Rbm38 Reduces the Transcription Elongation Defect of the SMEK2 Gene Caused by Splicing Deficiency

 ,
,  ,
,  and
and 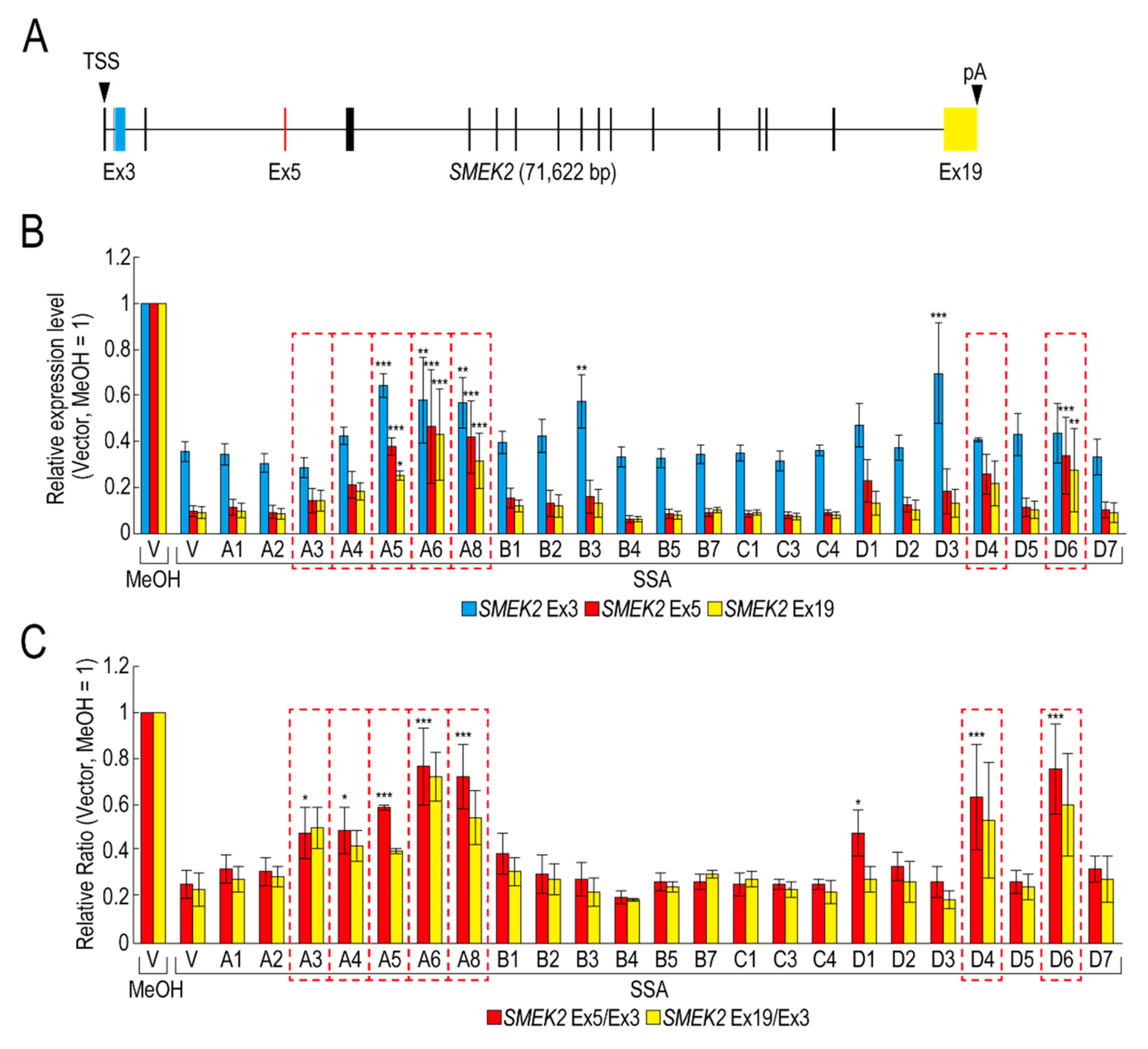{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Some RBPs Reduce the Transcription Elongation Defect Caused by Splicing Deficiency
2.2. Rbm38 Binds to SMEK2 Intron 4 and Reduces the Transcription Elongation Defect
2.3. The N- and C-Terminal Regions of Rbm38 Are Important for Reducing the Transcription Elongation Defect
2.4. The RNA Binding Domain of Rbm38 Is Important for Reducing the Transcription Elongation Defect
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. RNA Preparation and Quantitative RT-PCR
4.3. Antibodies
4.4. Plasmid Construction
4.5. Western Blotting
4.6. Biotinylated RNA Pull-Down Assays
4.7. Phosphatase Treatment
4.8. Immunofluorescence
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Rbm38 | RNA binding motif protein 38 |
| Rbm24 | RNA binding motif protein 24 |
| SMEK2 | Suppressor of MEK1 homolog 2 |
| CDK6 | Cyclin dependent kinase 6 |
| EGFR | Epidermal growth factor receptor |
| VEGFA | Vascular endothelial growth factor A |
| RNP | Ribonucleoprotein |
References
- Grzybowska, E.A. Human intronless genes: Functional groups, associated diseases, evolution, and mRNA processing in absence of splicing. Biochem. Biophys. Res. Commun. 2012, 424, 1–6. [Google Scholar] [CrossRef]
- Papasaikas, P.; Valcárcel, J. The Spliceosome: The Ultimate RNA Chaperone and Sculptor. Trends Biochem. Sci. 2016, 41, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kaida, D. Upregulation of p27 cyclin-dependent kinase inhibitor and a C-terminus truncated form of p27 contributes to G1 phase arrest. Sci. Rep. 2016, 6, 27829. [Google Scholar] [CrossRef]
- Bousquet-Antonelli, C.; Presutti, C.; Tollervey, D. Identification of a Regulated Pathway for Nuclear Pre-mRNA Turnover. Cell 2000, 102, 765–775. [Google Scholar] [CrossRef]
- Dziembowski, A.; Ventura, A.-P.; Rutz, B.; Caspary, F.; Faux, C.; Halgand, F.; Laprévote, O.; Séraphin, B. Proteomic analysis identifies a new complex required for nuclear pre-mRNA retention and splicing. EMBO J. 2004, 23, 4847–4856. [Google Scholar] [CrossRef]
- Galy, V.; Gadal, O.; Fromont-Racine, M.; Romano, A.; Jacquier, A.; Nehrbass, U. Nuclear Retention of Unspliced mRNAs in Yeast Is Mediated by Perinuclear Mlp1. Cell 2004, 116, 63–73. [Google Scholar] [CrossRef]
- Rutz, B.; Séraphin, B. A dual role for BBP/ScSF1 in nuclear pre-mRNA retention and splicing. EMBO J. 2000, 19, 1873–1886. [Google Scholar] [CrossRef]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Koga, M.; Hayashi, M.; Kaida, D. Splicing inhibition decreases phosphorylation level of Ser2 in Pol II CTD. Nucleic Acids Res. 2015, 43, 8258–8267. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Satoh, T.; Takasaki, I.; Kawamura, Y.; Yoshida, M.; Kaida, D. U2 snRNP Is Required for Expression of the 3′ End of Genes. PLoS ONE 2014, 9, e98015. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.-W.; Kaida, D.; Nishimura, S.; Matsuyama, A.; Yashiroda, Y.; Taoka, H.; Ishigami, K.; Watanabe, H.; Nakajima, H.; Tani, T.; et al. Inhibition of splicing and nuclear retention of pre-mRNA by spliceostatin A in fission yeast. Biochem. Biophys. Res. Commun. 2007, 364, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Dreyfuss, G.; Matunis, M.J.; Pinol-Roma, S.; Burd, C.G. hnRNP Proteins and the Biogenesis of mRNA. Ann. Rev. Biochem. 1993, 62, 289–321. [Google Scholar] [CrossRef] [PubMed]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.G.; Dreyfuss, G. Conserved structures and diversity of functions of RNA-binding proteins. Science 1994, 265, 615–621. [Google Scholar] [CrossRef]
- Cléry, A.; Blatter, M.; Allain, F.H. RNA recognition motifs: Boring? Not quite. Curr. Opin. Struct. Biol. 2008, 18, 290–298. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef]
- Cho, S.J.; Zhang, J.; Chen, X. RNPC1 modulates the RNA-binding activity of, and cooperates with, HuR to regulate p21 mRNA stability. Nucleic Acids Res. 2010, 38, 2256–2267. [Google Scholar] [CrossRef]
- Martin-Tumasz, S.; Richie, A.C.; Clos, L.J., 2nd; Brow, D.A.; Butcher, S.E. A novel occluded RNA recognition motif in Prp24 unwinds the U6 RNA internal stem loop. Nucleic Acids Res. 2011, 39, 7837–7847. [Google Scholar] [CrossRef]
- van den Hoogenhof, M.M.G.; Van Der Made, I.; Beqqali, A.; De Groot, N.E.; Damanafshan, A.; Van Oort, R.J.; Pinto, Y.M.; Creemers, E.E. The RNA-binding protein Rbm38 is dispensable during pressure overload-induced cardiac remodeling in mice. PLoS ONE 2017, 12, e0184093. [Google Scholar] [CrossRef]
- Heinicke, L.A.; Nabet, B.; Shen, S.; Jiang, P.; Van Zalen, S.; Cieply, B.; Russell, J.E.; Xing, Y.; Carstens, R.P. The RNA Binding Protein RBM38 (RNPC1) Regulates Splicing during Late Erythroid Differentiation. PLoS ONE 2013, 8, e78031. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Yan, W.; Chen, X. RNPC1, an RNA-binding protein and a target of the p53 family, is required for maintaining the stability of the basal and stress-induced p21 transcript. Genes Dev. 2006, 20, 2961–2972. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-J.; Jung, Y.-S.; Zhang, J.; Chen, X. The RNA-binding Protein RNPC1 Stabilizes the mRNA Encoding the RNA-binding Protein HuR and Cooperates with HuR to Suppress Cell Proliferation. J. Biol. Chem. 2012, 287, 14535–14544. [Google Scholar] [CrossRef] [PubMed]
- Leveille, N.; Elkon, R.; Davalos, V.; Manoharan, V.; Hollingworth, D.; Oude Vrielink, J.; le Sage, C.; Melo, C.A.; Horlings, H.M.; Wesseling, J.; et al. Selective inhibition of microRNA accessibility by RBM38 is required for p53 activity. Nat. Commun. 2011, 2, 513. [Google Scholar] [CrossRef]
- Xu, E.; Zhang, J.; Chen, X. MDM2 expression is repressed by the RNA-binding protein RNPC1 via mRNA stability. Oncogene 2013, 32, 2169–2178. [Google Scholar] [CrossRef][Green Version]
- Yan, W.; Zhang, J.; Zhang, Y.; Jung, Y.-S.; Chen, X. p73 Expression Is Regulated by RNPC1, a Target of the p53 Family, via mRNA Stability. Mol. Cell. Biol. 2012, 32, 2336–2348. [Google Scholar] [CrossRef]
- Zhang, J.; Cho, S.-J.; Shu, L.; Yan, W.; Guerrero, T.; Kent, M.; Skorupski, K.; Chen, H.; Chen, X. Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev. 2011, 25, 1528–1543. [Google Scholar] [CrossRef]
- Zhang, J.; Jun Cho, S.; Chen, X. RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc. Natl. Acad. Sci. USA 2010, 107, 9614–9619. [Google Scholar] [CrossRef]
- Kuwasako, K.; Takahashi, M.; Unzai, S.; Tsuda, K.; Yoshikawa, S.; He, F.; Kobayashi, N.; Güntert, P.; Shirouzu, M.; Ito, T.; et al. RBFOX and SUP-12 sandwich a G base to cooperatively regulate tissue-specific splicing. Nat. Struct. Mol. Biol. 2014, 21, 778–786. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, M.; Qian, Y.; Xu, E.; Zhang, J.; Chen, X. Rbm24, an RNA-binding Protein and a Target of p53, Regulates p21 Expression via mRNA Stability. J. Biol. Chem. 2014, 289, 3164–3175. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.; Zhang, J.; Zhang, M.; Jiang, Y.; Cho, S.-J.; Chen, X. RNA-Binding Protein RBM24 Regulates p63 Expression via mRNA Stability. Mol. Cancer Res. 2014, 12, 359–369. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, M.; Zhang, Y.; Xu, E.; Mohibi, S.; De Anda, D.M.; Jiang, Y.; Zhang, J.; Chen, X. Rbm24, a target of p53, is necessary for proper expression of p53 and heart development. Cell Death Differ. 2018, 25, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Ohe, K.; Yoshida, M.; Nakano-Kobayashi, A.; Hosokawa, M.; Sako, Y.; Sakuma, M.; Okuno, Y.; Usui, T.; Ninomiya, K.; Nojima, T.; et al. RBM24 promotes U1 snRNP recognition of the mutated 5′ splice site in the IKBKAP gene of familial dysautonomia. RNA 2017, 23, 1393–1403. [Google Scholar] [CrossRef]
- Shao, M.; Lu, T.; Zhang, C.; Zhang, Y.Z.; Kong, S.H.; Shi, D.L. Rbm24 controls poly(A) tail length and translation efficiency of crystallin mRNAs in the lens via cytoplasmic polyadenylation. Proc. Natl. Acad. Sci. USA 2020, 117, 7245–7254. [Google Scholar] [CrossRef]
- Qian, K.; Li, M.; Wang, J.; Zhang, M.; Wang, M. Structural basis for mRNA recognition by human RBM38. Biochem. J. 2020, 477, 161–172. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muraoka, S.; Fukumura, K.; Hayashi, M.; Kataoka, N.; Mayeda, A.; Kaida, D. Rbm38 Reduces the Transcription Elongation Defect of the SMEK2 Gene Caused by Splicing Deficiency. Int. J. Mol. Sci. 2020, 21, 8799. https://doi.org/10.3390/ijms21228799
Muraoka S, Fukumura K, Hayashi M, Kataoka N, Mayeda A, Kaida D. Rbm38 Reduces the Transcription Elongation Defect of the SMEK2 Gene Caused by Splicing Deficiency. International Journal of Molecular Sciences. 2020; 21(22):8799. https://doi.org/10.3390/ijms21228799
Chicago/Turabian StyleMuraoka, Shintaro, Kazuhiro Fukumura, Megumi Hayashi, Naoyuki Kataoka, Akila Mayeda, and Daisuke Kaida. 2020. "Rbm38 Reduces the Transcription Elongation Defect of the SMEK2 Gene Caused by Splicing Deficiency" International Journal of Molecular Sciences 21, no. 22: 8799. https://doi.org/10.3390/ijms21228799
APA StyleMuraoka, S., Fukumura, K., Hayashi, M., Kataoka, N., Mayeda, A., & Kaida, D. (2020). Rbm38 Reduces the Transcription Elongation Defect of the SMEK2 Gene Caused by Splicing Deficiency. International Journal of Molecular Sciences, 21(22), 8799. https://doi.org/10.3390/ijms21228799