Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series


Abstract
1. Introduction
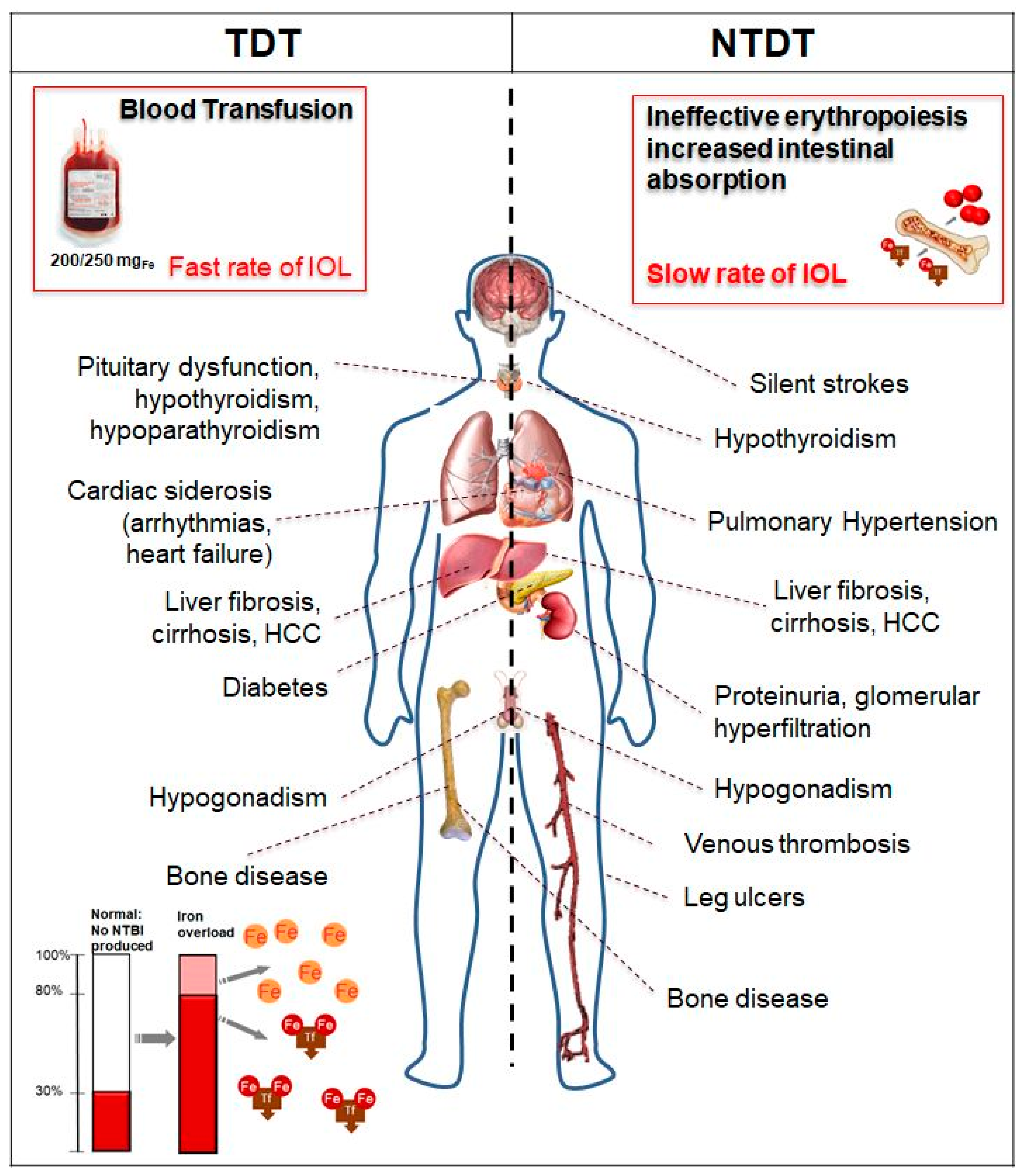
2. Pathophysiology of Iron Overload in Thalassemia
2.1. Case 1: Severe Multiorgan Siderosis in TDT
2.2. Case 2: ICT in TDT with Renal Function Alteration and Serum Ferritin < 500 μg/L
2.3. Comments on Cases 1 and 2
2.4. Case 3: Liver Iron Overload in NTDT
2.5. Comments on Case 3
3. Diagnosis and Monitoring Iron Overload
4. Iron Toxicity and Chelation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TDT | Transfusion-dependent thalassemia |
| NTDT | Non-transfusion-dependent thalassemia |
| MRI | Magnetic resonance imaging |
| IOL | Iron overload |
| Hb | Hemoglobin |
| RBC | Red blood cell |
| LCI | Labile cellular iron |
| NBTI | Non-transferrin-bound iron |
| LPI | Labile plasma iron |
| ROS | Reactive oxygen species |
| PRBC | Packed red blood cell |
| DFO | Deferoxamine |
| LVEF | Left ventricular ejection fraction |
| LIC | Liver iron concentration |
| Dw | Dry weight |
| IGT | Impaired glucose tolerance |
| DFP | Deferiprone |
| DFX | Deferasirox |
| GFR | Glomerular filtration rate |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| ICT | Iron chelation therapy |
| HCV | Hepatitis C virus |
References
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Modell, B. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 2008, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Taher, A.T.; Cappellini, M.D. How I manage medical complications of β-thalassemia in adults. Blood 2018, 132, 1781–1791. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Cohen, A.; Porter, J.; Taher, A.; Viprakasit, V. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT), 3rd ed.; Cappellini, M.D., Cohen, A., Porter, J., Taher, A., Viprakasit, V., Eds.; Thalassaemia International Federation: Nicosia, Cyprus, 2014. [Google Scholar]
- Taher, A.; Musallam, K.; Cappellini, M. Guidelines for the Management of Non-Transfusion Dependent Thalassaemias, 2nd ed.; Thalassaemia International Federation: Nicosia, Cyprus, 2017. [Google Scholar]
- Angelucci, E.; Matthes-Martin, S.; Baronciani, D.; Bernaudin, F.; Bonanomi, S.; Cappellini, M.D.; Dalle, J.-H.; Di Bartolomeo, P.; De Heredia, C.D.; Dickerhoff, R.; et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: Indications and management recommendations from an international expert panel. Haematologica 2014, 99, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; Rugolotto, S.; De Stefano, P.; Zhao, H.; Cappellini, M.D.; Del Vecchio, G.C.; Romeo, M.A.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004, 89, 1187–1193. [Google Scholar] [PubMed]
- Borgna-Pignatti, C.; Cappellini, M.D.; De Stefano, P.; Del Vecchio, G.C.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; Origa, R.; Piga, A.; Romeo, M.A.; et al. Survival and Complications in Thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Modell, B.; A Letsky, E.; Flynn, D.M.; Peto, R.; Weatherall, D.J. Survival and desferrioxamine in thalassaemia major. BMJ 1982, 284, 1081–1084. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Cappellini, M.D.; De Stefano, P.; Del Vecchio, G.C.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; Piga, A.; Romeo, M.A.; Zhao, H.; et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood 2006, 107, 3733–3737. [Google Scholar] [CrossRef]
- Anderson, L.J.; Westwood, M.A.; Holden, S.; Davis, B.; Prescott, E.; Wonke, B.; Porter, J.B.; Walker, J.M.; Pennell, D.J. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: A prospective study using T2* cardiovascular magnetic resonance. Br. J. Haematol. 2004, 127, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, L.J.; Panigrahy, A.; Mittelman, S.D.; Hyderi, A.; Dongelyan, A.; Coates, T.D.; Wood, J.C. Pituitary iron and volume predict hypogonadism in transfusional iron overload. Am. J. Hematol. 2012, 87, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.; Roughton, M.; Porter, J.; Walker, J.; Tanner, M.; Patel, J.; Wu, D.; Taylor, J.; Westwood, M.; Anderson, L.; et al. Cardiac T2* Magnetic Resonance for Prediction of Cardiac Complications in Thalassemia Major. Circulation 2009, 120, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.; Sheppard, M.; Carpenter, J.-P.; Anderson, L.; He, T.; Pierre, T.S.; Galanello, R.; Catani, G.; Wood, J.; Fucharoen, S.; et al. Post-mortem study of the association between cardiac iron and fibrosis in transfusion dependent anaemia. J. Cardiovasc. Magn. Reson. 2017, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, L.J.; Papudesi, J.; Coates, T.D.; Wood, J.C. Pancreatic iron loading predicts cardiac iron loading in thalassemia major. Blood 2009, 114, 4021–4026. [Google Scholar] [CrossRef]
- Wood, J.C. Estimating tissue iron burden: Current status and future prospects. Br. J. Haematol. 2015, 170, 15–28. [Google Scholar] [CrossRef]
- Wood, J.C. Guidelines for quantifying iron overload. Hematology 2014, 2014, 210–215. [Google Scholar] [CrossRef]
- Modell, B.; Khan, M.; Darlison, M.; A Westwood, M.; Ingram, D.; Pennell, D.J. Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2008, 10, 42. [Google Scholar] [CrossRef]
- Cazzola, M.; De Stefano, P.; Ponchio, L.; Locatelli, F.; Beguin, Y.; Dessì, C.; Barella, S.; Cao, A.; Galanello, R. Relationship between transfusion regimen and suppression of erythropoiesis in β-thalassaemia major. Br. J. Haematol. 1995, 89, 473–478. [Google Scholar] [CrossRef]
- Piga, A.; Serra, M.; Longo, F.; Forni, G.; Quarta, G.; Cappellini, M.D.; Galanello, R. Changing patterns of splenectomy in transfusion-dependent thalassemia patients. Am. J. Hematol. 2011, 86, 808–810. [Google Scholar] [CrossRef]
- Franchini, M.; Forni, G.L.; Marano, G.; Cruciani, M.; Mengoli, C.; Pinto, V.; De Franceschi, L.; Venturelli, D.; Casale, M.; Amerini, M.; et al. Red blood cell alloimmunisation in transfusion-dependent thalassaemia: A systematic review. Blood Transfus. 2019, 17, 4–15. [Google Scholar]
- Franchini, M.; Forni, G.L.; Liumbruno, G.M. Is there a standard-of-care for transfusion therapy in thalassemia? Curr. Opin. Hematol. 2017, 24, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Origa, R.; Piga, A.; Quarta, G.; Forni, G.L.; Longo, F.; Melpignano, A.; Galanello, R. Pregnancy and β-thalassemia: An Italian multicenter experience. Haematologica 2010, 95, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Pinto, V.; Poggi, M.; Russo, R.; Giusti, A.; Forni, G.L. Management of the aging beta-thalassemia transfusion-dependent population—The Italian experience. Blood Rev. 2019, 38, 100594. [Google Scholar] [CrossRef] [PubMed]
- Coates, T.D.; Carson, S.; Wood, J.C.; Berdoukas, V. Management of iron overload in hemoglobinopathies: What is the appropriate target iron level? Ann. N. Y. Acad. Sci. 2016, 1368, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Puliyel, M.; Mainous, A.G.; Berdoukas, V.; Coates, T.D. Iron toxicity and its possible association with treatment of Cancer: Lessons from hemoglobinopathies and rare, transfusion-dependent anemias. Free. Radic. Biol. Med. 2015, 79, 343–351. [Google Scholar] [CrossRef]
- Chung, W.-S.; Lin, C.-L.; Lin, C.-L.; Kao, C.-H. Thalassaemia and risk of cancer: A population-based cohort study. J. Epidemiol. Community Health 2015, 69, 1066–1070. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Garani, M.C.; Forni, G.L.; Cappellini, M.D.; Cassinerio, E.; Fidone, C.; Spadola, V.; Maggio, A.; Pantalone, G.R.; Piga, A.; et al. Hepatocellular carcinoma in thalassaemia: An update of the Italian Registry. Br. J. Haematol. 2014, 167, 121–126. [Google Scholar] [CrossRef]
- Poggi, M.; Sorrentino, F.; Pascucci, C.; Monti, S.; Lauri, C.; Bisogni, V.; Toscano, V.; Cianciulli, P. Malignancies in β-Thalassemia Patients: First Description of Two Cases of Thyroid Cancer and Review of the Literature. Hemoglobin 2011, 35, 439–446. [Google Scholar] [CrossRef]
- Ginzburg, Y.; Rivella, S. β-thalassemia: A model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 2011, 118, 4321–4330. [Google Scholar] [CrossRef]
- Origa, R.; Galanello, R.; Ganz, T.; Giagu, N.; Maccioni, L.; Faa, G.; Nemeth, E. Liver iron concentrations and urinary hepcidin in -thalassemia. Haematology 2007, 92, 583–588. [Google Scholar] [CrossRef]
- Coates, T.D. Physiology and pathophysiology of iron in hemoglobin-associated diseases. Free. Radic. Biol. Med. 2014, 72, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Saliba, A.N. Iron overload in thalassemia: Different organs at different rates. Hematology 2017, 2017, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Coates, T.D. Iron overload in transfusion-dependent patients. Hematology 2019, 2019, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Farmaki, K.; Tzoumari, I.; Pappa, C.; Chouliaras, G.; Berdoukas, V. Normalisation of total body iron load with very intensive combined chelation reverses cardiac and endocrine complications of thalassaemia major. Br. J. Haematol. 2010, 148, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Kolnagou, A.; Kontoghiorghes, G.J. New golden era of chelation therapy in thalassaemia: The achievement and maintenance of normal range body iron stores. Br. J. Haematol. 2010, 150, 489–490, author reply 491. [Google Scholar] [CrossRef] [PubMed]
- Coates, T.D.; Wood, J.C. How we manage iron overload in sickle cell patients. Br. J. Haematol. 2017, 177, 703–716. [Google Scholar] [CrossRef]
- Camaschella, C.; Pagani, A.; Nai, A.; Silvestri, L. The mutual control of iron and erythropoiesis. Int. J. Lab. Hematol. 2016, 38, 20–26. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; Wood, J.C.; Cappellini, M.D. Magnetic resonance evaluation of hepatic and myocardial iron deposition in transfusion-independent thalassemia intermedia compared to regularly transfused thalassemia major patients. Am. J. Hematol. 2010, 85, 288–290. [Google Scholar] [CrossRef]
- Origa, R.; Barella, S.; Argiolas, G.M.; Bina, P.; Agus, A.; Galanello, R. No evidence of cardiac iron in 20 never- or minimally-transfused patients with thalassemia intermedia. Haematologica 2008, 93, 1095–1096. [Google Scholar] [CrossRef]
- Forni, G.L.; Perrotta, S.; Giusti, A.; Quarta, G.; Pitrolo, L.; Cappellini, M.D.; D’Ascola, D.G.; Pignatti, C.B.; Rigano, P.; Filosa, A.; et al. Neridronate improves bone mineral density and reduces back pain in β-thalassaemia patients with osteoporosis: Results from a phase 2, randomized, parallel-arm, open-label study. Br. J. Haematol. 2012, 158, 274–282. [Google Scholar] [CrossRef]
- Voskaridou, E.; Ladis, V.; Kattamis, A.; Hassapopoulou, E.; Economou, M.; Kourakli, A.; Maragkos, K.; Kontogianni, K.; Lafioniatis, S.; Vrettou, E.; et al. A national registry of haemoglobinopathies in Greece: Deducted demographics, trends in mortality and affected births. Ann. Hematol. 2012, 91, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.-P.; Roughton, M.; Pennell, D.J. The Myocardial Iron in Thalassemia (MINT) Investigators International survey of T2* cardiovascular magnetic resonance in -thalassemia major. Haematologica 2013, 98, 1368–1374. [Google Scholar] [CrossRef] [PubMed]
- Pennell, D.J.; Udelson, J.E.; Arai, A.E.; Bozkurt, B.; Cohen, A.R.; Galanello, R.; Hoffman, T.M.; Kiernan, M.S.; Lerakis, S.; Piga, A.; et al. American Heart Association Committee on Heart Failure and Transplantation of the Council on Clinical Cardiology and Council on Cardiovascular Radiology and Imaging. Cardiovascular Function and Treatment in β-Thalassemia Major: A consensus statement from t. Circulation 2013, 128, 281–308. [Google Scholar] [CrossRef] [PubMed]
- Forni, G.L.; Puntoni, M.; Boeri, E.; Terenzani, L.; Balocco, M. The influence of treatment in specialized centers on survival of patients with thalassemia major. Am. J. Hematol. 2009, 84, 317–318. [Google Scholar] [CrossRef]
- Pennell, D.J.; Berdoukas, V.; Karagiorga, M.; Ladis, V.; Piga, A.; Aessopos, A.; Gotsis, E.D.; Tanner, M.A.; Smith, G.C.; Westwood, M.A.; et al. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood 2006, 107, 3738–3744. [Google Scholar] [CrossRef]
- Lal, A.; Porter, J.; Sweeters, N.; Ng, V.; Evans, P.; Neumayr, L.; Kurio, G.; Harmatz, P.; Vichinsky, E. Combined chelation therapy with deferasirox and deferoxamine in thalassemia. Blood Cells Mol. Dis. 2013, 50, 99–104. [Google Scholar] [CrossRef]
- Aydinok, Y.; Kattamis, A.; Cappellini, M.D.; El-Beshlawy, A.; Origa, R.; Elalfy, M.; Kilinç, Y.; Perrotta, S.; Karakas, Z.; Viprakasit, V.; et al. Deferasirox–Deferoxamine Combination Therapy Reduces Cardiac Iron With Rapid Liver Iron Removal In Patients With Severe Transfusional Iron Overload (HYPERION). Blood 2013, 122, 2257. [Google Scholar] [CrossRef]
- Voskaridou, E.; Christoulas, D.; Terpos, E. Successful chelation therapy with the combination of deferasirox and deferiprone in a patient with thalassaemia major and persisting severe iron overload after single-agent chelation therapies. Br. J. Haematol. 2011, 154, 654–656. [Google Scholar] [CrossRef]
- Angelucci, E.; Brittenham, G.M.; McLaren, C.E.; Ripalti, M.; Baronciani, D.; Giardini, C.; Galimberti, M.; Polchi, P.; Lucarelli, G. Correction: Hepatic Iron Concentration and Total Body Iron Stores in Thalassemia Major. N. Engl. J. Med. 2000, 343, 327–331. [Google Scholar] [CrossRef]
- Cohen, A.R.; Glimm, E.; Porter, J.B.; Suarez-Kurtz, G.; Perini, J.A.; Silva-Assunção, E.; Struchiner, C.J. Effect of transfusional iron intake on response to chelation therapy in β-thalassemia major. Blood 2008, 111, 583–587. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Origa, R.; Ponti, M.L.; Filosa, A.; Lanza, A.G.; Piga, A.; Saracco, G.M.; Pinto, V.; Picciotto, A.; Rigano, P.; Madonia, S.; et al. Treatment of hepatitis C virus infection with direct-acting antiviral drugs is safe and effective in patients with hemoglobinopathies. Am. J. Hematol. 2017, 92, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Forni, G.L.; Derchi, G. Typical manifestation of acute congestive heart failure in patients with Thalassaemia major causing diagnostic delay in the emergency room. Eur. J. Hear. Fail. 2003, 5, 607–608. [Google Scholar] [CrossRef]
- Derchi, G.; Formisano, F.; Balocco, M.; Galanello, R.; Bina, P.; Dessì, C.; Piga, A.; Donato, G.; Cappellini, M.D.; Cassinerio, E.; et al. Clinical management of cardiovascular complications in patients with thalassaemia major: A large observational multicenter study. Eur. J. Echocardiogr. 2011, 12, 242–246. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Roos, M.; Gasser, T.; Fortini, M.; Raiola, G.; Galati, M.C.; Italian Working Group on Endocrine Complications in Non-Endocrine Diseases. Impact of long-term iron chelation therapy on growth and endocrine functions in thalassaemia. J. Pediatr. Endocrinol. Metab. 2006, 19, 471–480. [Google Scholar] [PubMed]
- Casale, M.; Citarella, S.; Filosa, A.; De Michele, E.; Palmieri, F.; Ragozzino, A.; Amendola, G.; Pugliese, U.; Tartaglione, I.; Della Rocca, F.; et al. Endocrine function and bone disease during long-term chelation therapy with deferasirox in patients with β-thalassemia major. Am. J. Hematol. 2014, 89, 1102–1106. [Google Scholar] [CrossRef]
- Giusti, A.; Pinto, V.; Forni, G.L.; Pilotto, A. Management of beta-thalassemia-associated osteoporosis. Ann. N. Y. Acad. Sci. 2016, 1368, 73–81. [Google Scholar] [CrossRef]
- Tanner, M.; Galanello, R.; Dessi, C.; Smith, G.; Westwood, M.; Agus, A.; Roughton, M.; Assomull, R.; Nair, S.; Walker, J.; et al. A Randomized, Placebo-Controlled, Double-Blind Trial of the Effect of Combined Therapy With Deferoxamine and Deferiprone on Myocardial Iron in Thalassemia Major Using Cardiovascular Magnetic Resonance. Circulation 2007, 115, 1876–1884. [Google Scholar] [CrossRef]
- Farmaki, K.; Angelopoulos, N.; Anagnostopoulos, G.; Gotsis, E.; Rombopoulos, G.; Tolis, G. Effect of enhanced iron chelation therapy on glucose metabolism in patients with β-thalassaemia major. Br. J. Haematol. 2006, 134, 438–444. [Google Scholar] [CrossRef]
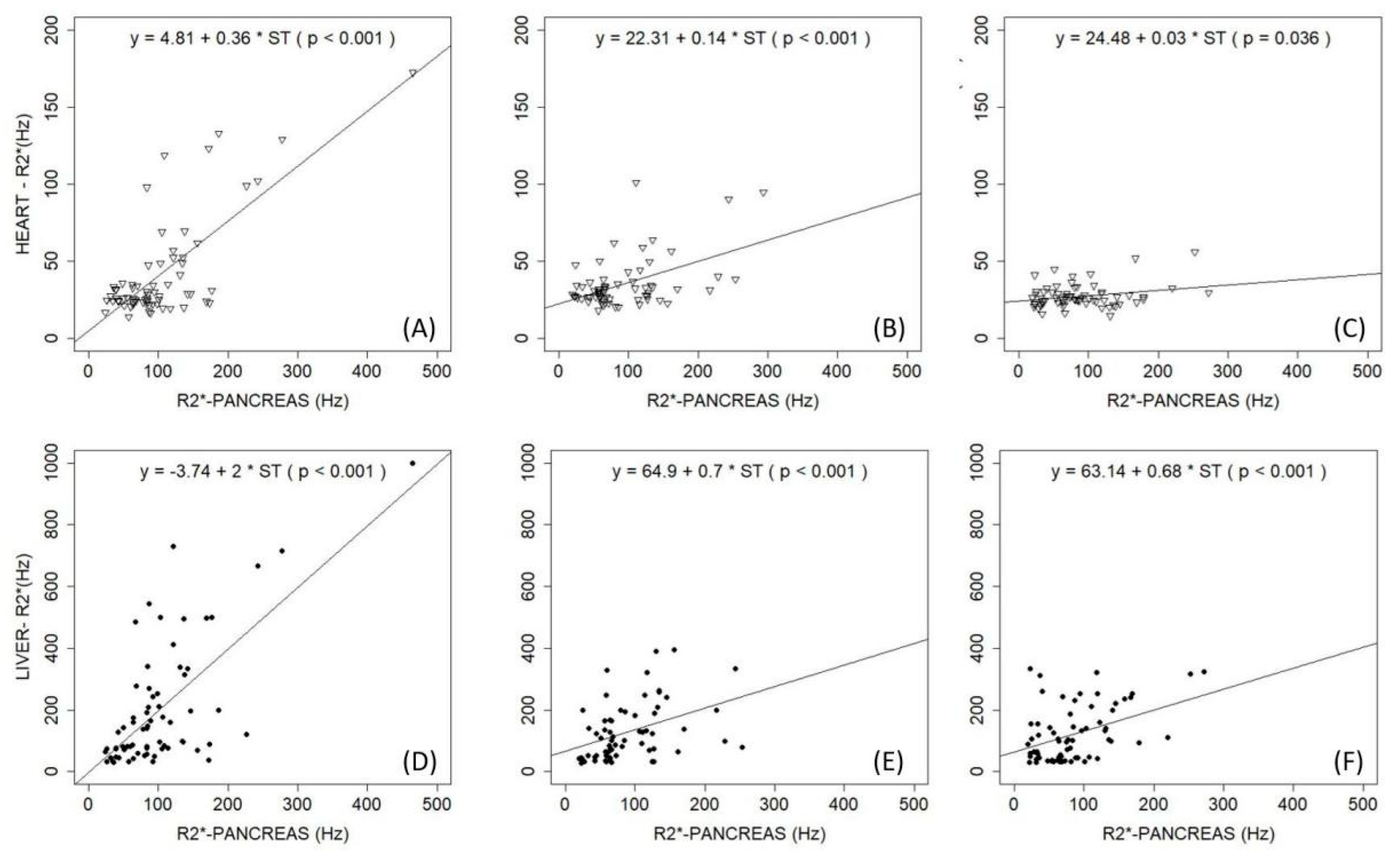
- Pinto, V.M.; Bacigalupo, L.; Gianesin, B.; Balocco, M.; De Franceschi, L.; Malagò, R.; Wood, J.; Forni, G.L. Lack of correlation between heart, liver and pancreas MRI-R2*: Results from long-term follow-up in a cohort of adult β-thalassemia major patients. Am. J. Hematol. 2018, 93, E79–E82. [Google Scholar] [CrossRef]
- Baldan, A.; Giusti, A.; Bosi, C.; Malaventura, C.; Musso, M.; Forni, G.L.; Volpato, S.; Zuliani, G.; Borgna-Pignatti, C. Klotho, a new marker for osteoporosis and muscle strength in β-thalassemia major. Blood Cells Mol. Dis. 2015, 55, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Castro-Mollo, M.; Martinez, M.R.; Feola, M.; Gumerova, A.A.; Casu, C.; E Fleming, R.; Rivella, S.; Yuen, T.; Zaidi, M.; Ginzburg, Y. Erythroferrone Regulates Bone Remodeling in β-Thalassemia. Blood 2019, 134 (Suppl. 1), 2. [Google Scholar] [CrossRef]
- Rafat, C.; Fakhouri, F.; Ribeil, J.-A.; Delarue, R.; Le Quintrec, M. Fanconi Syndrome Due to Deferasirox. Am. J. Kidney Dis. 2009, 54, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Origa, R.; Piga, A.; Tartaglione, I.; Della Corte, G.; Forni, G.L.; Bruederle, A.; Castiglioni, C.; Han, J. Renal safety under long-course deferasirox therapy in iron overloaded transfusion-dependent β-thalassemia and other anemias. Am. J. Hematol. 2018, 93, E172–E175. [Google Scholar] [CrossRef]
- Piga, A.; Fracchia, S.; Lai, M.E.; Cappellini, M.D.; Hirschberg, R.; Habr, D.; Wegener, A.; Bouillaud, E.; Forni, G.L. Deferasirox effect on renal haemodynamic parameters in patients with transfusion-dependent β thalassaemia. Br. J. Haematol. 2015, 168, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Scaramellini, N.; Arighi, C.; Marcon, A.; Consonni, D.; Cassinerio, E.; Graziadei, G.; Cappellini, M.D.; Motta, I. Iron Chelation and Ferritin below 500 Mcg/L in Transfusion Dependent Thalassemia: Beyond the Limits of Clinical Trials. Blood 2019, 134 (Suppl. 1), 3542. [Google Scholar] [CrossRef]
- Kew, M. Hepatic iron overload and hepatocellular carcinoma. Cancer Lett. 2009, 286, 38–43. [Google Scholar] [CrossRef]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004, 127, S79–S86. [Google Scholar] [CrossRef]
- Olynyk, J.; Pierre, T.S.; Britton, R.S.; Brunt, E.M.; Bacon, B.R. Duration of Hepatic Iron Exposure Increases the Risk of Significant Fibrosis in Hereditary Hemochromatosis: A New Role for Magnetic Resonance Imaging. Am. J. Gastroenterol. 2005, 100, 837–841. [Google Scholar] [CrossRef]
- Olivieri, N.F. Progression of iron overload in sickle cell disease. Semin. Hematol. 2001, 38, 57–62. [Google Scholar] [CrossRef]
- Matta, B.N.; Musallam, K.M.; Maakaron, J.E.; Koussa, S.; Taher, A.T. A killer revealed: 10-year experience with beta-thalassemia intermedia. Hematology 2014, 19, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Pennell, D.J.; Porter, J.B.; Cappellini, M.D.; Chan, L.L.; El-Beshlawy, A.; Aydinok, Y.; Ibrahim, H.; Li, C.-K.; Viprakasit, V.; Elalfy, M.S.; et al. Deferasirox for up to 3 years leads to continued improvement of myocardial T2* in patients with β-thalassemia major. Haematologica 2012, 97, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Pennell, D.J.; Porter, J.B.; Piga, A.; Lai, Y.; El-Beshlawy, A.; Belhoul, K.M.; Elalfy, M.; Yesilipek, A.; Kilinç, Y.; Lawniczek, T.; et al. A 1-year randomized controlled trial of deferasirox vs deferoxamine for myocardial iron removal in β-thalassemia major (CORDELIA). Blood 2014, 123, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Bejaoui, M.; Agaoglu, L.; Canatan, D.; Capra, M.; Cohen, A.; Drelichman, G.; Economou, M.; Fattoum, S.; Kattamis, A.; et al. Iron chelation with deferasirox in adult and pediatric patients with thalassemia major: Efficacy and safety during 5 years’ follow-up. Blood 2011, 118, 884–893. [Google Scholar] [CrossRef]
- Piga, A.; Longo, F.; Musallam, K.M.; Cappellini, M.D.; Forni, G.L.; Quarta, G.; Chiavilli, F.; Commendatore, F.; Mulas, S.; Caruso, V.; et al. Assessment and management of iron overload in β-thalassaemia major patients during the 21st century: A real-life experience from the Italian Webthal project. Br. J. Haematol. 2013, 161, 872–883. [Google Scholar] [CrossRef]
- Deugnier, Y.; Turlin, B.; Ropert, M.; Cappellini, M.D.; Porter, J.B.; Giannone, V.; Zhang, Y.; Griffel, L.; Brissot, P. Improvement in Liver Pathology of Patients With β-Thalassemia Treated With Deferasirox for at Least 3 Years. Gastroenterology 2011, 141, 1202–1211.e3. [Google Scholar] [CrossRef]
- Fischer, R.; Piga, A.; Harmatz, P.; Nielsen, P. Monitoring Long-Term Efficacy of Iron Chelation Treatment with Biomagnetic Liver Susceptometry. Ann. N. Y. Acad. Sci. 2005, 1054, 350–357. [Google Scholar] [CrossRef]
- Gianesin, B.; Zefiro, D.; Musso, M.; Rosa, A.; Bruzzone, C.; Balocco, M.; Carrara, P.; Bacigalupo, L.; Banderali, S.; Rollandi, G.A.; et al. Measurement of liver iron overload: Noninvasive calibration of MRI-R2* by magnetic iron detector susceptometer. Magn. Reson. Med. 2012, 67, 1782–1786. [Google Scholar] [CrossRef]
- Marinelli, M.; Gianesin, B.; Balocco, M.; Beruto, P.; Bruzzone, C.; Carrara, P.; Gallusi, P.; Macco, A.; Musso, M.; Oliveri, E.; et al. Total Iron-Overload Measurement in the Human Liver Region by the Magnetic Iron Detector. IEEE Trans. Biomed. Eng. 2010, 57, 2295–2303. [Google Scholar] [CrossRef]
- Bacigalupo, L.; Paparo, F.; Zefiro, D.; Viberti, C.M.; Cevasco, L.; Gianesin, B.; Pinto, V.M.; Rollandi, G.A.; Wood, J.C.; Forni, G.L. Comparison between different software programs and post-processing techniques for the MRI quantification of liver iron concentration in thalassemia patients. La Radiol. Med. 2016, 121, 751–762. [Google Scholar] [CrossRef]
- Carpenter, J.-P.; He, T.; Kirk, P.; Roughton, M.; Anderson, L.J.; De Noronha, S.V.; Baksi, A.J.; Sheppard, M.N.; Porter, J.B.; Walker, J.M.; et al. Calibration of myocardial T2 and T1 against iron concentration. J. Cardiovasc. Magn. Reson. 2014, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.-P.; He, T.; Kirk, P.; Roughton, M.; Anderson, L.J.; De Noronha, S.V.; Sheppard, M.N.; Porter, J.B.; Walker, J.M.; Wood, J.C.; et al. On T2* Magnetic Resonance and Cardiac Iron. Circulation 2011, 123, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Derchi, G.; Dessì, C.; Bina, P.; Cappellini, M.D.; Piga, A.; Perrotta, S.; Tartaglione, I.; Giuditta, M.; Longo, F.; Origa, R.; et al. Risk factors for heart disease in transfusion-dependent thalassemia: Serum ferritin revisited. Intern. Emerg. Med. 2019, 14, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Porter, J.B.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Galanello, R.; Karakas, Z.; Lawniczek, T.; et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood 2012, 120, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Viprakasit, V.; Ajlan, A.; Aydinok, Y.; Al Ebadi, B.A.A.; Dewedar, H.; Ibrahim, A.S.; Ragab, L.; Trad, O.; Wataify, A.S.; Wong, L.L.L.; et al. MRI for the diagnosis of cardiac and liver iron overload in patients with transfusion-dependent thalassemia: An algorithm to guide clinical use when availability is limited. Am. J. Hematol. 2018, 93, E135–E137. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, E.; Muretto, P.; Nicolucci, A.; Baronciani, D.; Erer, B.; Gaziev, J.; Ripalti, M.; Sodani, P.; Tomassoni, S.; Visani, G.; et al. Effects of iron overload and hepatitis C virus positivity in determining progression of liver fibrosis in thalassemia following bone marrow transplantation. Blood 2002, 100, 17–21. [Google Scholar] [CrossRef]
- Piga, A.; Galanello, R.; Forni, G.L.; Cappellini, M.D.; Origa, R.; Zappu, A.; Donato, G.; Bordone, E.; Lavagetto, A.; Zanaboni, L.; et al. Randomized phase II trial of deferasirox (Exjade, ICL670), a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassemia patients with transfusional iron overload. Haematologica 2006, 91, 873–880. Available online: http://www.ncbi.nlm.nih.gov/pubmed/16818273 (accessed on 31 October 2020).
- Bentley, A.; Gillard, S.; Spino, M.; Connelly, J.; Tricta, F. Cost–Utility Analysis of Deferiprone for the Treatment of β-Thalassaemia Patients with Chronic Iron Overload: A UK Perspective. PharmacoEconomics 2013, 31, 807–822. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals UK Ltd. Exjade® 125 mg, 250 mg, 500 mg Dispersible Tablets Summary of Product Characteristics. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000670/human_med_000780.jsp&mid=WC0b01ac058001d124 (accessed on 1 March 2018).
- Novartis Pharmaceuticals Corporation. Exjade® (Deferasirox) Tablets, for Oral Suspension Prescribing Information. Available online: https://www.accessdata.fda.gov/%0Adrugsatfda_docs/label/2016/021882s024lbl.pdf%0A (accessed on 1 March 2018).
- Novartis Pharma Stein AG. Desferal® (Deferoxamine Mesilate) Product Label. Available online: http://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?%0Aevent=overview.process&ApplNo=016267%0A (accessed on 1 March 2018).
- ApoPharma Inc. Ferriprox® (Deferiprone) Tablets for Oral Use Prescribing Information. Available online: http://www.accessdata.fda.gov/scripts/cder/daf/%0Aindex.cfm?event=overview.process&ApplNo=021825 (accessed on 1 March 2018).
- Angelucci, E.; Capasso, M.; Della Porta, M.G.; Forni, G.L.; Girelli, M.; Oliva, E.N.; Pilo, F.; Clavio, M.; Riva, M.; Pelizzari, A.; et al. A Multicenter, Italian Trial of Early Iron Chelation Therapy with Low Dose Deferasirox (Exjade®) in Patients with Low/Intermediate-1 Risk MDS at the Beginning of Transfusional Story. Blood 2019, 134 (Suppl. 1), 4256. [Google Scholar] [CrossRef]
- Aydinok, Y.; Kattamis, A.; Cappellini, M.D.; El-Beshlawy, A.; Origa, R.; Elalfy, M.; Kilinç, Y.; Perrotta, S.; Karakas, Z.; Viprakasit, V.; et al. Effects of deferasirox-deferoxamine on myocardial and liver iron in patients with severe transfusional iron overload. Blood 2015, 125, 3868–3877. [Google Scholar] [CrossRef]
- Aydinok, Y.; Ulger, Z.; Nart, D.; Terzi, A.; Cetiner, N.; Ellis, G.; Zimmermann, A.; Manz, C. A randomized controlled 1-year study of daily deferiprone plus twice weekly desferrioxamine compared with daily deferiprone monotherapy in patients with thalassemia major. Haematologica 2007, 92, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Di Maggio, R.; Maggio, A. The new era of chelation treatments: Effectiveness and safety of 10 different regimens for controlling iron overloading in thalassaemia major. Br. J. Haematol. 2017, 178, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Pinto, V.M.; Balocco, M.; Quintino, S.; Bacigalupo, L.; Gianesin, B.; Rizzi, M.; Malagò, R.; De Franceschi, L.; Forni, G.L. Daily alternating deferasirox and deferiprone therapy successfully controls iron accumulation in untreatable transfusion-dependent thalassemia patients. Am. J. Hematol. 2018, 93, E338–E340. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, E.; Pilo, F. Management of iron overload before, during, and after hematopoietic stem cell transplantation for thalassemia major. Ann. N. Y. Acad. Sci. 2016, 1368, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Motta, I.; Salvatori, M.; Fraquelli, M.; Marcon, A.; Taher, A.T.; Cappellini, M.D. Longitudinal changes in serum ferritin levels correlate with measures of hepatic stiffness in transfusion-independent patients with β-thalassemia intermedia. Blood Cells Mol. Dis. 2012, 49, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Karimi, M.; Rachmilewitz, E.A. Cerebral infarction in β-thalassemia intermedia: Breaking the silence. Thromb. Res. 2012, 130, 695–702. [Google Scholar] [CrossRef]
- Musallam, K.M.; Cappellini, M.D.; Daar, S.; Karimi, M.; El-Beshlawy, A.; Graziadei, G.; Magestro, M.; Wulff, J.; Pietri, G.; Taher, A.T. Serum ferritin level and morbidity risk in transfusion-independent patients with β-thalassemia intermedia: The ORIENT study. Haematologica 2014, 99, e218–e221. [Google Scholar] [CrossRef] [PubMed]
- Derchi, G.; Galanello, R.; Bina, P.; Cappellini, M.D.; Piga, A.; Lai, M.-E.; Quarta, A.; Casu, G.; Perrotta, S.; Pinto, V.; et al. Prevalence and Risk Factors for Pulmonary Arterial Hypertension in a Large Group of β-Thalassemia Patients Using Right Heart Catheterization. Circulation 2014, 129, 338–345. [Google Scholar] [CrossRef]
- Taher, A.; Vichinsky, E.; Musallam, K.; Cappellini, M.; Viprakasit, V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT); Thalassaemia International Federation: Nicosia, Cyprus, 2013. [Google Scholar]
- Saliba, A.N.; Musallam, K.M.; Cappellini, M.D.; Graziadei, G.; Daar, S.; Viprakasit, V.; Taher, A.T. Serum ferritin values between 300 and 800 ng/mL in nontransfusion-dependent thalassemia: A probability curve to guide clinical decision making when MRI is unavailable. Am. J. Hematol. 2017, 92, E35–E37. [Google Scholar] [CrossRef]
- Brittenham, G.M.; Griffith, P.M.; Nienhuis, A.W.; McLaren, C.E.; Young, N.S.; Tucker, E.E.; Allen, C.J.; Farrell, D.E.; Harris, J.W. Efficacy of Deferoxamine in Preventing Complications of Iron Overload in Patients with Thalassemia Major. N. Engl. J. Med. 1994, 331, 567–573. [Google Scholar] [CrossRef]
- DeLea, T.E.; Edelsberg, J.; Sofrygin, O.; Thomas, S.K.; Baladi, J.-F.; Phatak, P.D.; Coates, T.D. Consequences and costs of noncompliance with iron chelation therapy in patients with transfusion-dependent thalassemia: A literature review. Transfusion 2007, 47, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Gabutti, V.; Piga, A. Results of Long-Term Iron-Chelating Therapy. Acta Haematol. 1996, 95, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Origa, R.; Danjou, F.; Cossa, S.; Matta, G.; Bina, P.; Dessì, C.; DeFraia, E.; Foschini, M.L.; Leoni, G.; Morittu, M.; et al. Impact of heart magnetic resonance imaging on chelation choices, compliance with treatment and risk of heart disease in patients with thalassaemia major. Br. J. Haematol. 2013, 163, 400–403. [Google Scholar] [CrossRef]
- Baronciani, D.; Casale, M.; De Franceschi, L.; Graziadei, G.; Longo, F.; Origa, R.; Pinto, V.M.; Rigano, P.; Marchetti, M.; Gigante, A.; et al. Selecting ß-Thalassemia Patients for Gene Therapy: A Decision-Making Algorithm. Blood 2019, 134 (Suppl. 1), 972. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | 2015 | 2016 | 2017 | 2018 | |
|---|---|---|---|---|---|
| Pre-transfusion hemoglobin (g/dL) | >9.5 | 9.5 | 9.6 | 9.8 | 9.7 |
| Ferritin * (μg/L) | <200 | 8760 | 2450 | 864 | 451 |
| Transaminases * | NV | 3 × ULN | NV | NV | NV |
| LIC (mg/g dw) | <1.5 | 24 | 9 | 4 | 3.8 |
| MRI-T2* Heart (ms) | >20 | 8.3 | 16 | 24 | 26 |
| LVEF (%) | >55 | >55 | >55 | >55 | >55 |
| MRI-T2* Pancreas (ms) | >20 | 8.1 | 12.1 | 16 | 21 |
| ICT | DFP (90 mg/kg) DFO (50 mg/kg) | DFP (90 mg/kg) DFO (50 mg/kg) | DFX dispersible tablet (28 mg/kg) | DFX film coated tablet (14 mg/kg) | |
| Iron intake (mg/kg/day) | 0.3–0.6 | 0.5 | 0.5 | 0.5 | 0.5 |
| Target | T0 | After 3 Months | After 6 Months | |
|---|---|---|---|---|
| Pre-transfusion hemoglobin (g/dL) | >9.5 | 9.8 | 9.8 | 9.7 |
| Ferritin (μg/L) | <200 | 420 * | 415 | 402 |
| Transaminases | NV | 3 × ULN | NV | NV |
| Creatinine (mg/dL) | <1.1 | 1.2 | 0.8 | 0.7 |
| CrCl (mL/min) | >60 | 54 | 81 | 92 |
| Iron intake (mg/kg/day) | 0.3–0.6 | 0.4 | 0.4 | 0.4 |
| ICT | DFX film-coated tablet (14 mg/kg) | DFX film-coated tablet (14 mg/kg) | DFX film-coated tablet (14 mg/kg) | |
| NSAIDs | Yes | No | No |
| Chelator | DFO | DFP | DFX |
|---|---|---|---|
| Structure |  |  |  |
| Molecular weight | 560 | 139 | 373 |
| First clinically available | 1968 | 1999 | 2005 |
| Administration route | Parental (subcutaneous or intravenous) | Oral (tablets or solution) | Oral (dispersible or film-coated tablets) |
| Administration frequency | 8–12 h, 5–7 days per week; continuous infusion over 24 h in heart failure | Every 8 h, TID | Once daily, ongoing evaluations on BID dosing |
| Plasma half-life | 30 min | 3 h | 8–16 h |
| Route of iron excretion | Urinary and fecal | Urinary | Fecal |
| Recommended dose | 30–60 mg/g per day | 75–100 mg/kg per day | 20–40 mg/kg per day (dispersible tablets) or 14–28 mg/kg per day (film-coated tablets) |
| Main adverse event | Reaction at site of infusion, severe allergic reactions, bone abnormalities, growth failure, auditory (hearing loss), ophthalmologic (retinal damage), Yersinia infection | Gastrointestinal, arthralgia, transient increase in liver enzymes, neutropenia, agranulocytosis | Increased GFR and serum creatinine, proteinuria, rare renal failure, increased liver enzymes, rare liver failure, skin rash, gastrointestinal, rare gastrointestinal bleeding |
| Pregnancy | Contraindicated (can be used only at the end of the second trimester in patients with severe heart and liver IOL) | Contraindicated | Contraindicated |
| Licensed use—TDT | Treatment of chronic IOL resulting from transfusion-dependent anemia | Treatment of transfusional IOL in TDT where DFO is contraindicated or inadequate | US: Treatment of transfusional iron overload in patients 2 years or older Europe: Treatment of transfusional iron overload in patients 6 years and older, and when DFO is contraindicated or inadequate, in patients 2–5 years old |
| Licensed use—NTDT | No sufficient data, commonly used in clinical practice | Off-label | US: Treatment of chronic iron overload in patients 10 years of age and older with LIC ≥5 mg/g dry weight liver and SF ≥300 μg/L Europe: Treatment of chronic iron overload in patients 10 years of age and older with LIC ≥5 mg/g dry weight liver and/or SF ≥800 μg/L |
| Cost/year (£) | 5584 | 5519 | 23,179 |
| Tests | Baseline | 1st Month | 6th Month | Every 6 Months |
|---|---|---|---|---|
| Nephrology visit | X | |||
| General functional indices | ||||
| Creatinine | X | X | X | X |
| Urine test | X | X | X | X |
| Cystatin C | X | X * | X | X |
| Proteinuria/creatininuria (mg/g) | X | X ** | X | |
| Tubular functional indices | ||||
| β2-microglobulin/creatininuria (μg/g) | X | X | ||
| Calciuria/creatininuria (mg/g) | X | X | ||
| Phosphaturia/creatininuria (mg/g) | X | X | ||
| NGAL/creatininuria (μg/g) | X | X | ||
| Venous blood gas analysis | X | X *** | ||
| Glomerular functional indices | ||||
| Albuminuria/creatininuria (mg/g) | X | X ** | X |
| Method | Advantages | Limitations |
|---|---|---|
| Magnetic resonance imaging (MRI) |
|
|
Timing:
| ||
| Serum ferritin |
|
|
Timing:
| ||
| Transferrin saturation |
|
|
| NTBI/LPI |
|
|
| Liver biopsy |
|
|
| Biosusceptometry |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, V.M.; Forni, G.L. Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series. Int. J. Mol. Sci. 2020, 21, 8771. https://doi.org/10.3390/ijms21228771
Pinto VM, Forni GL. Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series. International Journal of Molecular Sciences. 2020; 21(22):8771. https://doi.org/10.3390/ijms21228771
Chicago/Turabian StylePinto, Valeria Maria, and Gian Luca Forni. 2020. "Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series" International Journal of Molecular Sciences 21, no. 22: 8771. https://doi.org/10.3390/ijms21228771
APA StylePinto, V. M., & Forni, G. L. (2020). Management of Iron Overload in Beta-Thalassemia Patients: Clinical Practice Update Based on Case Series. International Journal of Molecular Sciences, 21(22), 8771. https://doi.org/10.3390/ijms21228771