Administration of Human MSC-Derived Extracellular Vesicles for the Treatment of Primary Sclerosing Cholangitis: Preclinical Data in MDR2 Knockout Mice


 ,
,  , , and
, , and 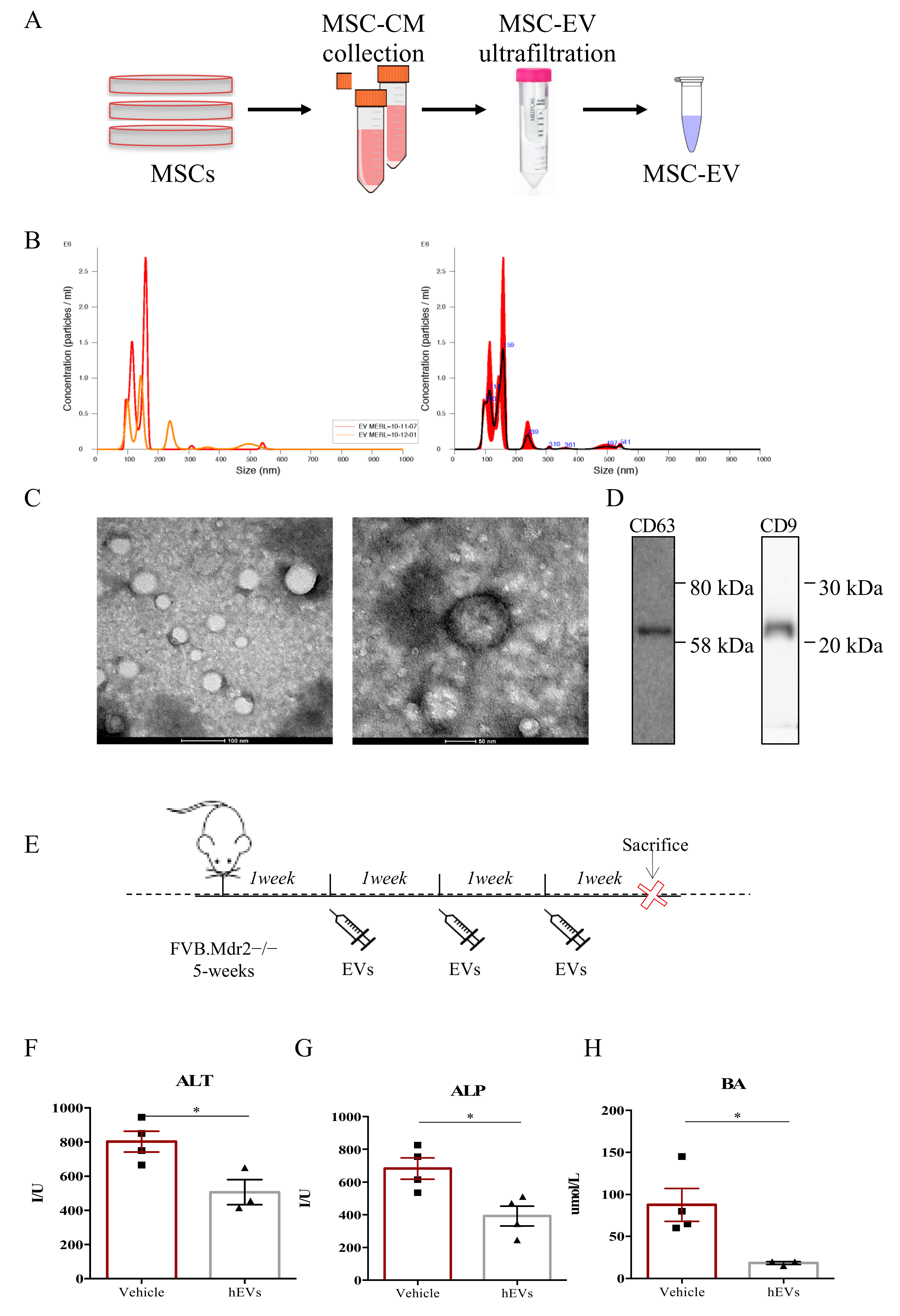{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. MSC-Derived EVs Ameliorate the Clinical Outcome in a Mouse Model of Human PSC
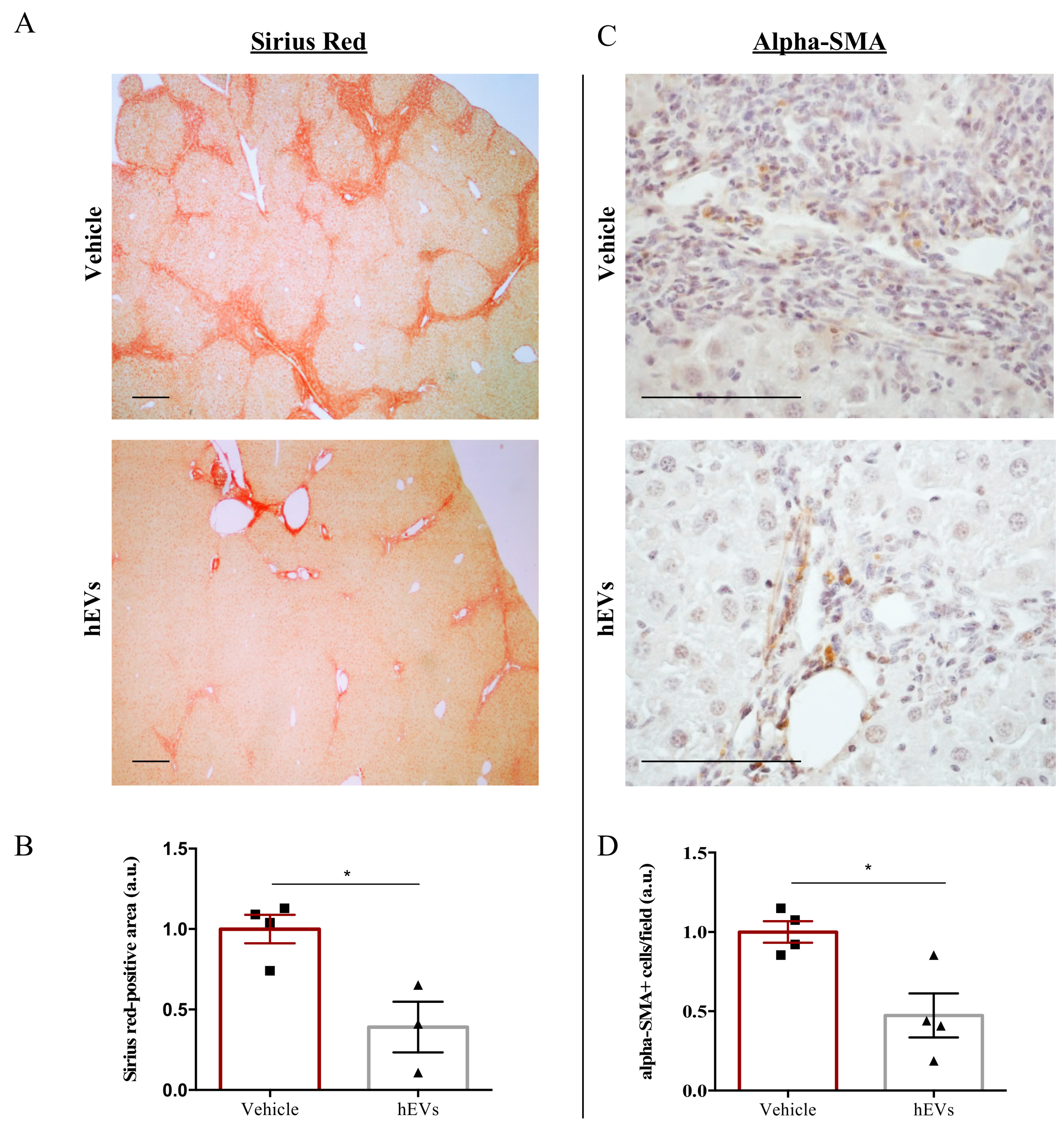
2.2. EVs Have Anti-Fibrotic Properties in the FVB.Mdr2−/− Mouse Model
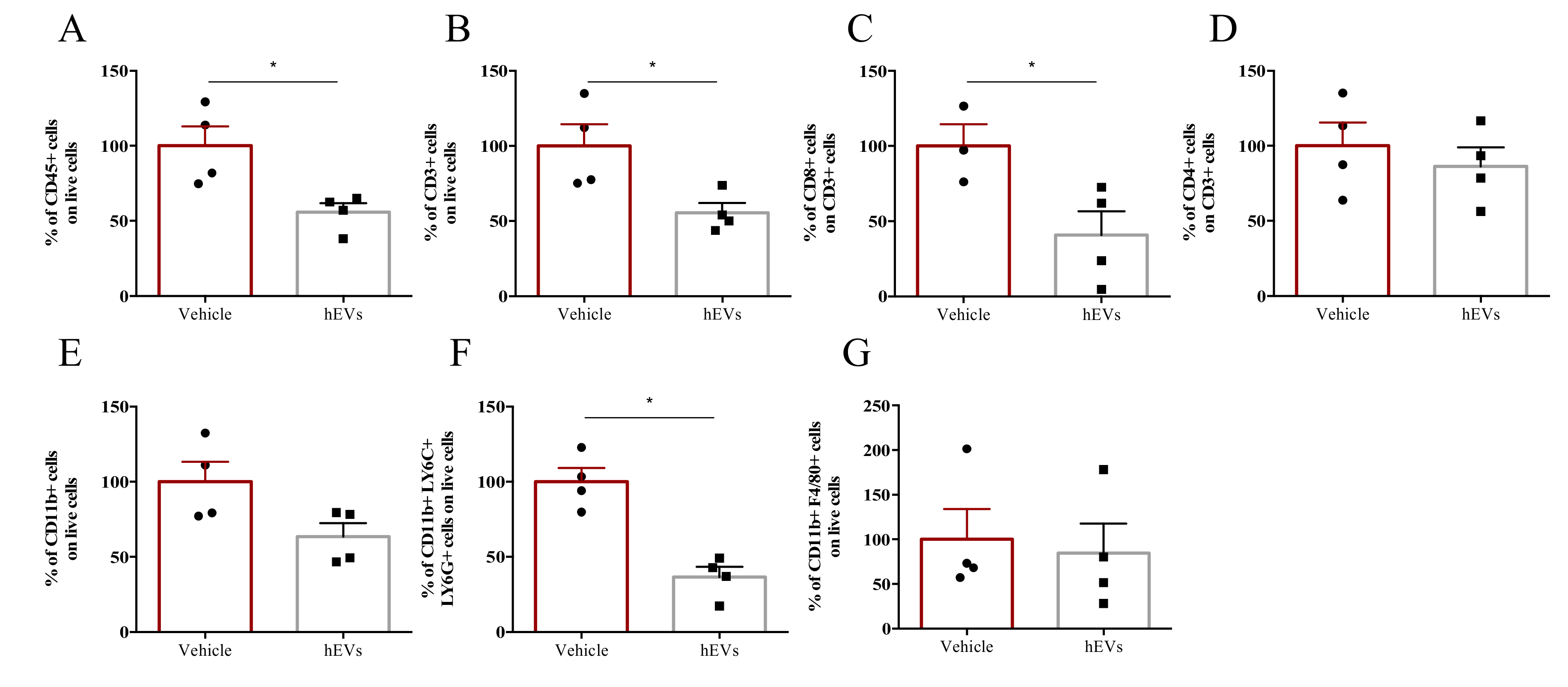
2.3. EVs Reduce Liver Immune Infiltration in the FVB.Mdr2−/− Mouse Model
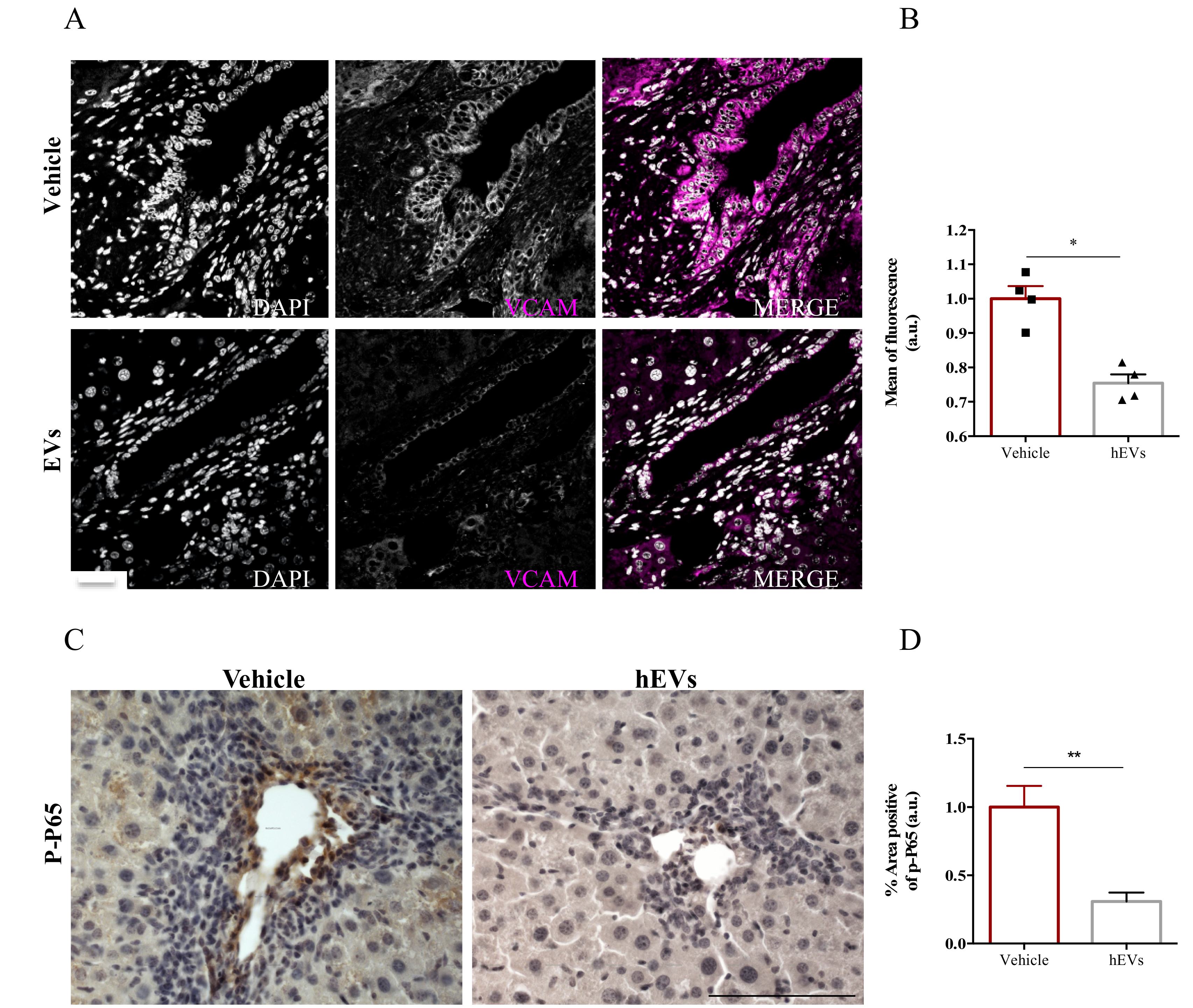
2.4. EVs Reduce Liver Expression of VCAM1 in the FVB.Mdr2−/− Mice
3. Discussion
4. Materials and Methods
4.1. Isolation of Human MSCs
4.2. Isolation and Characterization of Extracellular Vesicles (EVs)
4.3. Animal Experimentation
4.4. In Vivo Treatments
4.5. FACS Analysis of Liver Infiltrate
4.6. Immunohistochemical Analyses
4.7. Bone Marrow Derived Macrophages (BMDM) and RT-PCR Real Time
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PSC | Primary sclerosing cholangitis |
| EVs | Extracellular vesicles |
| MSCs | Mesenchymal stromal cells |
| ALP | Alkaline phosphatase |
| BA | Bile acid |
| ALT | Alanine aminotransferase |
| GVHD | Graft versus host disease |
| NASH | Non-alcoholic steatohepatitis |
| LN | Lymph node |
References
- Eaton, J.E.; Talwalkar, J.A.; Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology 2013, 145, 521–536. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. Primary Sclerosing Cholangitis. N. Eng. J. Med. 2016, 375, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.; Cullen, S. Etiopathogenesis of primary sclerosing cholangitis. World J. Gastroenterol. 2008, 14, 3350–3359. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [PubMed]
- Aron, J.H.; Bowlus, C.L. The immunobiology of primary sclerosing cholangitis. Semin. Immunopathol. 2009, 31, 383–397. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, S.; Shi, X.; Cao, H.; Li, L. A pooled analysis of mesenchymal stem cell-based therapy for liver disease. Stem Cell Res. Ther. 2018, 9, 72. [Google Scholar] [CrossRef]
- Borger, V.; Bremer, M.; Ferrer-Tur, R.; Gockeln, L.; Stambouli, O.; Becic, A.; Giebel, B. Mesenchymal Stem/Stromal Cell-Derived Extracellular Vesicles and Their Potential as Novel Immunomodulatory Therapeutic Agents. Int. J. Mol. Sci. 2017, 18, 1450. [Google Scholar] [CrossRef]
- Katsuda, T.; Kosaka, N.; Takeshita, F.; Ochiya, T. The therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Proteomics 2013, 13, 1637–1653. [Google Scholar] [CrossRef]
- Riazifar, M.; Pone, E.J.; Lotvall, J.; Zhao, W. Stem Cell Extracellular Vesicles: Extended Messages of Regeneration. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 125–154. [Google Scholar] [CrossRef]
- McDaniel, K.; Wu, N.; Zhou, T.; Huang, L.; Sato, K.; Venter, J.; Ceci, L.; Chen, D.; Ramos-Lorenzo, S.; Invernizzi, P.; et al. Amelioration of Ductular Reaction by Stem Cell Derived Extracellular Vesicles in MDR2 knockout mice via let-7 microRNA. Hepatology 2019, 69, 2562–2578. [Google Scholar] [PubMed]
- Mauad, T.H.; van Nieuwkerk, C.M.; Dingemans, K.P.; Smit, J.J.; Schinkel, A.H.; Notenboom, R.G.; van den Bergh Weerman, M.A.; Verkruisen, R.P.; Groen, A.K.; Oude Elferink, R.P.; et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am. J. Pathol. 1994, 145, 1237–1245. [Google Scholar] [PubMed]
- Popov, Y.; Patsenker, E.; Fickert, P.; Trauner, M.; Schuppan, D. Mdr2 (Abcb4)-/-mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J. Hepatol. 2005, 43, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Zollner, G.; Fuchsbichler, A.; Stumptner, C.; Weiglein, A.H.; Lammert, F.; Marschall, H.U.; Tsybrovskyy, O.; Zatloukal, K.; Denk, H.; et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology 2002, 123, 1238–1251. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed]
- Rupert, D.L.M.; Claudio, V.; Lasser, C.; Bally, M. Methods for the physical characterization and quantification of extracellular vesicles in biological samples. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3164–3179. [Google Scholar] [CrossRef]
- de Vries, E.M.G.; Farkkila, M.; Milkiewicz, P.; Hov, J.R.; Eksteen, B.; Thorburn, D.; Chazouilleres, O.; Pares, A.; Nygard, S.; Gilja, O.H.; et al. Enhanced liver fibrosis test predicts transplant-free survival in primary sclerosing cholangitis, a multi-centre study. Liver Int. 2017, 37, 1554–1561. [Google Scholar] [CrossRef]
- Fickert, P.; Pollheimer, M.J.; Beuers, U.; Lackner, C.; Hirschfield, G.; Housset, C.; Keitel, V.; Schramm, C.; Marschall, H.U.; Karlsen, T.H.; et al. Characterization of animal models for primary sclerosing cholangitis (PSC). J. Hepatol. 2014, 60, 1290–1303. [Google Scholar] [CrossRef]
- Jones, H.; Hargrove, L.; Kennedy, L.; Meng, F.; Graf-Eaton, A.; Owens, J.; Alpini, G.; Johnson, C.; Bernuzzi, F.; Demieville, J.; et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2(-/-) mice. Hepatology 2016, 64, 1202–1216. [Google Scholar] [CrossRef]
- Meng, F.; Kennedy, L.; Hargrove, L.; Demieville, J.; Jones, H.; Madeka, T.; Karstens, A.; Chappell, K.; Alpini, G.; Sybenga, A.; et al. Ursodeoxycholate inhibits mast cell activation and reverses biliary injury and fibrosis in Mdr2(-/-) mice and human primary sclerosing cholangitis. Lab. Investig. 2018, 98, 1465–1477. [Google Scholar] [CrossRef]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth muscle alpha actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS ONE 2013, 8, e77166. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Morini, S.; Ginanni Corradini, S.; Franchitto, A.; Merli, M.; Siciliano, M.; Gentili, F.; Onetti Muda, A.; Berloco, P.; Rossi, M.; et al. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig. Liver Dis. 2005, 37, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, N.; Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Nassiri, S.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight 2019, 4, e128060. [Google Scholar] [CrossRef] [PubMed]
- Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Mitsialis, S.A.; Kourembanas, S. Macrophage Immunomodulation: The Gatekeeper for Mesenchymal Stem Cell Derived-Exosomes in Pulmonary Arterial Hypertension? Int. J. Mol. Sci. 2018, 19, 2534. [Google Scholar] [CrossRef]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Lai, P.; Weng, J.; Guo, L.; Chen, X.; Du, X. Novel insights into MSC-EVs therapy for immune diseases. Biomark. Res. 2019, 7, 6. [Google Scholar] [CrossRef]
- Munir, H.; McGettrick, H.M. Mesenchymal Stem Cell Therapy for Autoimmune Disease: Risks and Rewards. Stem Cells Dev. 2015, 24, 2091–2100. [Google Scholar] [CrossRef]
- Alfaifi, M.; Eom, Y.W.; Newsome, P.N.; Baik, S.K. Mesenchymal stromal cell therapy for liver diseases. J. Hepatol. 2018, 68, 1272–1285. [Google Scholar] [CrossRef]
- Zanotti, L.; Sarukhan, A.; Dander, E.; Castor, M.; Cibella, J.; Soldani, C.; Trovato, A.E.; Ploia, C.; Luca, G.; Calvitti, M.; et al. Encapsulated mesenchymal stem cells for in vivo immunomodulation. Leukemia 2013, 27, 500–503. [Google Scholar] [CrossRef]
- Zanotti, L.; Angioni, R.; Cali, B.; Soldani, C.; Ploia, C.; Moalli, F.; Gargesha, M.; D’Amico, G.; Elliman, S.; Tedeschi, G.; et al. Mouse mesenchymal stem cells inhibit high endothelial cell activation and lymphocyte homing to lymph nodes by releasing TIMP-1. Leukemia 2016, 30, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Rad, F.; Ghorbani, M.; Mohammadi Roushandeh, A.; Habibi Roudkenar, M. Mesenchymal stem cell-based therapy for autoimmune diseases: Emerging roles of extracellular vesicles. Mol. Biol. Rep. 2019, 46, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Huang, B.Q.; Ward, C.J.; Gradilone, S.A.; Banales, J.M.; Masyuk, T.V.; Radtke, B.; Splinter, P.L.; LaRusso, N.F. Biliary exosomes influence cholangiocyte regulatory mechanisms and proliferation through interaction with primary cilia. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G990–G999. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Huang, H.; Zhang, Z.; Wang, F.S. The role of neutrophils in the development of liver diseases. Cell Mol. Immunol. 2014, 11, 224–231. [Google Scholar] [CrossRef]
- Alkhouri, N.; Morris-Stiff, G.; Campbell, C.; Lopez, R.; Tamimi, T.A.; Yerian, L.; Zein, N.N.; Feldstein, A.E. Neutrophil to lymphocyte ratio: A new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int. 2012, 32, 297–302. [Google Scholar] [CrossRef]
- Ibusuki, R.; Uto, H.; Arima, S.; Mawatari, S.; Setoguchi, Y.; Iwashita, Y.; Hashimoto, S.; Maeda, T.; Tanoue, S.; Kanmura, S.; et al. Transgenic expression of human neutrophil peptide-1 enhances hepatic fibrosis in mice fed a choline-deficient, L-amino acid-defined diet. Liver Int. 2013, 33, 1549–1556. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M.; Haigh, W.G.; Van Rooyen, D.M.; Brooling, J.; Heydet, D.; Nolan, C.J.; Teoh, N.C.; Farrell, G.C. Dietary modification dampens liver inflammation and fibrosis in obesity-related fatty liver disease. Obesity 2013, 21, 1189–1199. [Google Scholar] [CrossRef]
- Rensen, S.S.; Bieghs, V.; Xanthoulea, S.; Arfianti, E.; Bakker, J.A.; Shiri-Sverdlov, R.; Hofker, M.H.; Greve, J.W.; Buurman, W.A. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS ONE 2012, 7, e52411. [Google Scholar] [CrossRef]
- Penz-Osterreicher, M.; Osterreicher, C.H.; Trauner, M. Fibrosis in autoimmune and cholestatic liver disease. Best Pract. Res. Clin. Gastroenterol. 2011, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Cho, G.S.; Han, C.; Park, D.H.; Park, H.K.; Woo, D.H.; Kim, J.H. Current Understanding of Stem Cell and Secretome Therapies in Liver Diseases. Tissue Eng. Regen. Med. 2017, 14, 653–665. [Google Scholar] [PubMed]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Investig. 2013, 123, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, M.J.; Cai, Y.; Wang, W.; Jiang, J.X.; Varga, Z.V.; Feng, D.; Pacher, P.; Kunos, G.; Torok, N.J.; et al. Neutrophil-Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 399–413. [Google Scholar]
- Crispe, I.N.; Dao, T.; Klugewitz, K.; Mehal, W.Z.; Metz, D.P. The liver as a site of T-cell apoptosis: Graveyard, or killing field? Immunol. Rev. 2000, 174, 47–62. [Google Scholar] [CrossRef]
- Bartholdy, C.; Marker, O.; Thomsen, A.R. Migration of activated CD8(+) T lymphocytes to sites of viral infection does not require endothelial selectins. Blood 2000, 95, 1362–1369. [Google Scholar]
- Wong, J.; Johnston, B.; Lee, S.S.; Bullard, D.C.; Smith, C.W.; Beaudet, A.L.; Kubes, P. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J. Clin. Investig. 1997, 99, 2782–2790. [Google Scholar]
- John, B.; Crispe, I.N. Passive and active mechanisms trap activated CD8+ T cells in the liver. J. Immunol. 2004, 172, 5222–5229. [Google Scholar]
- Lautenschlager, I.; Hockerstedt, K.; Taskinen, E.; von Willebrand, E. Expression of adhesion molecules and their ligands in liver allografts during cytomegalovirus (CMV) infection and acute rejection. Transpl. Int. 1996, 9 (Suppl. S1), S213–S215. [Google Scholar] [CrossRef]
- Volpes, R.; Van Den Oord, J.J.; Desmet, V.J. Vascular adhesion molecules in acute and chronic liver inflammation. Hepatology 1992, 15, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, M.N.; Menter, D.G.; Konstantopoulos, K.; Nicolson, G.L.; McIntire, L.V. Integrin alpha4beta1/VCAM-1 pathway mediates primary adhesion of RAW117 lymphoma cells to hepatic sinusoidal endothelial cells under flow. Clin. Exp. Metastasis 1999, 17, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Ibbotson, G.C.; Doig, C.; Kaur, J.; Gill, V.; Ostrovsky, L.; Fairhead, T.; Kubes, P. Functional alpha4-integrin: A newly identified pathway of neutrophil recruitment in critically ill septic patients. Nat. Med. 2001, 7, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K. Endothelial nuclear factor kappaB in obesity and aging: Is endothelial nuclear factor kappaB a master regulator of inflammation and insulin resistance? Circulation 2012, 125, 1081–1083. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-kappaB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, M.P.; Xu, B.; Fan, F.; Lu, S.F.; Pan, M.; Wu, H.S. A study of regulatory effects of TLR4 and NF-kappaB on primary biliary cholangitis. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3951–3959. [Google Scholar]
- Yang, J.; Liu, X.X.; Fan, H.; Tang, Q.; Shou, Z.X.; Zuo, D.M.; Zou, Z.; Xu, M.; Chen, Q.Y.; Peng, Y.; et al. Extracellular Vesicles Derived from Bone Marrow Mesenchymal Stem Cells Protect against Experimental Colitis via Attenuating Colon Inflammation, Oxidative Stress and Apoptosis. PLoS ONE 2015, 10, e0140551. [Google Scholar] [CrossRef]
- McAuley, D.F.; Curley, G.F.; Hamid, U.I.; Laffey, J.G.; Abbott, J.; McKenna, D.H.; Fang, X.; Matthay, M.A.; Lee, J.W. Clinical grade allogeneic human mesenchymal stem cells restore alveolar fluid clearance in human lungs rejected for transplantation. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L809–L815. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Maffioli, E.; Nonnis, S.; Angioni, R.; Santagata, F.; Cali, B.; Zanotti, L.; Negri, A.; Viola, A.; Tedeschi, G. Proteomic analysis of the secretome of human bone marrow-derived mesenchymal stem cells primed by pro-inflammatory cytokines. J. Proteom. 2017, 166, 115–126. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 153750. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angioni, R.; Calì, B.; Vigneswara, V.; Crescenzi, M.; Merino, A.; Sánchez-Rodríguez, R.; Liboni, C.; Hoogduijn, M.J.; Newsome, P.N.; Muraca, M.; et al. Administration of Human MSC-Derived Extracellular Vesicles for the Treatment of Primary Sclerosing Cholangitis: Preclinical Data in MDR2 Knockout Mice. Int. J. Mol. Sci. 2020, 21, 8874. https://doi.org/10.3390/ijms21228874
Angioni R, Calì B, Vigneswara V, Crescenzi M, Merino A, Sánchez-Rodríguez R, Liboni C, Hoogduijn MJ, Newsome PN, Muraca M, et al. Administration of Human MSC-Derived Extracellular Vesicles for the Treatment of Primary Sclerosing Cholangitis: Preclinical Data in MDR2 Knockout Mice. International Journal of Molecular Sciences. 2020; 21(22):8874. https://doi.org/10.3390/ijms21228874
Chicago/Turabian StyleAngioni, Roberta, Bianca Calì, Vasanthy Vigneswara, Marika Crescenzi, Ana Merino, Ricardo Sánchez-Rodríguez, Cristina Liboni, Martin J. Hoogduijn, Philip Noel Newsome, Maurizio Muraca, and et al. 2020. "Administration of Human MSC-Derived Extracellular Vesicles for the Treatment of Primary Sclerosing Cholangitis: Preclinical Data in MDR2 Knockout Mice" International Journal of Molecular Sciences 21, no. 22: 8874. https://doi.org/10.3390/ijms21228874
APA StyleAngioni, R., Calì, B., Vigneswara, V., Crescenzi, M., Merino, A., Sánchez-Rodríguez, R., Liboni, C., Hoogduijn, M. J., Newsome, P. N., Muraca, M., Russo, F. P., & Viola, A. (2020). Administration of Human MSC-Derived Extracellular Vesicles for the Treatment of Primary Sclerosing Cholangitis: Preclinical Data in MDR2 Knockout Mice. International Journal of Molecular Sciences, 21(22), 8874. https://doi.org/10.3390/ijms21228874