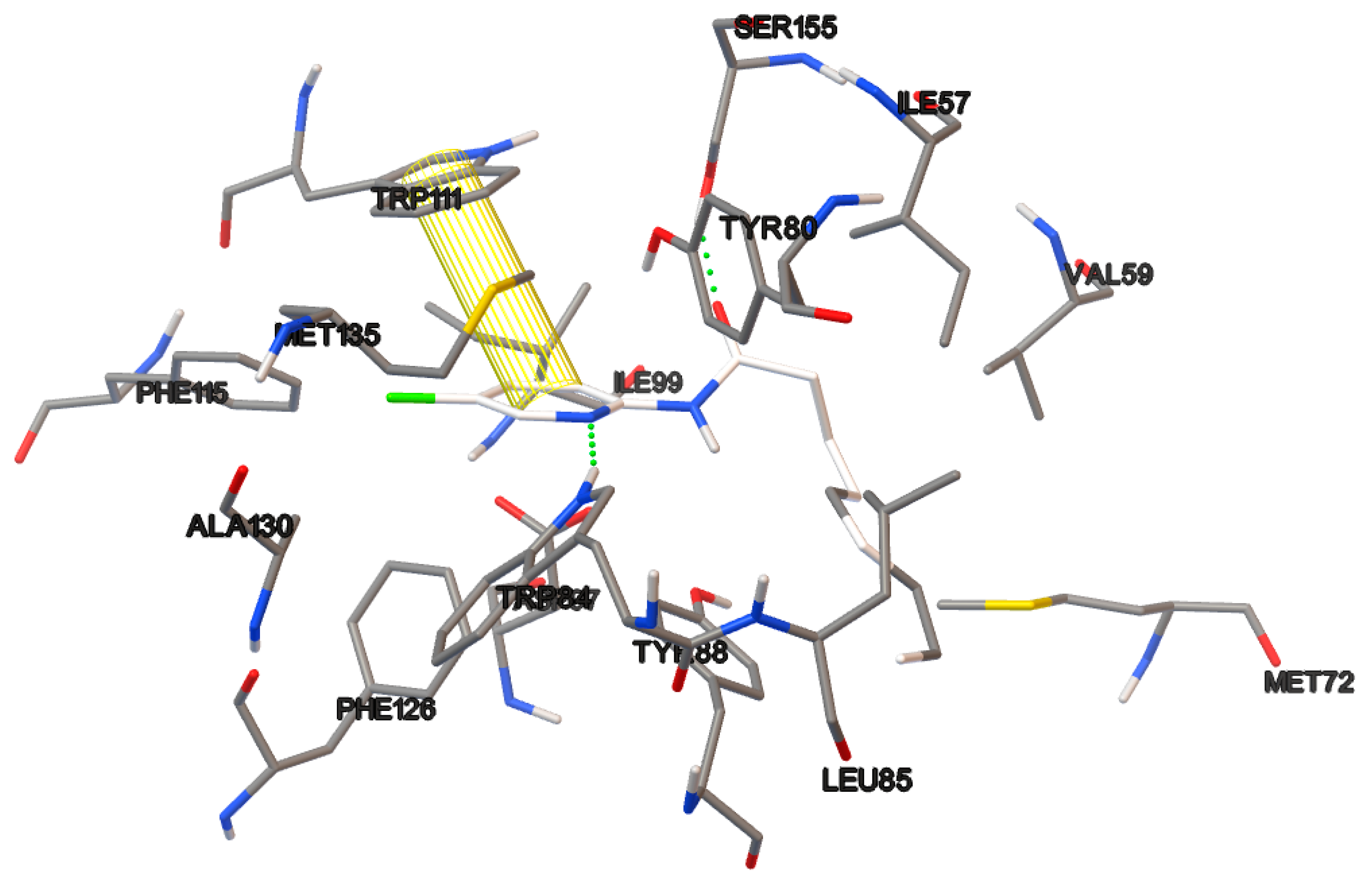
6.14. Modeling and Optimization of Ligands.
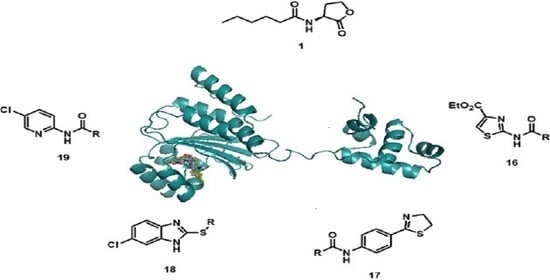
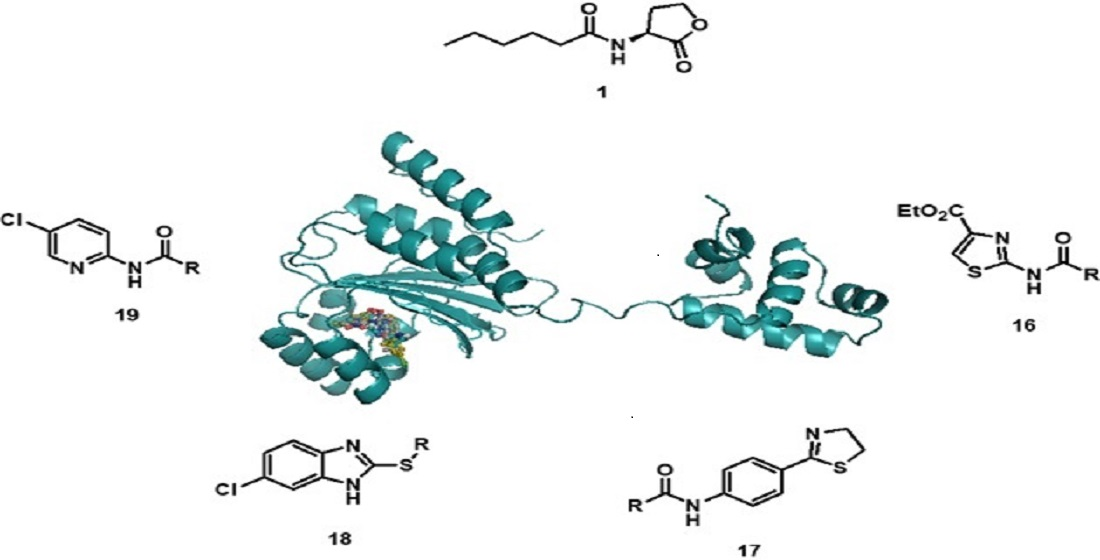
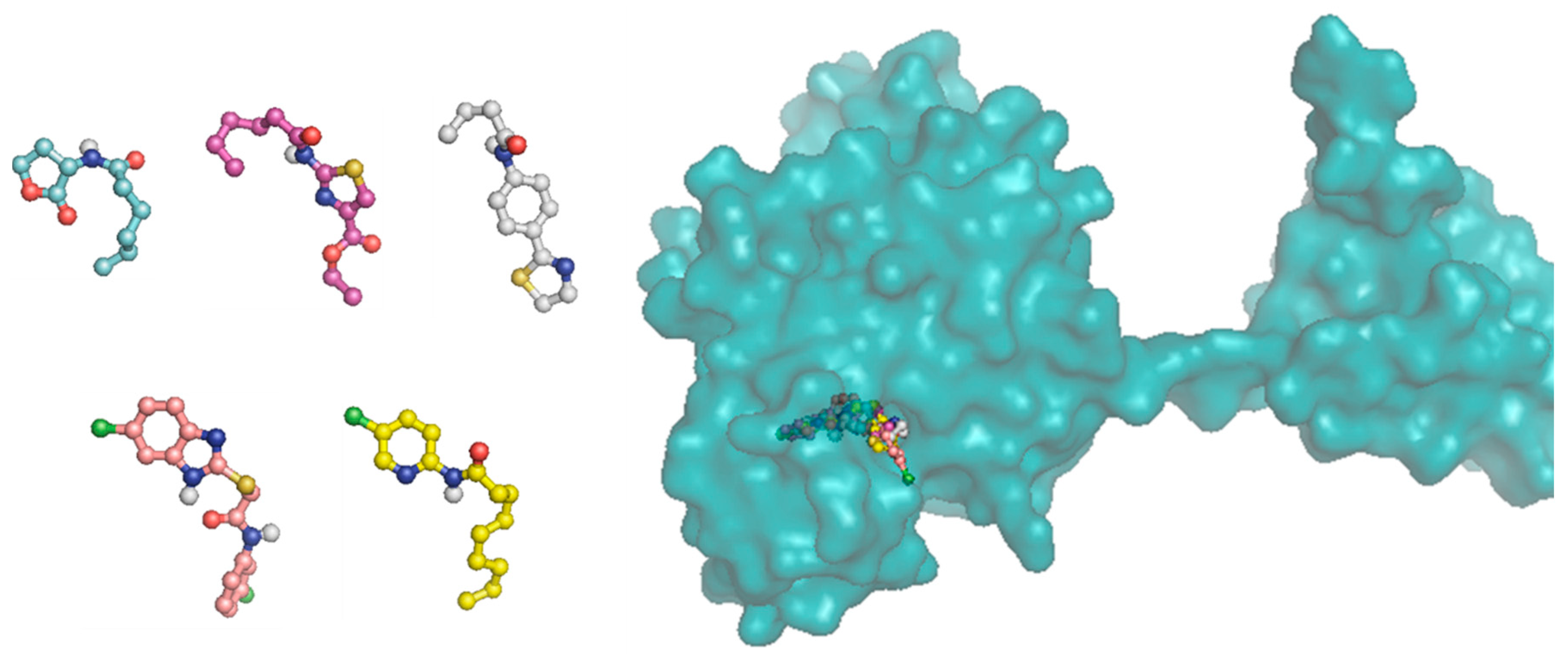
Docking studies of the target compounds were carried out on the CviR protein (PDB code: 3QP6).
Prior to docking, the ligands were drawn with the ACD/ ChemSketch program [
51]. A geometric pre-optimization was created in 3D maintaining the stereochemistry. The 3D structures were submitted to a geometric and energetic optimization at the AM1 (Austin Model 1) semiempirical level using the Gaussian 09 program [
52]. The output files were saved as (PDB).
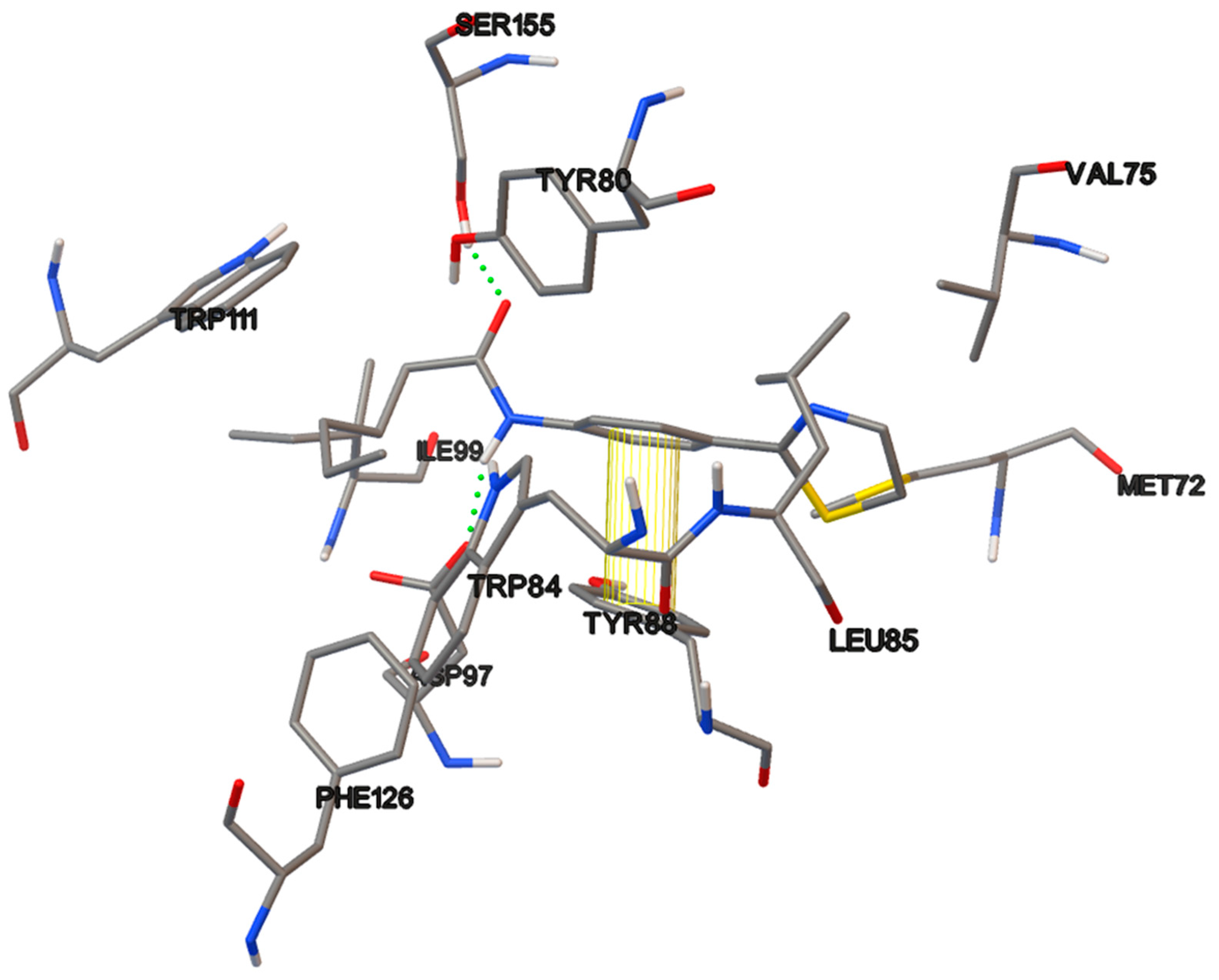
6.15. Molecular Docking
The docking studies were performed on the AutoDock 4.2 program, which maintains the macromolecule rigid while allowing flexibility in the ligand. Polar hydrogens were added to the ligand, and protein atoms and partial charges were assigned to the receptor (Kollman) and ligand (Gasteiger). Blind docking was achieve using a grid box of 126 Å3 with a grid spacing of 0.375 Å3 using the Hybrid Genetic Algorithm of Lamarckian with an initial population of 100 randomized individuals and a maximum number of energy evaluations of 107. The results of these docking simulations experiments were examined on AutoDockTools version 1.5.2 and PyMol visualizer to describe the non-bond interaction from the ligand-protein complex.










 , 15 min; then NBS, ACN,
, 15 min; then NBS, ACN,  , 10 min.
, 10 min.












{kind=link}
{kind=link}
{kind=link}
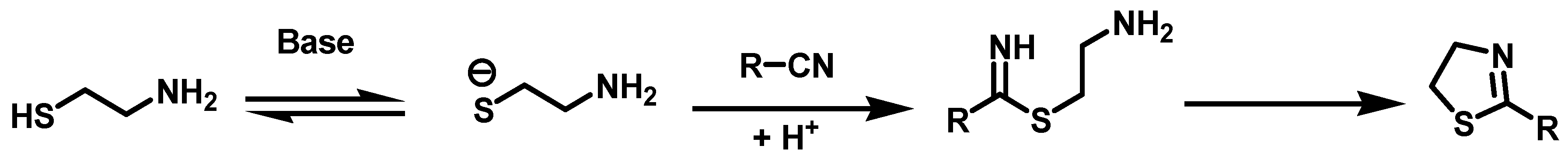{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
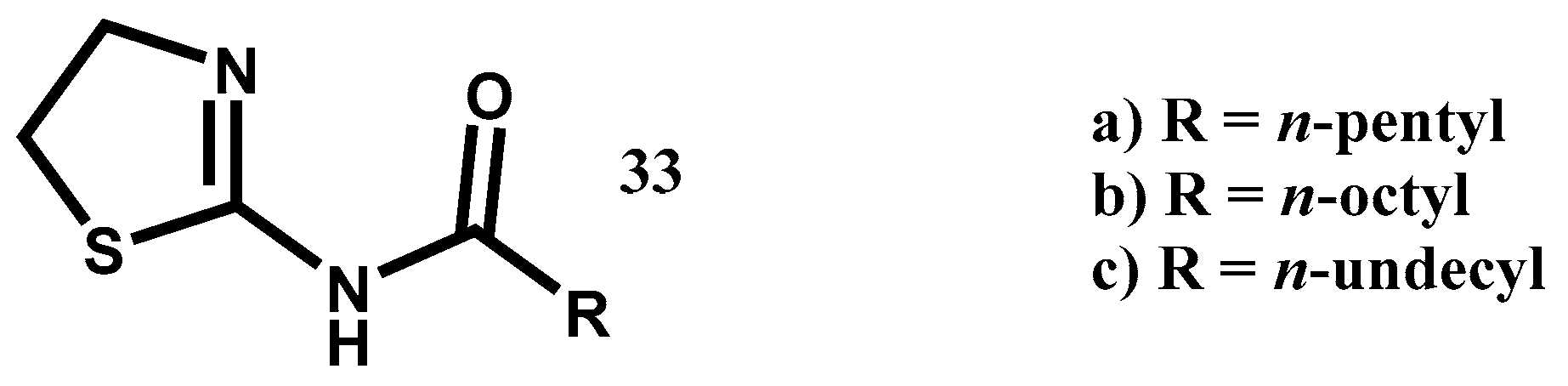{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





