Establishment and Characterization of Immortalized Miniature Pig Pancreatic Cell Lines Expressing Oncogenic K-RasG12D

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
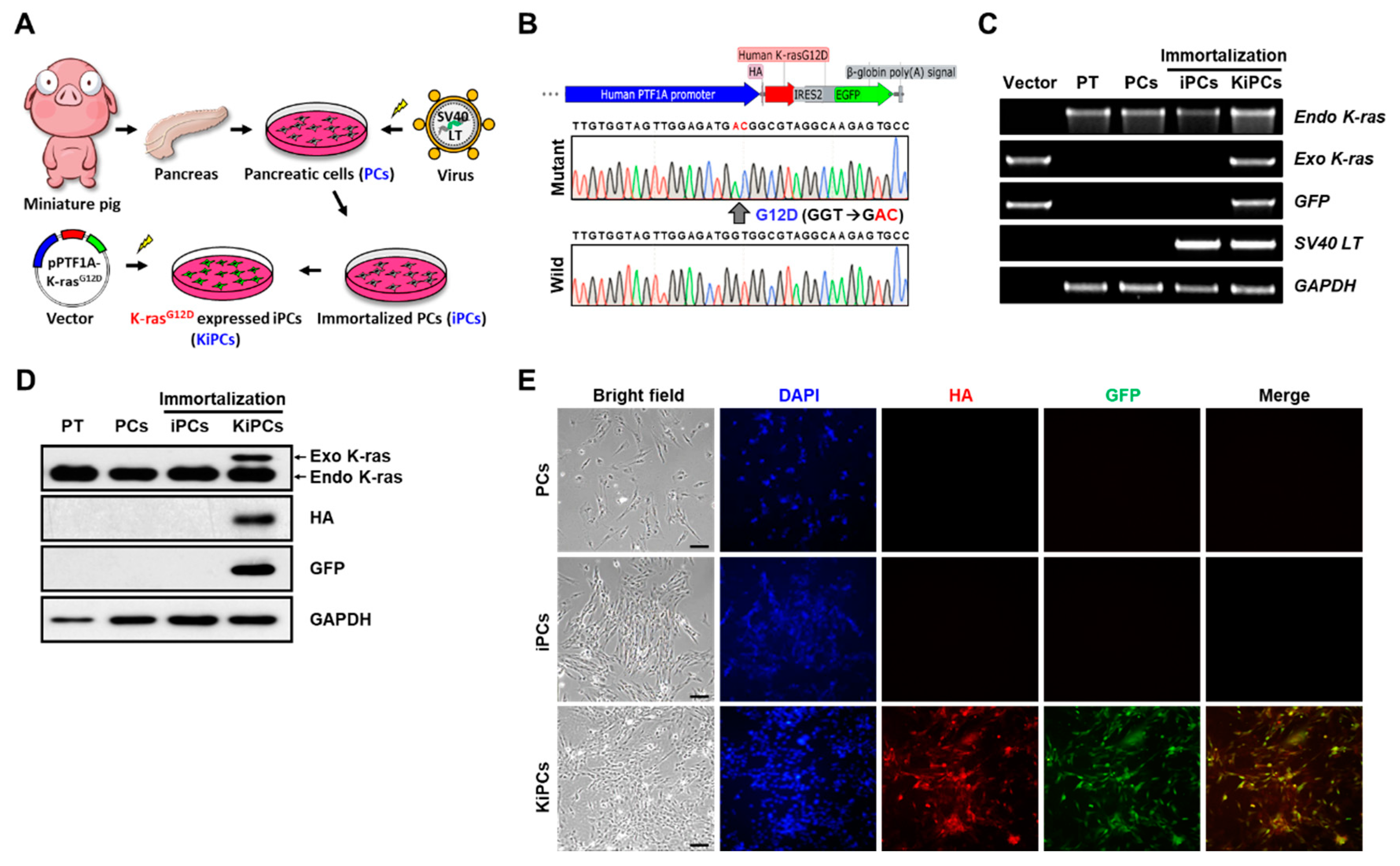
2.1. Generation of a Miniature Pig PC Line Expressing K-rasG12D under the Control of the PTF1A Promoter
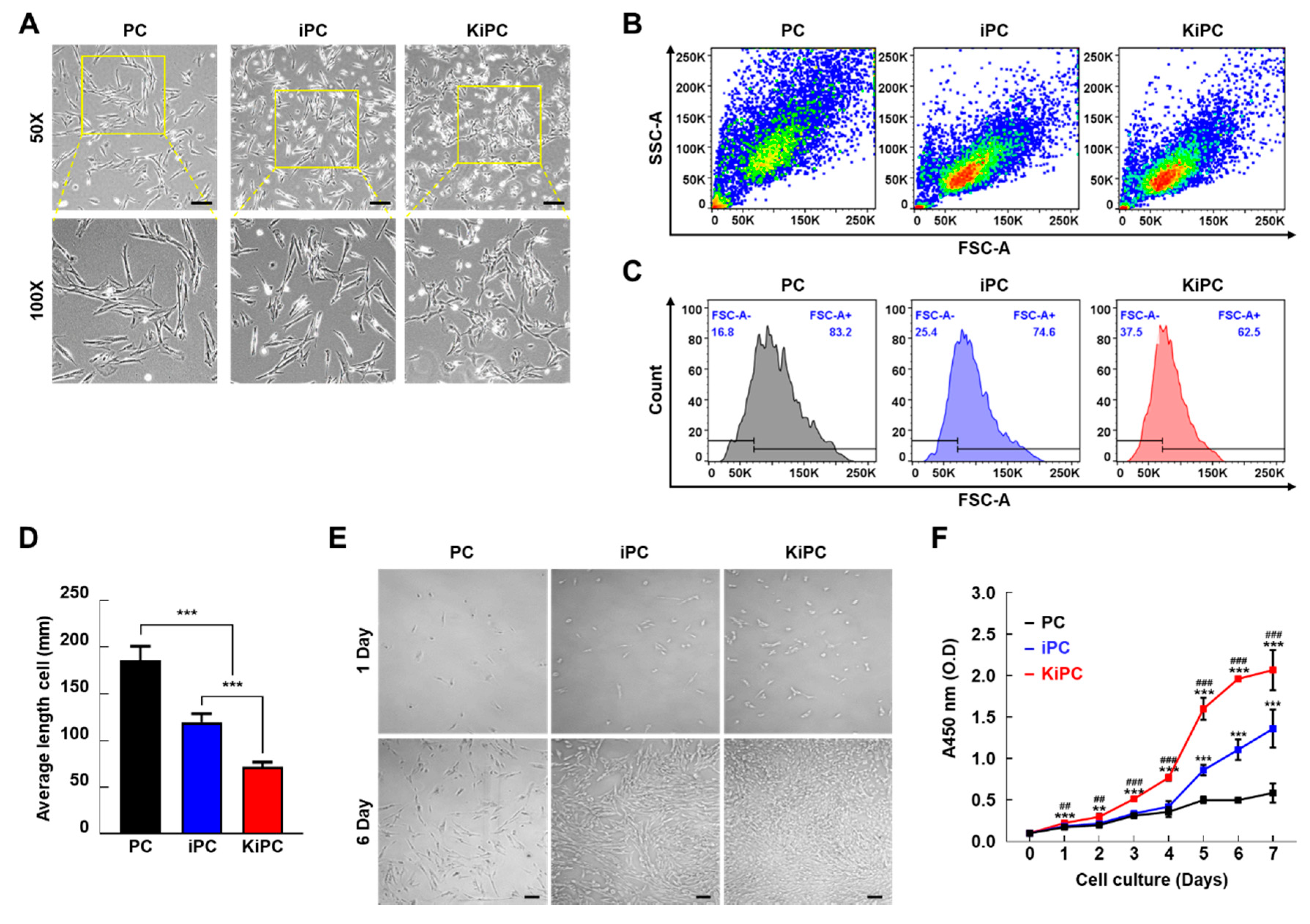
2.2. Cellular Morphology and Growth Characteristics of Miniature Pig KiPCs
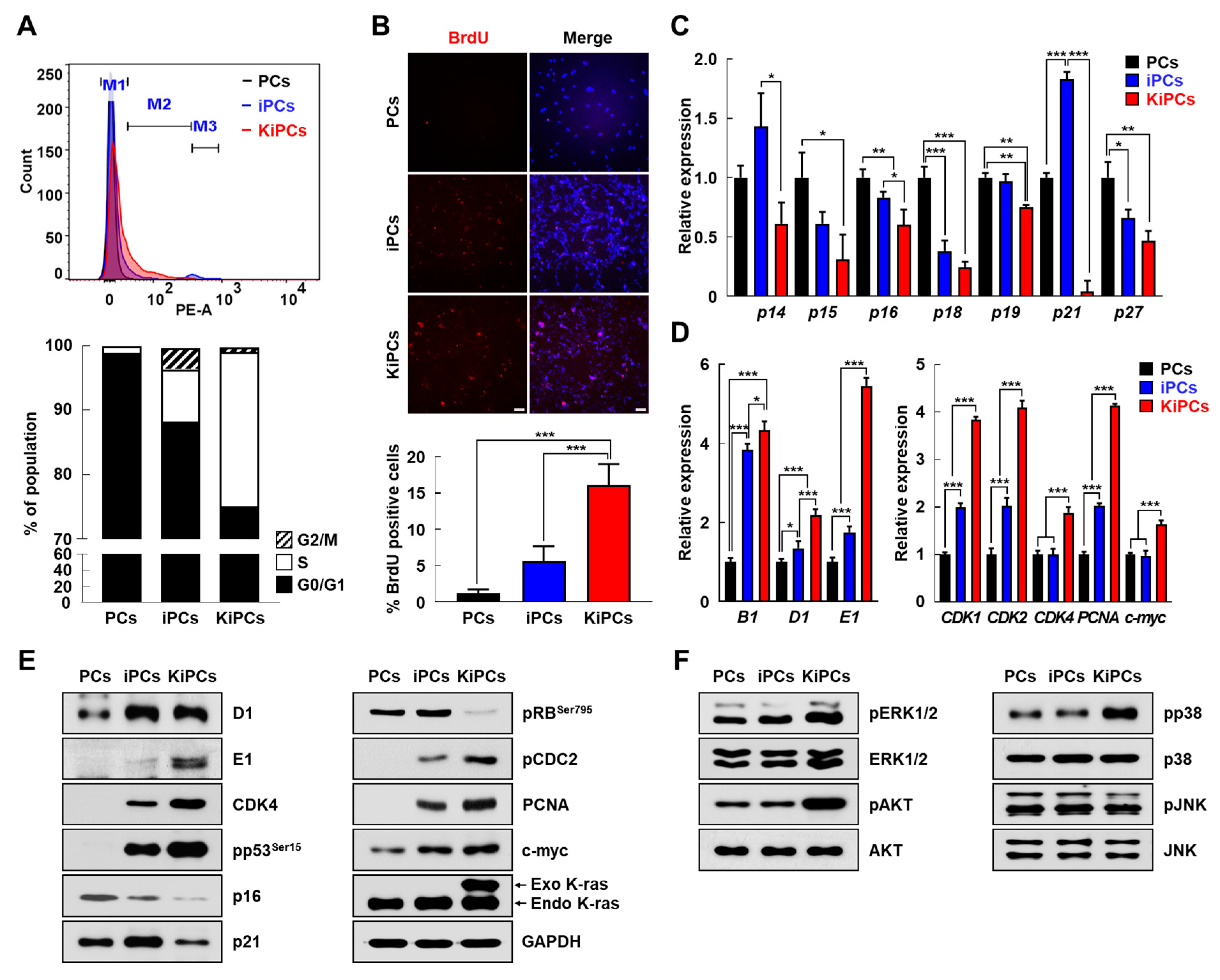
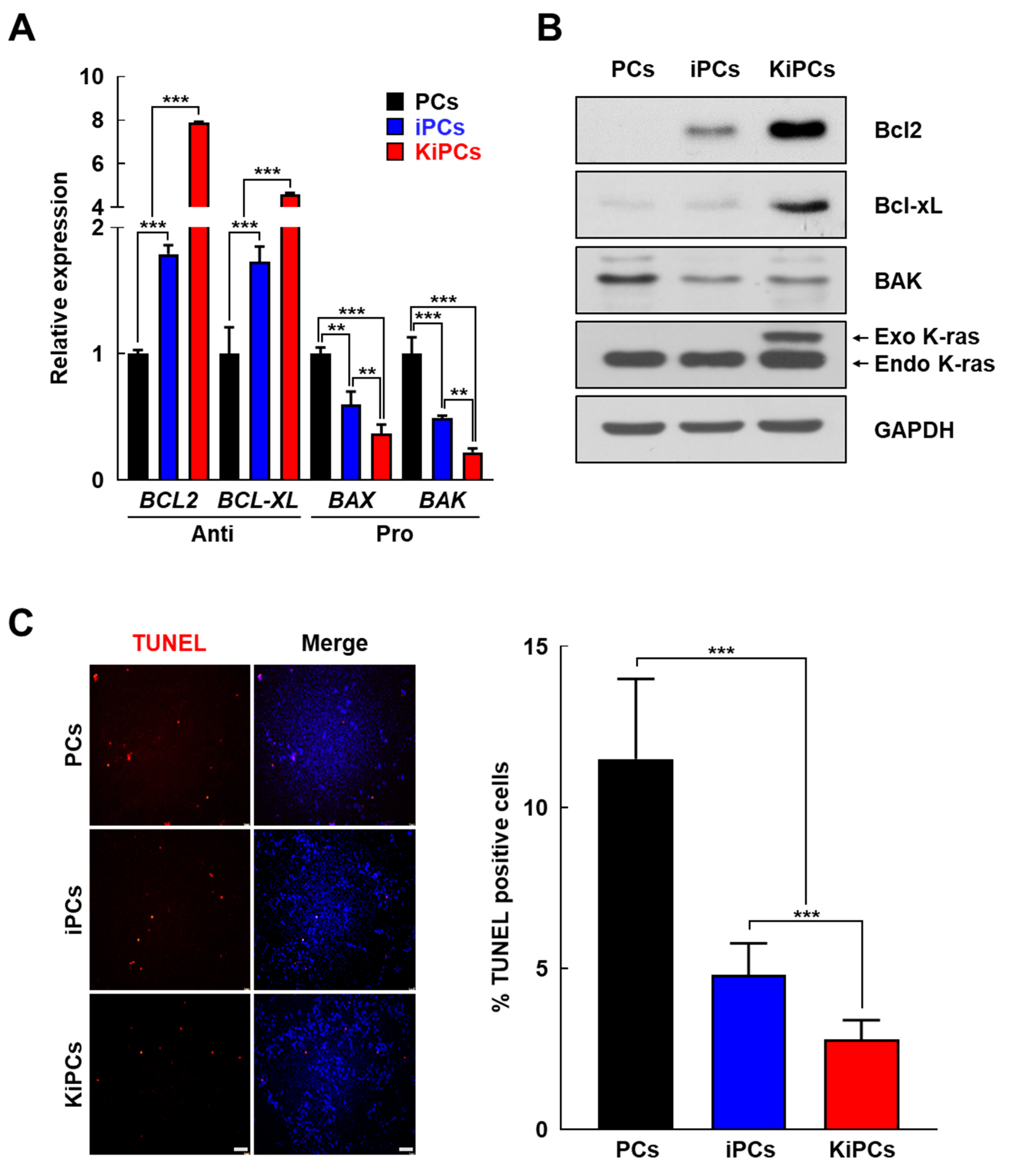
2.3. Altered Expression Patterns of Cell Cycle and Apoptosis-Associated Genes in KiPCs
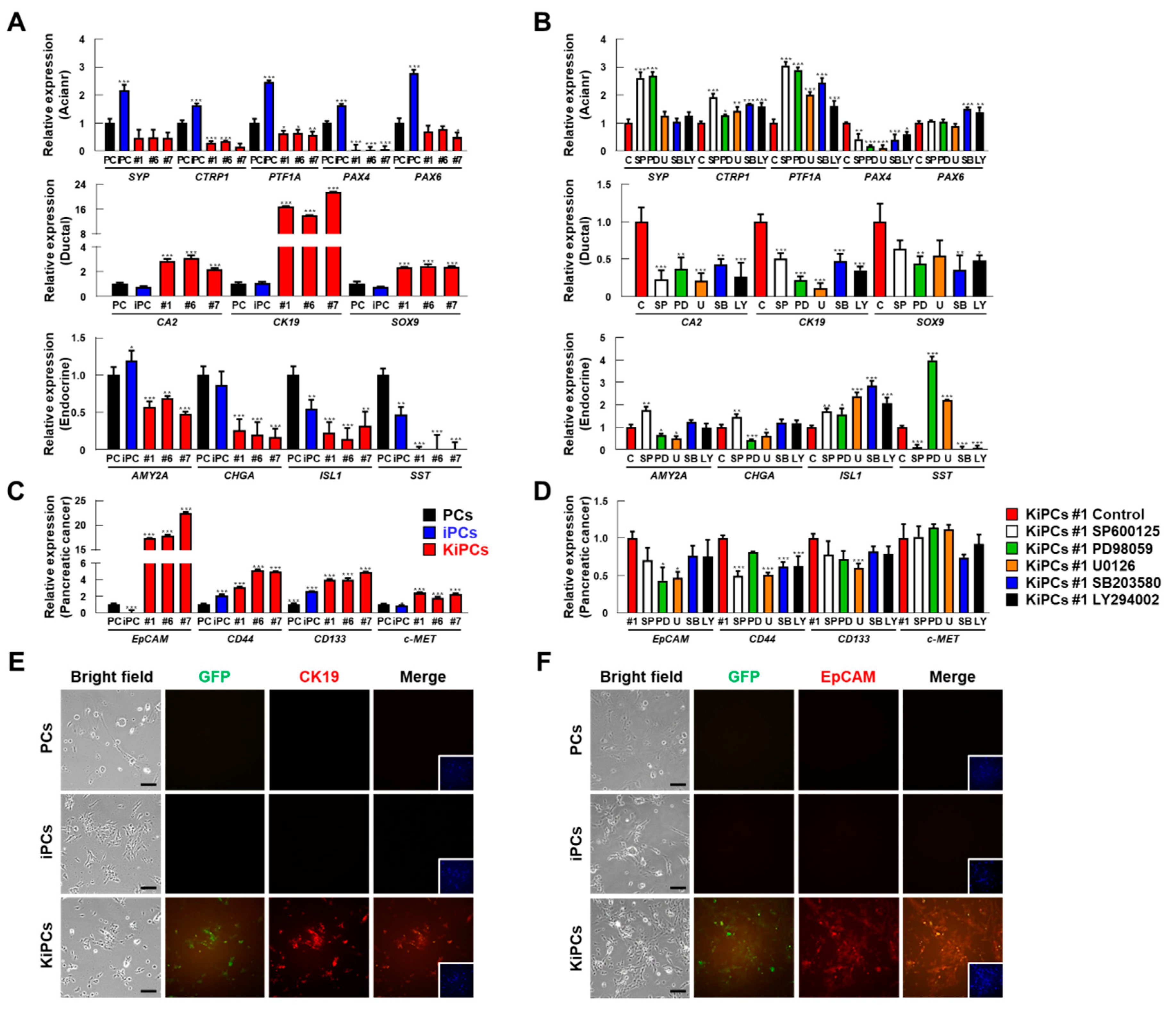
2.4. Robust Induction of Pancreatic Ductal and Cancer Cell Markers in KiPCs
3. Discussion
4. Materials and Methods
4.1. Preparation of Miniature Pig Primary Pancreatic Cells
4.2. Generation of Miniature Pig Pancreatic Cell Lines
4.3. Immunocytochemistry
4.4. BrdU Incorporation Assay
4.5. TUNEL Assay
4.6. Semi-Quantitative RT-PCR and qPCR
4.7. Western Blot Analysis
4.8. Cell-Proliferation Assay
4.9. Anchorage-Independent Growth Assay
4.10. Flow Cytometry Assay
4.11. Somatic Cell Nuclear Transfer (SCNT)
4.12. ELISA Assay
4.13. Positron Emission Tomography and Computed Tomography (PET/CT) Image
4.14. Ultrasound Image
4.15. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic ductal adenocaricnoma |
| ADM | Acinar-to-ductal metaplasia |
| PanIN | Pancreatic intraepithelial neoplasia |
| IPMN | Intraductal papillary mucinous neoplasm |
| PTF1A | Pancreatic-specific transcription factor 1 alpha |
| PDX1 | Pancreatic and duodenal homeobox 1 |
| SA-β-gal | Senescence-associated-β-galactosidase |
| EGFP | Enhanced green fluorescent protein |
| PT | Pancreatic tissue |
| PCs | Pancreatic cells |
| iPCs | Immortalized pancreatic cells |
| KiPCs | K-rasG12D expressing of immortalized pancreatic cells |
| FSC | Forward scatter |
| SSC | Side scatter |
| FACS | Fluorescence-activated cell sorting |
| CCK | Cell Counting Kit |
| CDK | Cyclin-dependent kinase |
| MAPK | Mitogen-activated protein kinase |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| JNK | c-Jun N-terminal kinases |
| PCNA | Proliferating cell nuclear antigen |
| EpCAM | Epithelial cell adhesion molecule |
| CK19-9 | Cytokeratin-19 |
| AMY | Amylase |
| CPA1 | Carboxypeptidase A1 |
| GCG | Glucagon |
| PAX | Paired box |
| CEL | Carboxyl ester lipase |
| Ctrp1 | C1q and TNF related 1 |
| CTSB | Cathepsin B |
| CTSC | Cathepsin C |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end-labeling |
| qRT-PCR | Quantitative Real-Time polymerase chain reaction |
| Exo | Exogenous |
| Endo | Endogenous |
| SCNT | Somatic cell nuclear transfer |
| FBS | Fetal bovine serum |
| BSA | Bovine serum albumin |
| IVC | in vitro culture |
| S.D | Standard deviation |
References
- Kaur, S.; Baine, M.J.; Jain, M.; Sasson, A.R.; Batra, S.K. Early diagnosis of pancreatic cancer: Challenges and new developments. Biomark. Med. 2012, 6, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.P.; Sacks, G.D.; Rochefort, M.M.; Donahue, T.R.; Reber, H.A.; Tomlinson, J.S.; Dawson, D.W.; Eibl, G.; Hines, O.J. Long-term survival in patients with pancreatic ductal adenocarcinoma. Surgery 2016, 159, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar] [PubMed]
- Cascinu, S.; Falconi, M.; Valentini, V.; Jelic, S.; Group, E.G.W. Pancreatic cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21 (Suppl. 5), v55–v58. [Google Scholar] [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef]
- Murphy, S.J.; Hart, S.N.; Lima, J.F.; Kipp, B.R.; Klebig, M.; Winters, J.L.; Szabo, C.; Zhang, L.; Eckloff, B.W.; Petersen, G.M.; et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 2013, 145, 1098–1109. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, J.; Nipper, M.; Wang, P. Ductal vs. acinar? Recent insights into identifying cell lineage of pancreatic ductal adenocarcinoma. Ann. Pancreat. Cancer 2019, 2. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P., 4th; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef]
- De La, O.J.; Emerson, L.L.; Goodman, J.L.; Froebe, S.C.; Illum, B.E.; Curtis, A.B.; Murtaugh, L.C. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 18907–18912. [Google Scholar] [CrossRef]
- Habbe, N.; Shi, G.; Meguid, R.A.; Fendrich, V.; Esni, F.; Chen, H.; Feldmann, G.; Stoffers, D.A.; Konieczny, S.F.; Leach, S.D.; et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl. Acad. Sci. USA 2008, 105, 18913–18918. [Google Scholar] [CrossRef] [PubMed]
- Lampreht Tratar, U.; Horvat, S.; Cemazar, M. Transgenic Mouse Models in Cancer Research. Front. Oncol. 2018, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Walrath, J.C.; Hawes, J.J.; Van Dyke, T.; Reilly, K.M. Genetically engineered mouse models in cancer research. Adv. Cancer Res. 2010, 106, 113–164. [Google Scholar] [PubMed]
- de Jong, M.; Maina, T. Of mice and humans: Are they the same?—Implications in cancer translational research. J. Nucl. Med. 2010, 51, 501–504. [Google Scholar] [CrossRef]
- Bailey, K.L.; Carlson, M.A. Porcine Models of Pancreatic Cancer. Front. Oncol. 2019, 9, 144. [Google Scholar] [CrossRef]
- Andren-Sandberg, A. Pancreatic cancer: Animal model and molecular biology. N. Am. J. Med. Sci. 2011, 3, 441–450. [Google Scholar] [CrossRef]
- Kong, K.; Guo, M.; Liu, Y.; Zheng, J. Progress in Animal Models of Pancreatic Ductal Adenocarcinoma. J. Cancer 2020, 11, 1555–1567. [Google Scholar] [CrossRef]
- Perleberg, C.; Kind, A.; Schnieke, A. Genetically engineered pigs as models for human disease. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Meurens, F.; Summerfield, A.; Nauwynck, H.; Saif, L.; Gerdts, V. The pig: A model for human infectious diseases. Trends Microbiol. 2012, 20, 50–57. [Google Scholar] [CrossRef]
- Kalla, D.; Kind, A.; Schnieke, A. Genetically Engineered Pigs to Study Cancer. Int. J. Mol. Sci. 2020, 21, 488. [Google Scholar] [CrossRef]
- Watson, A.L.; Carlson, D.F.; Largaespada, D.A.; Hackett, P.B.; Fahrenkrug, S.C. Engineered Swine Models of Cancer. Front. Genet. 2016, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Schook, L.B.; Collares, T.V.; Hu, W.; Liang, Y.; Rodrigues, F.M.; Rund, L.A.; Schachtschneider, K.M.; Seixas, F.K.; Singh, K.; Wells, K.D.; et al. A Genetic Porcine Model of Cancer. PLoS ONE 2015, 10, e0128864. [Google Scholar] [CrossRef] [PubMed]
- Sanclemente, M.; Francoz, S.; Esteban-Burgos, L.; Bousquet-Mur, E.; Djurec, M.; Lopez-Casas, P.P.; Hidalgo, M.; Guerra, C.; Drosten, M.; Musteanu, M.; et al. c-RAF Ablation Induces Regression of Advanced Kras/Trp53 Mutant Lung Adenocarcinomas by a Mechanism Independent of MAPK Signaling. Cancer Cell 2018, 33, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Greer, R.L.; Staley, B.K.; Liou, A.; Hebrok, M. Numb regulates acinar cell dedifferentiation and survival during pancreatic damage and acinar-to-ductal metaplasia. Gastroenterology 2013, 145, 1088–1097. [Google Scholar] [CrossRef]
- Leach, S.D. Mouse models of pancreatic cancer: The fur is finally flying! Cancer. Cell. 2004, 5, 7–11. [Google Scholar] [CrossRef]
- Mazur, P.K.; Siveke, J.T. Genetically engineered mouse models of pancreatic cancer: Unravelling tumour biology and progressing translational oncology. Gut 2012, 61, 1488–1500. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Dolensek, J.; Rupnik, M.S.; Stozer, A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015, 7, e1024405. [Google Scholar] [CrossRef]
- Elferink, R.O.; Beuers, U. Are pigs more human than mice? J. Hepatol. 2009, 50, 838–841. [Google Scholar] [CrossRef]
- Principe, D.R.; Overgaard, N.H.; Park, A.J.; Diaz, A.M.; Torres, C.; McKinney, R.; Dorman, M.J.; Castellanos, K.; Schwind, R.; Dawson, D.W.; et al. KRAS(G12D) and TP53(R167H) Cooperate to Induce Pancreatic Ductal Adenocarcinoma in Sus scrofa Pigs. Sci. Rep. 2018, 8, 12548. [Google Scholar] [CrossRef]
- Campbell, P.M.; Groehler, A.L.; Lee, K.M.; Ouellette, M.M.; Khazak, V.; Der, C.J. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res. 2007, 67, 2098–2106. [Google Scholar] [CrossRef] [PubMed]
- Baek, E.; Noh, S.M.; Lee, G.M. Anti-Apoptosis Engineering for Improved Protein Production from CHO Cells. Methods Mol. Biol. 2017, 1603, 71–85. [Google Scholar] [PubMed]
- Krah, N.M.; De La, O.J.; Swift, G.H.; Hoang, C.Q.; Willet, S.G.; Chen Pan, F.; Cash, G.M.; Bronner, M.P.; Wright, C.V.; MacDonald, R.J.; et al. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015, 4, e07125. [Google Scholar] [CrossRef] [PubMed]
- Smit, V.T.; Boot, A.J.; Smits, A.M.; Fleuren, G.J.; Cornelisse, C.J.; Bos, J.L. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988, 16, 7773–7782. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Furukawa, T. Impacts of activation of the mitogen-activated protein kinase pathway in pancreatic cancer. Front. Oncol. 2015, 5, 23. [Google Scholar] [CrossRef]
- Yang, J.; Yi, N.; Zhang, J.; He, W.; He, D.; Wu, W.; Xu, S.; Li, F.; Fan, G.; Zhu, X.; et al. Generation and characterization of a hypothyroidism rat model with truncated thyroid stimulating hormone receptor. Sci. Rep. 2018, 8, 4004. [Google Scholar] [CrossRef]
- Gibbs, J.B.; Sigal, I.S.; Poe, M.; Scolnick, E.M. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc. Natl. Acad. Sci. USA 1984, 81, 5704–5708. [Google Scholar] [CrossRef]
- Wicki, A.; Herrmann, R.; Christofori, G. Kras in metastatic colorectal cancer. Swiss. Med. Wkly. 2010, 140, w13112. [Google Scholar] [CrossRef]
- Collins, M.A.; Yan, W.; Sebolt-Leopold, J.S.; Pasca di Magliano, M. MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology 2014, 146, 822–834. [Google Scholar] [CrossRef]
- Beel, S.; Kolloch, L.; Apken, L.H.; Jurgens, L.; Bolle, A.; Sudhof, N.; Ghosh, S.; Wardelmann, E.; Meisterernst, M.; Steinestel, K.; et al. kappaB-Ras and Ral GTPases regulate acinar to ductal metaplasia during pancreatic adenocarcinoma development and pancreatitis. Nat. Commun. 2020, 11, 3409. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Edlinger, M.; Saalfrank, A.; Flisikowski, K.; Tschukes, A.; Kurome, M.; Zakhartchenko, V.; Kessler, B.; Saur, D.; Kind, A.; et al. Viable pigs with a conditionally-activated oncogenic KRAS mutation. Transgenic Res. 2015, 24, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Berthelsen, M.F.; Callesen, M.M.; Ostergaard, T.S.; Liu, Y.; Li, R.; Callesen, H.; Dagnaes-Hansen, F.; Hamilton-Dutoit, S.; Jakobsen, J.E.; Thomsen, M.K. Pancreas specific expression of oncogenes in a porcine model. Transgenic Res. 2017, 26, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, F.W.; Melton, D.A. How to make a functional beta-cell. Development 2013, 140, 2472–2483. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.B.; Park, Y.H.; Choi, S.A.; Yang, H.J.; Jeong, P.S.; Cha, J.J.; Lee, S.; Lee, S.H.; Lee, J.H.; Sim, B.W.; et al. Real-time PCR quantification of spliced X-box binding protein 1 (XBP1) using a universal primer method. PLoS ONE 2019, 14, e0219978. [Google Scholar] [CrossRef]
- Reichert, M.; Rhim, A.D.; Rustgi, A.K. Culturing primary mouse pancreatic ductal cells. Cold Spring Harb. Protoc. 2015, 2015, 558–561. [Google Scholar] [CrossRef]
- Jeong, P.S.; Yoon, S.B.; Choi, S.A.; Song, B.S.; Kim, J.S.; Sim, B.W.; Park, Y.H.; Yang, H.J.; Mun, S.E.; Kim, Y.H.; et al. Iloprost supports early development of in vitro-produced porcine embryos through activation of the phosphatidylinositol 3-kinase/AKT signalling pathway. Reprod. Fertil. Dev. 2017, 29, 1306–1318. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.-J.; Song, B.-S.; Sim, B.-W.; Jung, Y.; Chae, U.; Lee, D.G.; Cha, J.-J.; Baek, S.-J.; Lim, K.S.; Choi, W.S.; et al. Establishment and Characterization of Immortalized Miniature Pig Pancreatic Cell Lines Expressing Oncogenic K-RasG12D. Int. J. Mol. Sci. 2020, 21, 8820. https://doi.org/10.3390/ijms21228820
Yang H-J, Song B-S, Sim B-W, Jung Y, Chae U, Lee DG, Cha J-J, Baek S-J, Lim KS, Choi WS, et al. Establishment and Characterization of Immortalized Miniature Pig Pancreatic Cell Lines Expressing Oncogenic K-RasG12D. International Journal of Molecular Sciences. 2020; 21(22):8820. https://doi.org/10.3390/ijms21228820
Chicago/Turabian StyleYang, Hae-Jun, Bong-Seok Song, Bo-Woong Sim, Yena Jung, Unbin Chae, Dong Gil Lee, Jae-Jin Cha, Seo-Jong Baek, Kyung Seob Lim, Won Seok Choi, and et al. 2020. "Establishment and Characterization of Immortalized Miniature Pig Pancreatic Cell Lines Expressing Oncogenic K-RasG12D" International Journal of Molecular Sciences 21, no. 22: 8820. https://doi.org/10.3390/ijms21228820
APA StyleYang, H.-J., Song, B.-S., Sim, B.-W., Jung, Y., Chae, U., Lee, D. G., Cha, J.-J., Baek, S.-J., Lim, K. S., Choi, W. S., Lee, H.-Y., Son, H.-C., Park, S.-H., Jeong, K.-J., Kang, P., Baek, S. H., Koo, B.-S., Kim, H.-N., Jin, Y. B., ... Kim, S.-U. (2020). Establishment and Characterization of Immortalized Miniature Pig Pancreatic Cell Lines Expressing Oncogenic K-RasG12D. International Journal of Molecular Sciences, 21(22), 8820. https://doi.org/10.3390/ijms21228820