SPInDel Analysis of the Non-Coding Regions of cpDNA as a More Useful Tool for the Identification of Rye (Poaceae: Secale) Species





Abstract
1. Introduction
2. Results
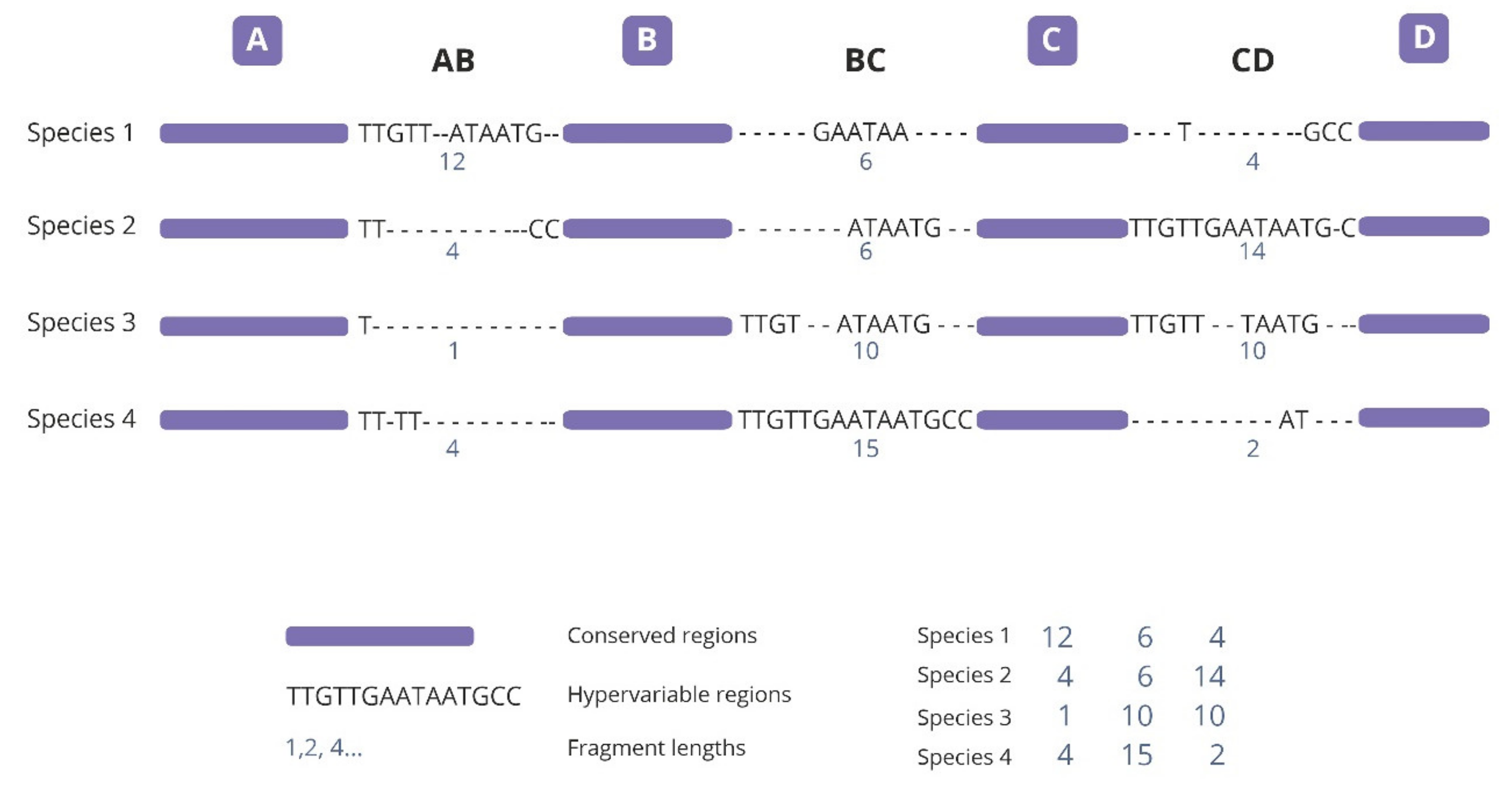
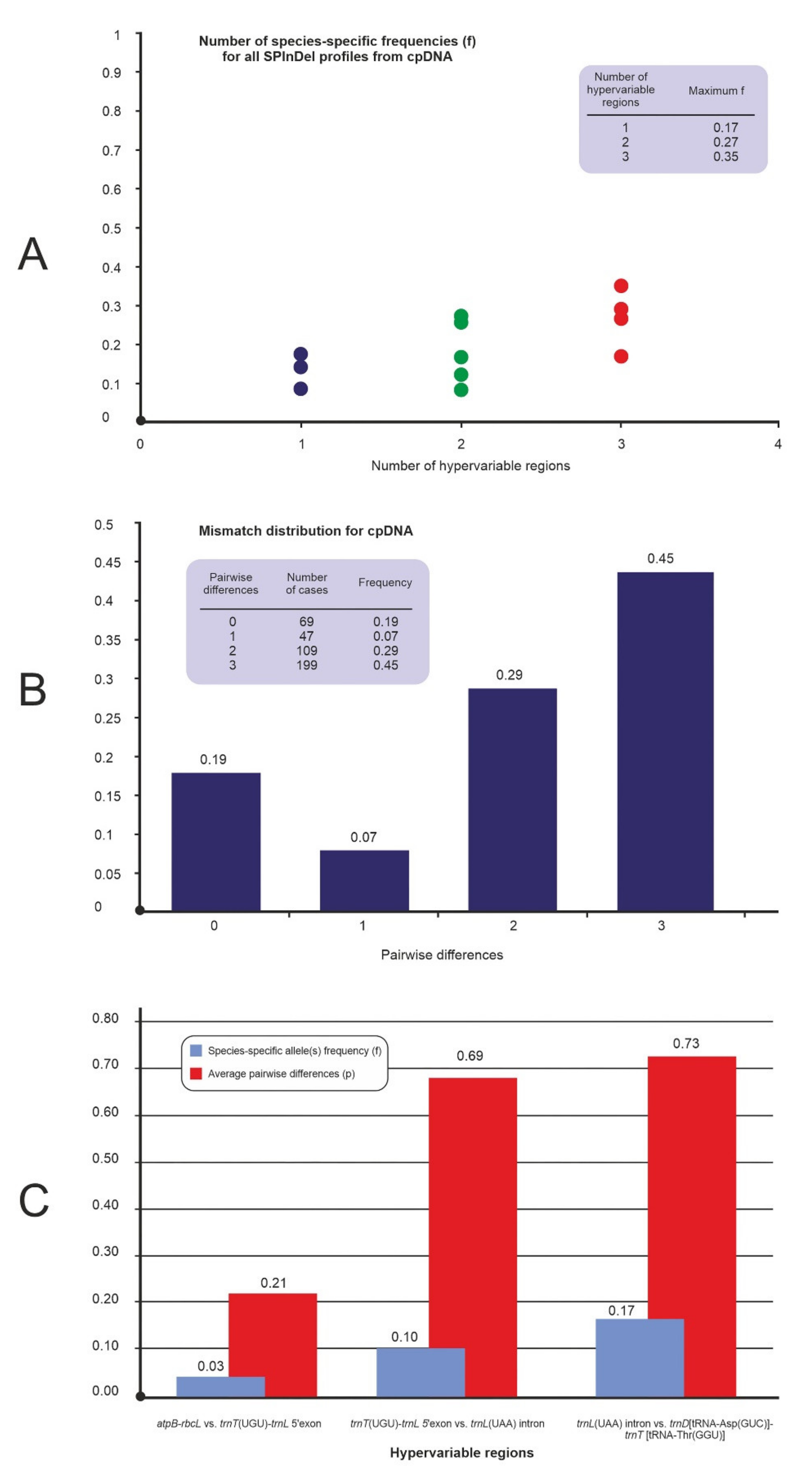
2.1. Analysis of Variable-Length cpDNA Sequences
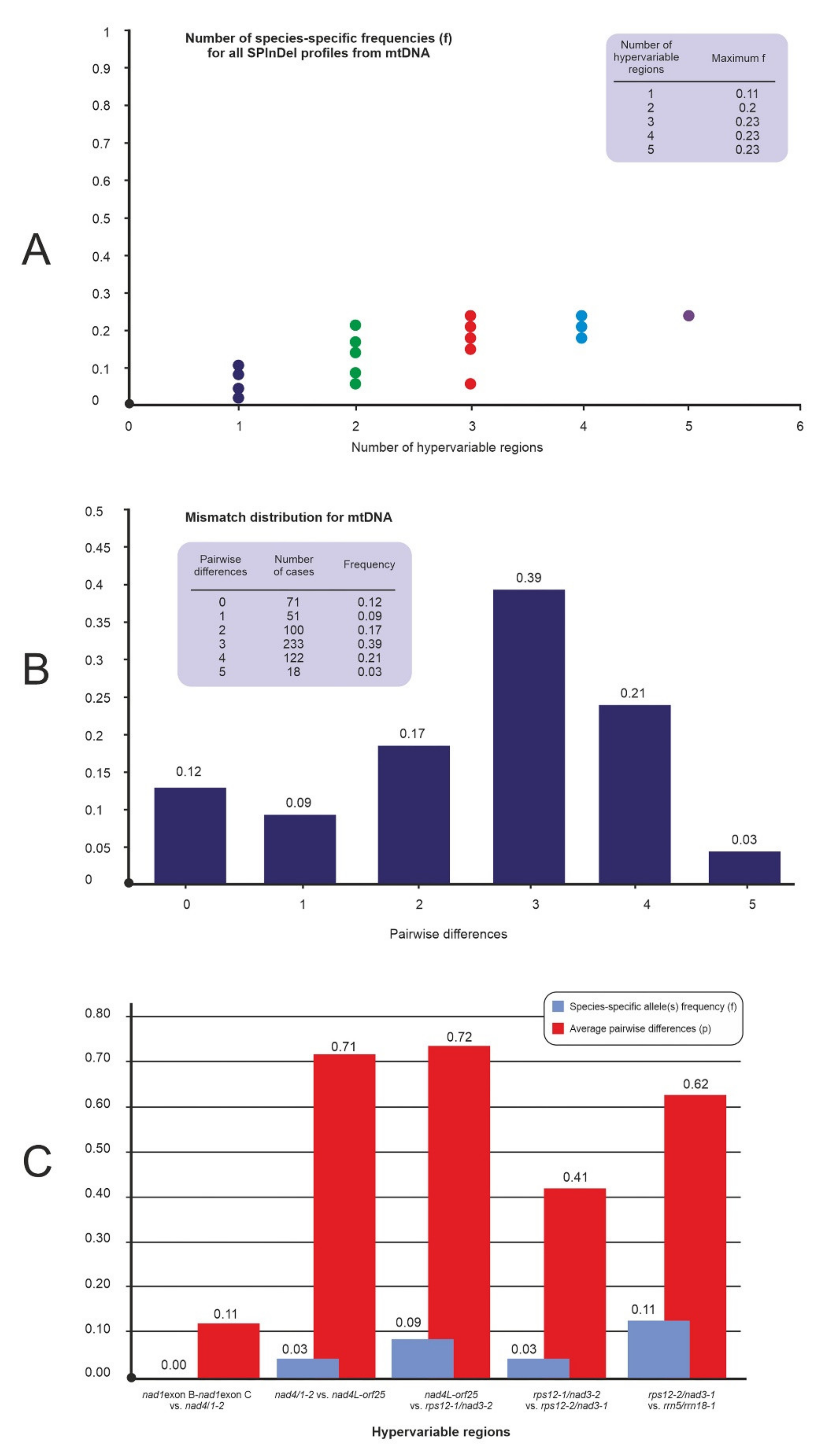
2.2. Analysis of Variable-Length mtDNA Sequences
3. Discussion
4. Materials and Methods
4.1. DNA Extraction, PCR Amplification, and DNA Sequencing
4.2. SPInDel Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Alptekin, B.; Langridge, P.; Budak, H. Abiotic stress miRNomes in the Triticeae. Funct. Integr. Genom. 2017, 17, 145–170. [Google Scholar] [CrossRef]
- Madej, L.J. Worldwide trends in rye growing and breeding. Vortraege Fuer Pflanz. 1996, 35, 1–6. [Google Scholar]
- Mago, R.; Spielmeyer, W.; Lawrence, G.; Lagudah, E.; Ellis, J.; Pryor, A. Identification and mapping of molecular markers linked to rust resistance genes located on chromosome 1RS of rye using wheat-rye translocation lines. Theor. Appl. Genet. 2002, 104, 1317–1324. [Google Scholar] [CrossRef]
- Hurni, S.; Brunner, S.; Buchmann, G.; Herren, G.; Jordan, T.; Krukowski, P.; Wicker, T.; Yahiaoui, N.; Mago, R.; Keller, B. Rye Pm8 and wheat Pm3 are orthologous genes and show evolutionary conservation of resistance function against powdery mildew. Plant J. 2013, 76, 957–969. [Google Scholar] [CrossRef]
- Broda, Z.; Tomkowiak, A.; Mikolajczyk, S.; Weigt, D.; Górski, F.; Kurasiak-Popowska, D. The genetic polymorphism between the wild species and cultivars of rye Secale cereale L. Acta Agrobot. 2016, 69. [Google Scholar] [CrossRef]
- Schlegel, R. Rye (Secale cereale L)—A younger crop plant with bright future. In Genetic Resources, Chromosome Engineering, and Crop Improvement: Vol II Cereals; Sing, R.J., Jauhar, P., Eds.; CRC Press: Boca Raton, FL, USA, 2006; pp. 365–394. [Google Scholar]
- Bartoš, J.; Paux, E.; Kofler, R.; Havránková, M.; Kopecký, D.; Suchánková, P.; Šafář, J.; Šimková, H.; Town, C.D.; Lelley, T.; et al. A first survey of the rye (Secale cereale) genome composition through BAC end sequencing of the short arm of chromosome 1R. BMC Plant Biol. 2008, 8, 95. [Google Scholar] [CrossRef]
- Flavell, R.B.; Bennett, M.D.; Smith, J.B.; Smith, D.B. Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem. Genet. 1974, 12, 257–269. [Google Scholar] [CrossRef]
- Roshevitz, R.Y. A monograph of the wild, weedy and cultivated species of rye. Acta Inst. Bot. Nomine Acad. Sci. USSR Ser. 1947, 1, 105–163. [Google Scholar]
- Delipavlov, D. Secale rhodopaeum Delipavlov—A new species of rye from the Rhodope Mountains. Dokl. Bolg. Akad. Nauk 1962, 15, 407–411. [Google Scholar]
- Frederiksen, S.; Petersen, G. Morphometrical analyses of Secale (Triticeae, Poaceae). Nord. J. Bot. 1997, 17, 185–198. [Google Scholar] [CrossRef]
- Frederiksen, S.; Petersen, G. A taxonomic revision of Secale (Triticeae, Poaceae). Nord. J. Bot. 1998, 18, 399–420. [Google Scholar] [CrossRef]
- GRIN National Plant Germplasm System ARS. Germplasm Resources Information Network (GRIN-Taxonomy); National Germplasm Resources Laboratory: Beltsville, MD, USA, 2019. Available online: http://npgsweb.ars-grin.gov/gringlobal/taxon/taxonomysearchcwr.aspx (accessed on 8 December 2020).
- Shang, H.-Y.; Wei, Y.-M.; Long, H.; Yan, Z.-H.; Zheng, Y.-L. Identification of LMW Glutenin-Like Genes from Secale sylvestre Host. Russ. J. Genet. 2005, 41, 1372–1380. [Google Scholar] [CrossRef]
- Hammer, K.; Skolimowska, E.; Knüpffer, H. Vorarbeiten zur monographischen Darstellung von Wildpflanzensortimenten: Secale L. Die Kult. 1987, 35, 135–177. [Google Scholar] [CrossRef]
- Singh, R.J.; Röbbelen, G. Identification by Giemsa technique of the translocations separating cultivated rye from three wild species of Secale. Chromosoma 1977, 59, 217–225. [Google Scholar] [CrossRef]
- Bennett, M.D.; Gustafson, J.P.; Smith, J.B. Variation in nuclear DNA in the genus Secale. Chromosoma 1977, 61, 149–176. [Google Scholar] [CrossRef]
- Murai, K.; Naiyu, X.T.K. Studies on the origin of crop species by restriction endonuclease analysis of organellar DNA. III. Chloroplast DNA variation and interspecific relationships in the genus Secale. Jpn. J. Genet. 1989, 64, 36–47. [Google Scholar] [CrossRef]
- Petersen, G.; Doebley, J.F. Chloroplast DNA variation in the genus Secale (Poaceae). Plant Syst. Evol. 1993, 187, 115–125. [Google Scholar] [CrossRef]
- Cuadrado, A.; Jouve, N. Evolutionary Trends of Different Repetitive DNA Sequences during Speciation in the Genus Secale. J. Hered. 2002, 93, 339–345. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, Z.; Li, G.; Liu, C.; Tang, Z.; Zhang, Y.; Ren, Z. Diversified chromosomal distribution of tandemly repeated sequences revealed evolutionary trends in Secale (Poaceae). Plant Syst. Evol. 2010, 287, 49–56. [Google Scholar] [CrossRef]
- Chikmawati, T.; Skovmand, B.; Gustafson, J.P. Phylogenetic relationships among Secale species revealed by amplified fragment length polymorphisms. Genome 2005, 48, 792–801. [Google Scholar] [CrossRef]
- Fowler, D.B.; Limin, A.E. Exploitable genetic variability for cold tolerance in commercially grown cereals. Can. J. Plant Sci. 1987, 67, 278. [Google Scholar]
- Cuadrado, A.; Schwarzacher, T. The chromosomal organization of simple sequence repeats in wheat and rye genomes. Chromosoma 1998, 107, 587–594. [Google Scholar] [CrossRef]
- Maraci, Ö.; Özkan, H.; Bilgin, R. Phylogeny and genetic structure in the genus Secale. PLoS ONE 2018, 13, e0200825. Available online: https://pubmed.ncbi.nlm.nih.gov/30024916 (accessed on 8 December 2020). [CrossRef]
- Hagenblad, J.; Oliveira, H.R.; Forsberg, N.E.G.; Leino, M.W. Geographical distribution of genetic diversity in Secale landrace and wild accessions. BMC Plant Biol. 2016, 16, 23. [Google Scholar] [CrossRef]
- Santos, E.; Matos, M.; Silva, P.; Figueiras Am Benito, C.; Pinto-Carnide, O. Molecular diversity and genetic relationships in Secale. J. Genet. 2016, 95, 273–281. [Google Scholar] [CrossRef]
- Al-Beyroutiová, M.; Sabo, M.; Sleziak, P.; Dušinský, R.; Birčák, E.; Hauptvogel, P.; Kilian, A.; Švec, M. Evolutionary relationships in the genus Secale revealed by DArTseq DNA polymorphism. Plant Syst. Evol. 2016, 302, 1083–1091. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Flavell, R.B. The structure, amount and chromosomal localisation of defined repeated DNA sequences in species of the genus Secale. Chromosoma 1982, 86, 613–641. [Google Scholar] [CrossRef]
- Doebley, J.; von Bothmer, R.; Larson, S. Chloroplast DNA Variation and the Phylogeny of Hordeum (Poaceae). Am. J. Bot. 1992, 79, 576–584. [Google Scholar] [CrossRef]
- Fuchs, J.; Kühne, M.; Schubert, I. Assignment of linkage groups to pea chromosomes after karyotyping and gene mapping by fluorescent in situ hybridization. Chromosoma 1998, 107, 272–276. [Google Scholar] [CrossRef]
- Cuadrado, A.; Jouve, N. Distribution of highly repeated DNA sequences in species of the genus Secale. Genome 1997, 40, 309–317. [Google Scholar] [CrossRef]
- Sidhu, J.S.; Ramakrishnan, S.M.; Ali, S.; Bernardo, A.; Bai, G.; Abdullah, S.; Girma Ayana, G.; Sehgal, S.K. Assessing the genetic diversity and characterizing genomic regions conferring Tan Spot resistance in cultivated rye. PLoS ONE 2019, 14, e0214519. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, C. Biologia Żyta; PWN: Warsaw, Poland, 1983. [Google Scholar]
- Palmer, J.D.; Jansen, R.K.; Michaels, H.J.; Chase, M.W.; Manhart, J.R. Chloroplast DNA Variation and Plant Phylogeny. Ann. Missouri. Bot. Gard. 1988, 75, 1180–1206. Available online: http://www.jstor.org/stable/2399279 (accessed on 8 December 2020). [CrossRef]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. Available online: http://www.pnas.org/content/91/15/6795.abstract (accessed on 8 December 2020). [CrossRef] [PubMed]
- Wang, X.-R.; Tsumura, Y.; Yoshimaru, H.; Nagasaka, K.; Szmidt, A.E. Phylogenetic relationships of Eurasian pines (Pinus, Pinaceae) based on chloroplast rbcL, matk, rpl20-rps18 spacer, and trnv intron sequences. Am. J. Bot. 1999, 86, 1742–1753. [Google Scholar] [CrossRef] [PubMed]
- Skuza, L.; Szućko, I.; Filip, E.; Strzała, T. Genetic diversity and relationship between cultivated, weedy and wild rye species as revealed by chloroplast and mitochondrial DNA non-coding regions analysis. PLoS ONE 2019, 14, e0213023. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W.; Fay, M.F. Barcoding of Plants and Fungi. Science 2009, 325, 682–683. Available online: http://science.sciencemag.org/content/325/5941/682.abstract (accessed on 8 December 2020). [CrossRef] [PubMed]
- Ford, C.S.; Ayres, K.L.; Toomey, N.; Haider, N.; Van Alphen Stahl, J.; Kelly, L.J.; Wikström, N.; Hollingsworth, P.M.; Duff, R.J.; Hoot, S.B.; et al. Selection of candidate coding DNA barcoding regions for use on land plants. Bot. J. Linn. Soc. 2009, 159, 1–11. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011, 6, e19254. Available online: https://pubmed.ncbi.nlm.nih.gov/21637336 (accessed on 26 May 2011). [CrossRef]
- Suo, Z.; Zhang, C.; Zheng, Y.; He, L.; Jin, X.; Hou, B.; Li, J. Revealing genetic diversity of tree peonies at micro-evolution level with hyper-variable chloroplast markers and floral traits. Plant Cell Rep. 2012, 31, 2199–2213. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, D.; Jin, X.; Li, R.; Lu, Q.; Suo, Z. Comparative analysis of the complete chloroplast genome sequences in psammophytic Haloxylon species (Amaranthaceae). PeerJ 2016, 4, e2699. [Google Scholar] [CrossRef]
- Wang, M.; Xie, X.; Yan, B.; Yan, X.; Luo, J.; Liu, Y.; Qiu, Q. The completed chloroplast genome of Ostrya trichocarpa. Conserv. Genet. Resour. 2018, 10, 579–581. [Google Scholar] [CrossRef]
- Xu, C.; Dong, W.; Li, W.; Lu, Y.; Xie, X.; Jin, X.; Shi, J.; He, K.; Suo, Z. Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front. Plant Sci. 2017, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Scarcelli, N.; Barnaud, A.; Eiserhardt, W.; Treier, U.A.; Seveno, M.; d’Anfray, A.; Vigouroux, Y.; Pintaud, J.-C. A Set of 100 Chloroplast DNA Primer Pairs to Study Population Genetics and Phylogeny in Monocotyledons. PLoS ONE 2011, 6, e19954. Available online: https://pubmed.ncbi.nlm.nih.gov/21637837 (accessed on 26 May 2011). [CrossRef] [PubMed]
- Patwardhan, A.; Ray, S.; Roy, A. Molecular markers in phylogenetic studies-a review. J. Phylogenetics Evol. Biol. 2014. [Google Scholar] [CrossRef]
- Middleton, C.P.; Senerchia, N.; Stein, N.; Akhunov, E.D.; Keller, B.; Wicker, T. Sequencing of chloroplast genomes from wheat, barley, rye and their relatives provides a detailed insight into the evolution of the Triticeae tribe. PLoS ONE 2014, 9, e85761. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: Retrospect and prospect. J. Biogeogr. 2009, 36, 3–15. [Google Scholar] [CrossRef]
- Yamane, K.; Kawahara, T. Intra- and interspecific phylogenetic relationships among diploid Triticum-Aegilops species (Poaceae) based on base-pair substitutions, indels, and microsatellites in chloroplast noncoding sequences. Am. J. Bot. 2005, 92, 1887–1898. [Google Scholar] [CrossRef]
- Gielly, L.; Taberlet, P. The use of chloroplast DNA to resolve plant phylogenies: Noncoding versus rbcL sequences. Mol. Biol. Evol. 1994, 11, 769–777. [Google Scholar] [CrossRef]
- Golenberg, E.M.; Clegg, M.T.; Durbin, M.L.; Doebley, J.; Ma, D.P. Evolution of a Noncoding Region of the Chloroplast Genome. Mol. Phylogenetics Evol. 1993, 2, 52–64. Available online: http://www.sciencedirect.com/science/article/pii/S1055790383710067 (accessed on 8 December 2020). [CrossRef]
- Morton, B.R.; Clegg, M.T. A chloroplast DNA mutational hotspot and gene conversion in a noncoding region near rbcL in the grass family (Poaceae). Curr. Genet. 1993, 24, 357–365. [Google Scholar] [CrossRef]
- Schilbert, H.M.; Rempel, A.; Pucker, B. Comparison of read mapping and variant calling tools for the analysis of plant NGS data. Plants 2020, 9, 439. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Gielly, L.; Miquel, C.; Valentini, A.; Vermat, T.; Corthier, G.; Brochmann, C.; Willerslev, E. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 2007, 35, e14. [Google Scholar] [CrossRef] [PubMed]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial Diversity in the Deep Sea and the Underexplored “Rare Biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. Available online: http://www.pnas.org/content/103/32/12115.abstract (accessed on 8 December 2020). [CrossRef] [PubMed]
- Zinger, L.; Gury, J.; Alibeu, O.; Rioux, D.; Gielly, L.; Sage, L.; Pompanon, F.; Geremia, R.A. CE-SSCP and CE-FLA, Simple and High-Throughput Alternatives for Fungal Diversity Studies. J. Microbiol. Methods 2008, 72, 42–53. Available online: http://www.sciencedirect.com/science/article/pii/S0167701207003478 (accessed on 8 December 2020). [CrossRef] [PubMed]
- Steinke, D.; Vences, M.; Salzburger, W.; Meyer, A. TaxI: A software tool for DNA barcoding using distance methods. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1975–1980. [Google Scholar] [CrossRef] [PubMed]
- Vences, M.; Thomas, M.; van der Meijden, A.; Chiari, Y.; Vieites, D.R. Comparative performance of the 16S rRNA gene in DNA barcoding of amphibians. Front. Zool. 2005, 2, 5. [Google Scholar] [CrossRef]
- Young, N.D.; Healy, J. GapCoder automates the use of indel characters in phylogenetic analysis. BMC Bioinform. 2003, 4, 6. [Google Scholar] [CrossRef]
- Neuman, J.A.; Isakov, O.; Shomron, N. Analysis of insertion–deletion from deep-sequencing data: Software evaluation for optimal detection. Brief. Bioinform. 2013, 14, 46–55. [Google Scholar] [CrossRef]
- Hsieh, H.-M.; Chiang, H.-L.; Tsai, L.-C.; Lai, S.-Y.; Huang, N.-E.; Linacre, A.; Lee, J.C.-I. Cytochrome b Gene for Species Identification of the Conservation Animals. Forensic. Sci. Int. 2001, 122, 7–18. Available online: http://www.sciencedirect.com/science/article/pii/S0379073801004030 (accessed on 8 December 2020). [CrossRef]
- Parson, W.; Pegoraro, K.; Niederstätter, H.; Föger, M.; Steinlechner, M. Species identification by means of the cytochrome b gene. Int. J. Legal. Med. 2000, 114, 23–28. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. London Ser. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.C. Plant Mitochondrial Genome Evolution Can Be Explained by DNA Repair Mechanisms. Genome Biol. Evol. 2013, 5, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Detlef Weigel, D.; Lynch, M. The Rate and Molecular Spectrum of Spontaneous Mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. Available online: http://science.sciencemag.org/content/327/5961/92.abstract (accessed on 8 December 2020). [CrossRef]
- Li, W. Molecular Evolution; Sinauer Associates, Inc.: Sunderland, MA, USA, 1997. [Google Scholar]
- Sang, T.; Crawford, D.J.; Stuessy, T.F. Chloroplast DNA Phylogeny, Reticulate Evolution, and Biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 1997, 84, 1120–1136. [Google Scholar] [CrossRef]
- Hardig, T.M.; Soltis, P.S.; Soltis, D.E. Diversification of the North American shrub genus Ceanothus (Rhamnaceae): Conflicting phylogenies from nuclear ribosomal DNA and chloroplast DNA. Am. J. Bot. 2000, 87, 108–123. [Google Scholar] [CrossRef]
- Schönenberger, J.; Conti, E. Molecular phylogeny and floral evolution of Penaeaceae, Oliniaceae, Rhynchocalycaceae, and Alzateaceae (Myrtales). Am. J. Bot. 2003, 90, 293–309. [Google Scholar] [CrossRef]
- Yamane, K.; Yasui, Y.; Ohnishi, O. Intraspecific cpDNA variations of diploid and tetraploid perennial buckwheat, Fagopyrum cymosum (Polygonaceae). Am. J. Bot. 2003, 90, 339–346. [Google Scholar] [CrossRef]
- Pereira, F.; Carneiro, J.; Matthiesen, R.; van Asch, B.; Pinto, N.; Gusmão, L.; Amori, A. Identification of species by multiplex analysis of variable-length sequences. Nucleic Acids Res. 2010, 38, e203. [Google Scholar] [CrossRef]
- Bonen, L.; Vogel, J. The Ins and Outs of Group II Introns. Trends Genet. 2001, 17, 322–331. Available online: http://www.sciencedirect.com/science/article/pii/S0168952501023241 (accessed on 8 December 2020). [CrossRef]
- Lambowitz, A.M.; Zimmerly, S. Mobile Group II Introns. Annu. Rev. Genet. 2004, 38, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Qiu, Y.-L.; Kuhlman, P.; Palmer, J.D. Explosive invasion of plant mitochondria by a group I intron. Proc. Natl. Acad. Sci. USA 1998, 95, 14244–14249. Available online: http://www.pnas.org/content/95/24/14244.abstract (accessed on 8 December 2020). [CrossRef] [PubMed]
- Cui, P.; Liu, H.; Lin, Q.; Ding, F.; Zhuo, G.; Hu, S.; Liu, D.; Yang, W.; Zhan, K.; Zhang, A.; et al. A Complete Mitochondrial Genome of Wheat (Triticum aestivum cv. Chinese Yumai), and Fast Evolving Mitochondrial Genes in Higher Plants. J. Genet. 2009, 88, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Demesure, B.; Sodzi, N.; Petit, R.J. A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol. Ecol. 1995, 4, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-W.; Lai, K.-N.; Tai, P.-Y.; Li, W.-H. Rates of Nucleotide Substitution in Angiosperm Mitochondrial DNA Sequences and Dates of Divergence between Brassica and Other Angiosperm Lineages. J. Mol. Evol. 1999, 48, 597–604. [Google Scholar] [CrossRef]
- Kubo, T.; Yamamoto, M.P.; Mikami, T. The nad4L-orf25 gene cluster is conserved and expressed in sugar beet mitochondria. Theor. Appl. Genet. 2000, 100, 214–220. [Google Scholar] [CrossRef]
- Carneiro, J.; Pereira, F.; Amorim, A. SPInDel: A multifunctional workbench for species identification using insertion/deletion variants. Mol. Ecol. Resour. 2012, 12, 1190–1195. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Genomic Region | Number of Conserved Regions | Number of Hypervariable Regions (n) | Average Number of Pairwise Differences (pGn) | Average Number of Pairwise Differences Per Hypervariable Region | Number of Species-Specific Profiles (Nsp) | Frequency of Species-Specific Profiles (fsp) | Number of Species-Shared Profiles | Number of Minimum Hypervariable Regions for Discrimination of All Species |
|---|---|---|---|---|---|---|---|---|
| cpDNA | ||||||||
| atpB-rbcL intergenic spacer | 6 | 2 | 1.02 | 0.91 | 1 | 0.91 | 4 | 4 |
| trnT(UGU)-trnL(UAA)5′exon intergenic spacer | 5 | 2 | 0.97 | 0.62 | 2 | 0.83 | 4 | 4 |
| trnL(UAA) intron intergenic spacer | 4 | 2 | 0.51 | 0.41 | 4 | 0.87 | 2 | 1 |
| trnD[tRNA-Asp(GUC)]-trnT[tRNA-Thr(GGU)] intergenic spacer | 4 | 1 | 0.07 | 0.02 | 3 | 0.72 | 3 | 2 |
| mtDNA | ||||||||
| nad1exon B-nad1exon C intron intergenic spacer | 4 | 1 | 0.06 | 0.01 | 2 | 0.89 | 2 | 2 |
| nad4/1-2 intergenic spacer | 4 | 2 | 0.27 | 0.23 | 1 | 0.72 | 2 | 2 |
| nad4L-orf25 intergenic spacer | 5 | 2 | 1.23 | 0.99 | 7 | 0.45 | 4 | 3 |
| rps12-1/nad3-2 intergenic spacer | 4 | 1 | 0.07 | 0.05 | 2 | 0.67 | 2 | 1 |
| rps12-2/nad3-1 intergenic spacer | 3 | 1 | 0.01 | 0.01 | 1 | 0.81 | 1 | 1 |
| rrn5/rrn18-1 intergenic spacer | 5 | 2 | 0.73 | 0.23 | 3 | 0.86 | 2 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skuza, L.; Filip, E.; Szućko, I.; Bocianowski, J. SPInDel Analysis of the Non-Coding Regions of cpDNA as a More Useful Tool for the Identification of Rye (Poaceae: Secale) Species. Int. J. Mol. Sci. 2020, 21, 9421. https://doi.org/10.3390/ijms21249421
Skuza L, Filip E, Szućko I, Bocianowski J. SPInDel Analysis of the Non-Coding Regions of cpDNA as a More Useful Tool for the Identification of Rye (Poaceae: Secale) Species. International Journal of Molecular Sciences. 2020; 21(24):9421. https://doi.org/10.3390/ijms21249421
Chicago/Turabian StyleSkuza, Lidia, Ewa Filip, Izabela Szućko, and Jan Bocianowski. 2020. "SPInDel Analysis of the Non-Coding Regions of cpDNA as a More Useful Tool for the Identification of Rye (Poaceae: Secale) Species" International Journal of Molecular Sciences 21, no. 24: 9421. https://doi.org/10.3390/ijms21249421
APA StyleSkuza, L., Filip, E., Szućko, I., & Bocianowski, J. (2020). SPInDel Analysis of the Non-Coding Regions of cpDNA as a More Useful Tool for the Identification of Rye (Poaceae: Secale) Species. International Journal of Molecular Sciences, 21(24), 9421. https://doi.org/10.3390/ijms21249421