The Cutaneous Wound Innate Immunological Microenvironment


Abstract
1. Wounding Induces an Immunological Disruption to the Skin Barrier
2. Cutaneous Wounds as Entry Site for Infectious Agents
3. Innate Immune Response to Acute Cutaneous Wounds: A Brief Overview
4. Sensing a Wound: Immune Microenvironment Is Dependent on Neuroimmune Signaling
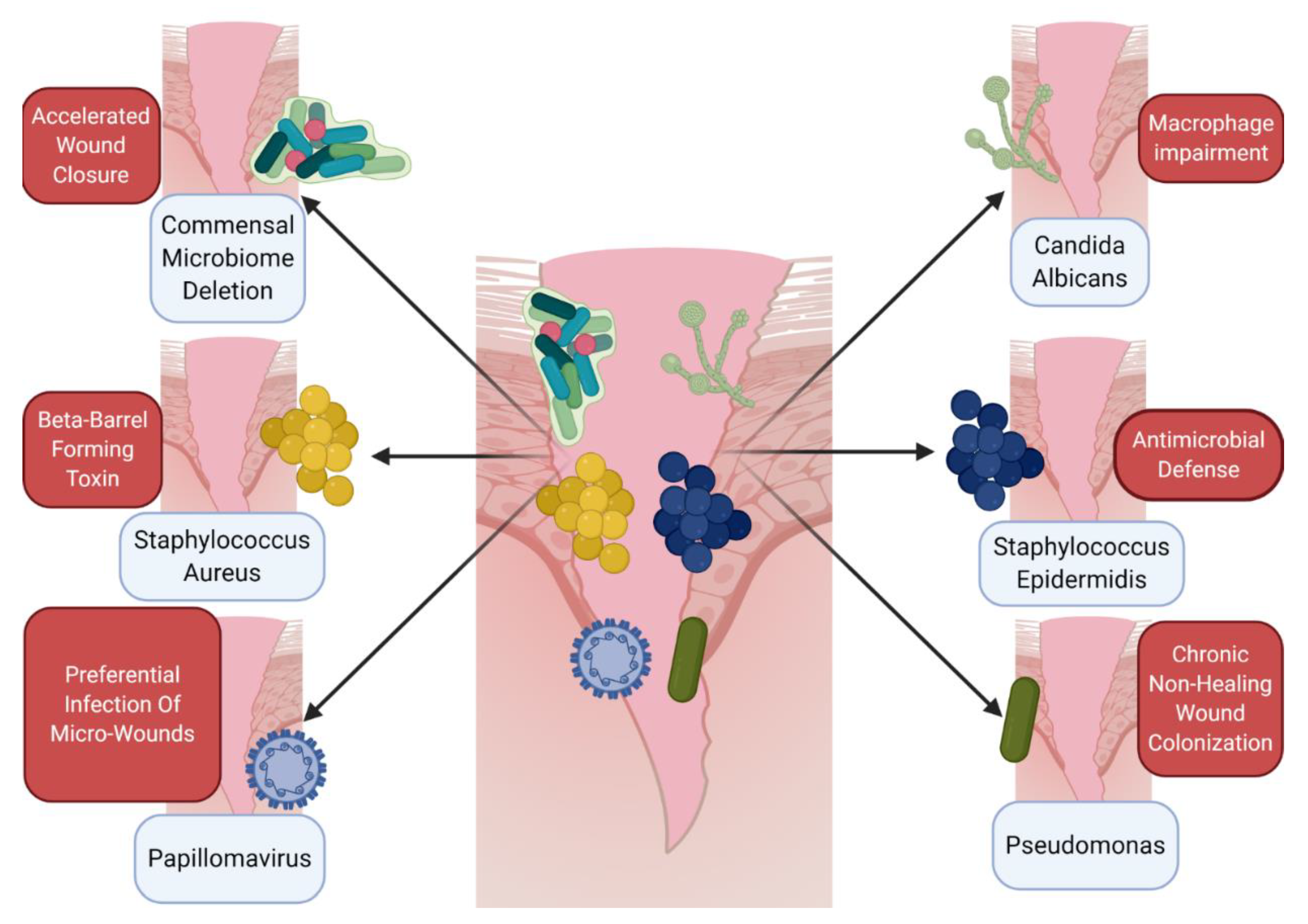
5. Host–Microbe Interactions in the Wound as a Component of the Immune Microenvironment
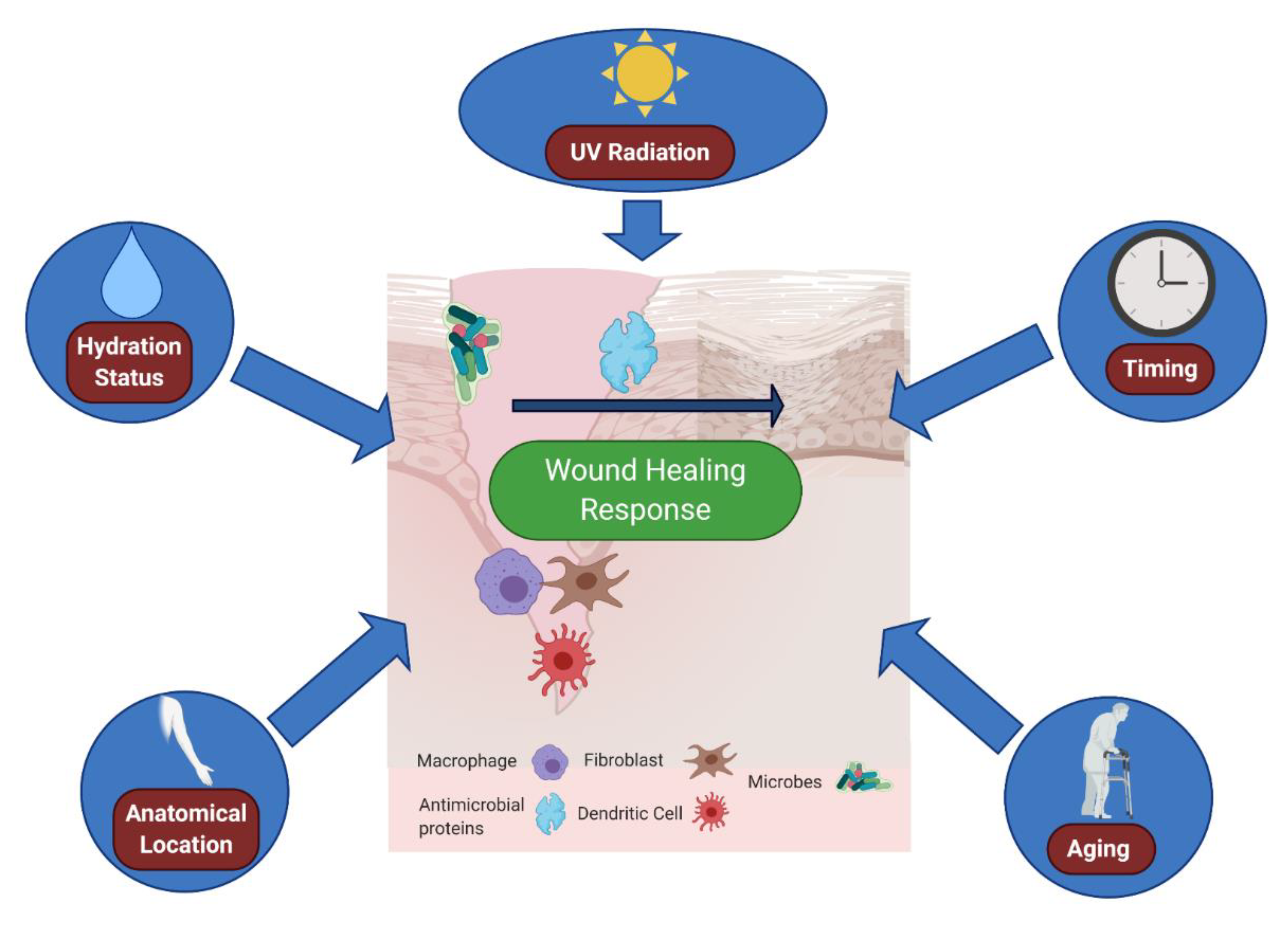
6. Environmental Effects: The Outside World Alters the Cutaneous Wound Environment

{kind=link}
{kind=link}
| Microenvironment Component | Outcome(s) | Reference(s) | |
|---|---|---|---|
| Internal | Neural Sensation | Denervated skin heals at slower rates; | [42] |
| TRPV1 nerve fibers activate host immune defenses | [44,45] | ||
| Internal | Wound Location | Immune cell numbers vary with body site | [80] |
| Internal | Age | Elderly Skin heals slower than younger skin; | [81] |
| Inflammation/repair spectrum is impaired in aged skin | [82] | ||
| External | Cutaneous Bacteria | Microbiome deletion potentiates wound closure; | [54] |
| Commensal microbes can promote antimicrobial defense; | [57] | ||
| Microbiome is altered in chronic, non-healing wounds | [64,65] | ||
| External | Cutaneous Fungus | Cutaneous fungal communities are predictive of wound healing time | [67] |
| External | Cutaneous Virus | IL-27 promotes antiviral defense and healing in cutaneous wounds | [47] |
| External | Moisture | Emollients can promote antibacterial defenses; | [76] |
| Skin moisture levels directly impact wound healing rate | [75] | ||
| External | UV Radiation | UVB radiation activates Type I interferon responses; | [83] |
| UVB radiation can directly stimulate wound healing | [84] | ||
| External | Time of Wound | Fibroblast migration and wound healing varies with time of wound | [85] |

7. An Aging Microenvironment
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gallo, R.L. Human Skin Is the Largest Epithelial Surface for Interaction with Microbes. J. Investig. Dermatol. 2017, 137, 1213–1214. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Eyerich, K.; Traidl-Hoffmann, C.; Biedermann, T. Cutaneous Barriers and Skin Immunity: Differentiating A Connected Network. Trends Immunol. 2018, 39, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.W.; Watkins, K.; Hewlett, A. Infection control through the ages. Am. J. Infect. Control. 2012, 40, 35–42. [Google Scholar] [CrossRef]
- Haagensen, C.D.; Lloyd, W.E.B. A Hundred Years of Medicine; Beard Books: Frederick, MD, USA, 1943. [Google Scholar]
- Mayon-White, R.T.; Ducel, G.; Kereselidze, T.; Tikomirov, E. An international survey of the prevalence of hospital-acquired infection. J. Hosp. Infect. 1988, 11, 43–48. [Google Scholar] [CrossRef]
- Garner, B.H.; Anderson, D.J. Surgical Site Infections: An Update. Infect. Dis. Clin. N. Am. 2016, 30, 909–929. [Google Scholar] [CrossRef]
- McKibben, L.; Horan, T.; Tokars, J.I.; Fowler, G.; Cardo, D.M.; Pearson, M.L.; Brennan, P.J.; The Healthcare Infection Control Practices Advisory Committee. Guidance on Public Reporting of Healthcare-Associated Infections: Recommendations of the Healthcare Infection Control Practices Advisory Committee. Am. J. Infect. Control. 2005, 33, 217–226. [Google Scholar] [CrossRef]
- Awad, S.S. Adherence to surgical care improvement project measures and post-operative surgical site infections. Surg. Infect. 2012, 13, 234–237. [Google Scholar] [CrossRef]
- Zimlichman, E.; Henderson, D.; Tamir, O.; Franz, C.; Song, P.; Yamin, C.K.; Keohane, C.; Denham, C.R.; Bates, D.W. Health care-associated infections: A meta-analysis of costs and financial impact on the US health care system. JAMA Intern. Med. 2013, 173, 2039–2046. [Google Scholar] [CrossRef]
- Sen, C.K. Human Wounds and Its Burden: An Updated Compendium of Estimates. Adv. Wound Care 2019, 8, 39–48. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Wlaschek, M.; Scharffetter-Kochanek, K. Pathogenesis of wound healing disorders in the elderly. JDDG J. Der Dtsch. Dermatol. Ges. 2017, 15, 255–275. [Google Scholar] [CrossRef]
- Toniolo, A.; Cassani, G.; Puggioni, A.; Rossi, A.; Colombo, A.; Onodera, T.; Ferrannini, E. The diabetes pandemic and associated infections: Suggestions for clinical microbiology. Rev. Med. Microbiol. 2019, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bowler, P.G.; Duerden, B.I.; Armstrong, D.G. Wound microbiology and associated approaches to wound management. Clin. Microbiol. Rev. 2001, 14, 244–269. [Google Scholar] [CrossRef] [PubMed]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363, eaat9691. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, M.G.; Holzer, K.J.; Carbone, J.T.; Salas-Wright, C.P. Arthropod Bites and Stings Treated in Emergency Departments: Recent Trends and Correlates. Wilderness Environ. Med. 2019, 30, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.; McDowell, R.H. Insect Bites. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Vasievich, M.P.; Villarreal, J.D.M.; Tomecki, K.J. Got the Travel Bug? A Review of Common Infections, Infestations, Bites, and Stings Among Returning Travelers. Am. J. Clin. Dermatol. 2016, 17, 451–462. [Google Scholar] [CrossRef]
- Hadfield, J.; Brito, A.F.; Swetnam, D.M.; Vogels, C.B.F.; Tokarz, R.E.; Andersen, K.G.; Smith, R.C.; Bedford, T.; Grubaugh, N.D. Twenty years of West Nile virus spread and evolution in the Americas visualized by Nextstrain. PLoS Pathog. 2019, 15, e1008042. [Google Scholar] [CrossRef]
- Bos, S.; Gadea, G.; Despres, P. Dengue: A growing threat requiring vaccine development for disease prevention. Pathog. Glob. Health 2018, 112, 294–305. [Google Scholar] [CrossRef]
- Hills, S.L.; Fischer, M.; Petersen, L.R. Epidemiology of Zika Virus Infection. J. Infect. Dis. 2017, 216, S868–S874. [Google Scholar] [CrossRef]
- Guo, S.; Dipietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef]
- MacLeod, A.S.; Mansbridge, J.N. The Innate Immune System in Acute and Chronic Wounds. Adv. Wound Care 2016, 5, 65–78. [Google Scholar] [CrossRef]
- Larouche, J.; Sheoran, S.; Maruyama, K.; Martino, M.M. Immune Regulation of Skin Wound Healing: Mechanisms and Novel Therapeutic Targets. Adv. Wound Care 2018, 7, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Sub-Cell. Biochem. 2017, 82, 405–456. [Google Scholar]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar] [PubMed]
- Ali, R.A.; Wuescher, L.M.; Worth, R.G. Platelets: Essential components of the immune system. Curr. Trends Immunol. 2015, 16, 65–78. [Google Scholar]
- Landén, N.X.; Li, D.; Ståhle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef]
- Munir, S.; Basu, A.; Maity, P.; Krug, L.; Haas, P.; Jiang, D.; Strauss, G.; Wlaschek, M.; Geiger, H.; Singh, K.; et al. TLR4-dependent shaping of the wound site by MSCs accelerates wound healing. EMBO Rep. 2020, 21, e48777. [Google Scholar] [CrossRef]
- Büchau, A.S.; Hassan, M.; Kukova, G.; Lewerenz, V.; Kellermann, S.; Würthner, J.U.; Wolf, R.; Walz, M.; Gallo, R.L.; Ruzicka, T. S100A15, an Antimicrobial Protein of the Skin: Regulation by E. coli through Toll-Like Receptor 4. J. Investig. Dermatol. 2007, 127, 2596–2604. [Google Scholar]
- Bdeir, K.; Gollomp, K.; Stasiak, M.; Mei, J.; Papiewska-Pajak, I.; Zhao, G.; Worthen, G.S.; Cines, D.B.; Poncz, M.; Kowalska, M.A. Platelet-Specific Chemokines Contribute to the Pathogenesis of Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2016, 56, 261–270. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 98–108. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Tonello, S.; Rizzi, M.; Migliario, M.; Rocchetti, V.; Renò, F. Low concentrations of neutrophil extracellular traps induce proliferation in human keratinocytes via NF-kB activation. J. Dermatol. Sci. 2017, 88, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Mirza, R.; Koh, T.J. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 2011, 56, 256–264. [Google Scholar] [CrossRef]
- Yan, J.; Tie, G.; Wang, S.; Tutto, A.; DeMarco, N.; Khair, L.; Fazzio, T.G.; Messina, L.M. Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat. Commun. 2018, 9, 33. [Google Scholar] [CrossRef]
- Paige, J.T.; Kremer, M.; Landry, J.; Hatfield, S.A.; Wathieu, D.; Brug, A.; Lightell, D.J.; Spiller, K.L.; Woods, T.C. Modulation of inflammation in wounds of diabetic patients treated with porcine urinary bladder matrix. Regen. Med. 2019, 14, 269–277. [Google Scholar] [CrossRef]
- Ashrafi, M.; Baguneid, M.; Bayat, A. The Role of Neuromediators and Innervation in Cutaneous Wound Healing. Acta Derm. Venereol. 2016, 96, 587–594. [Google Scholar] [CrossRef]
- Roosterman, D.; Goerge, T.; Schneider, S.W.; Bunnett, N.W.; Steinhoff, M. Neuronal Control of Skin Function: The Skin as a Neuroimmunoendocrine Organ. Physiol. Rev. 2006, 86, 1309–1379. [Google Scholar] [CrossRef]
- Fukai, T.; Takeda, A.; Uchinuma, E. Wound healing in denervated rat skin. Wound Repair Regen. 2005, 13, 175–180. [Google Scholar] [CrossRef]
- Souza, B.R.; Cardoso, J.F.; Amadeu, T.P.; Desmoulière, A.; Costa, A.M.A. Sympathetic denervation accelerates wound contraction but delays reepithelialization in rats. Wound Repair Regen. 2005, 13, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Riedl, M.S.; Yao, C.; Honda, C.N.; Vulchanova, L.; Kaplan, D.H. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 2015, 43, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Edwards, T.N.; Liu, A.W.; Hirai, T.; Jones, M.R.; Wu, J.; Li, Y.; Zhang, S.; Ho, J.; Davis, B.M.; et al. Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 2019, 178, 919–932.e14. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.S.; Hemmers, S.; Garijo, O.; Chabod, M.; Mowen, K.; Witherden, D.A.; Havran, W.L. Dendritic epidermal T cells regulate skin antimicrobial barrier function. J. Clin. Investig. 2013, 123, 4364–4374. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Suwanpradid, J.; Sanchez-Lagunes, R.; Choi, H.W.; Hoang, P.; Wang, D.; Abraham, S.N.; Macleod, A.S. IL-27 Facilitates Skin Wound Healing through Induction of Epidermal Proliferation and Host Defense. J. Investig. Dermatol. 2017, 137, 1166–1175. [Google Scholar] [CrossRef]
- Shook, B.; Xiao, E.; Kumamoto, Y.; Iwasaki, A.; Horsley, V. CD301b+ Macrophages Are Essential for Effective Skin Wound Healing. J. Investig. Dermatol. 2016, 136, 1885–1891. [Google Scholar] [CrossRef]
- Shin, H.; Kumamoto, Y.; Gopinath, S.; Iwasaki, A. CD301b+ dendritic cells stimulate tissue-resident memory CD8+ T cells to protect against genital HSV-2. Nat. Commun. 2016, 7, 13346. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Johnson, T.R.; Gómez, B.I.; McIntyre, M.K.; Dubick, M.A.; Christy, R.J.; Nicholson, S.E.; Burmeister, D.M. The Cutaneous Microbiome and Wounds: New Molecular Targets to Promote Wound Healing. Int. J. Mol. Sci. 2018, 19, 2699. [Google Scholar] [CrossRef]
- Gardiner, M.; Vicaretti, M.; Sparks, J.; Bansal, S.; Bush, S.; Liu, M.; Darling, A.; Harry, E.; Burke, C.M. A longitudinal study of the diabetic skin and wound microbiome. PeerJ 2017, 5, e3543. [Google Scholar] [CrossRef]
- Bartow-McKenney, C.; Hannigan, G.D.; Horwinski, J.; Hesketh, P.; Horan, A.D.; Mehta, S.; Grice, E.A. The microbiota of traumatic, open fracture wounds is associated with mechanism of injury. Wound Repair Regen. 2018, 26, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Canesso, M.C.C.; Vieira, A.T.; Castro, T.B.R.; Schirmer, B.G.A.; Cisalpino, D.; Martins, F.S.; Rachid, M.A.; Nicoli, J.R.; Teixeira, M.M.; Barcelos, L.S. Skin Wound Healing Is Accelerated and Scarless in the Absence of Commensal Microbiota. J. Immunol. 2014, 193, 5171. [Google Scholar] [CrossRef] [PubMed]
- Linehan, J.L.; Harrison, O.J.; Han, S.J.; Byrd, A.L.; Vujkovic-Cvijin, I.; Villarino, A.V.; Sen, S.K.; Shaik, J.; Smelkinson, M.; Tamoutounour, S.; et al. Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair. Cell 2018, 172, 784–796.e18. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Di Nardo, A.; Nakatsuji, T.; Leichtle, A.; Yang, Y.; Cogen, A.L.; Wu, Z.-R.; Hooper, L.V.; Schmidt, R.R.; von Aulock, S.; et al. Commensal bacteria regulate Toll-like receptor 3–dependent inflammation after skin injury. Nat. Med. 2009, 15, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Cogen, A.L.; Yamasaki, K.; Sanchez, K.M.; Dorschner, R.A.; Lai, Y.; MacLeod, D.T.; Torpey, J.W.; Otto, M.; Nizet, V.; Kim, J.E.; et al. Selective Antimicrobial Action Is Provided by Phenol-Soluble Modulins Derived from Staphylococcus epidermidis, a Normal Resident of the Skin. J. Investig. Dermatol. 2010, 130, 192–200. [Google Scholar] [CrossRef]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol 2015, 13, 529–543. [Google Scholar] [CrossRef]
- Uwamahoro, N.; Verma-Gaur, J.; Shen, H.-H.; Qu, Y.; Lewis, R.; Lu, J.; Bambery, K.; Masters, S.L.; Vince, J.E.; Naderer, T.; et al. The Pathogen Candida albicans Hijacks Pyroptosis for Escape from Macrophages. mBio 2014, 5, e00003-14. [Google Scholar] [CrossRef]
- Hufbauer, M.; Akgül, B. Molecular Mechanisms of Human Papillomavirus Induced Skin Carcinogenesis. Viruses 2017, 9, 187. [Google Scholar] [CrossRef]
- Williams, H.; Crompton, R.A.; Thomason, H.A.; Campbell, L.; Singh, G.; McBain, A.J.; Cruickshank, S.M.; Hardman, M.J. Cutaneous Nod2 Expression Regulates the Skin Microbiome and Wound Healing in a Murine Model. J. Investig. Dermatol. 2017, 137, 2427–2436. [Google Scholar] [CrossRef]
- Williams, H.; Campbell, L.; Crompton, R.A.; Singh, G.; McHugh, B.J.; Davidson, D.J.; McBain, A.J.; Cruickshank, S.M.; Hardman, M.J. Microbial Host Interactions and Impaired Wound Healing in Mice and Humans: Defining a Role for BD14 and NOD2. J. Investig. Dermatol. 2018, 138, 2264–2274. [Google Scholar] [CrossRef]
- Campbell, L.; Williams, H.; Crompton, R.A.; Cruickshank, S.M.; Hardman, M.J. Nod2 deficiency impairs inflammatory and epithelial aspects of the cutaneous wound-healing response. J. Pathol. 2013, 229, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Wolcott, R.D.; Hanson, J.D.; Rees, E.J.; Koenig, L.D.; Phillips, C.D.; Wolcott, R.A.; Cox, S.B.; White, J.S. Analysis of the chronic wound microbiota of 2963 patients by 16S rDNA pyrosequencing. Wound Repair Regen. 2016, 24, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Verbanic, S.; Shen, Y.; Lee, J.; Deacon, J.M.; Chen, I.A. Microbial predictors of healing and short-term effect of debridement on the microbiome of chronic wounds. NPJ Biofilms Microbiomes 2020, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Ruffin, M.; Brochiero, E. Repair Process Impairment by Pseudomonas aeruginosa in Epithelial Tissues: Major Features and Potential Therapeutic Avenues. Front. Cell. Infect. Microbiol. 2019, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Kalan, L.; Loesche, M.; Hodkinson, B.P.; Heilmann, K.; Ruthel, G.; Gardner, S.E.; Grice, E.A. Redefining the Chronic-Wound Microbiome: Fungal Communities Are Prevalent, Dynamic, and Associated with Delayed Healing. mBio 2016, 7, e01058-16. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Meisel, J.S.; Tyldsley, A.S.; Zheng, Q.; Hodkinson, B.P.; SanMiguel, A.J.; Minot, S.; Bushman, F.D.; Grice, E.A. The Human Skin Double-Stranded DNA Virome: Topographical and Temporal Diversity, Genetic Enrichment, and Dynamic Associations with the Host Microbiome. mBio 2015, 6, e01578-15. [Google Scholar] [CrossRef]
- Foulongne, V.; Sauvage, V.; Hebert, C.; Dereure, O.; Cheval, J.; Gouilh, M.A.; Pariente, K.; Segondy, M.; Burguière, A.; Manuguerra, J.C.; et al. Human skin microbiota: High diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS ONE 2012, 7, e38499. [Google Scholar] [CrossRef]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef]
- Hunter, C.A.; Kastelein, R. Interleukin-27: Balancing protective and pathological immunity. Immunity 2012, 37, 960–969. [Google Scholar] [CrossRef]
- Kwock, J.T.; Handfield, C.; Suwanpradid, J.; Hoang, P.; McFadden, M.J.; Labagnara, K.F.; Floyd, L.; Shannon, J.; Uppala, R.; Sarkar, M.K.; et al. IL-27 signaling activates skin cells to induce innate antiviral proteins and protects against Zika virus infection. Sci. Adv. 2020, 6, eaay3245. [Google Scholar] [CrossRef]
- Marks, R. The stratum corneum barrier: The final frontier. J. Nutr. 2004, 134, 2017s–2021s. [Google Scholar] [CrossRef] [PubMed]
- Sparr, E.; Millecamps, D.; Isoir, M.; Burnier, V.; Larsson, Å.; Cabane, B. Controlling the hydration of the skin though the application of occluding barrier creams. J. R. Soc. Interface 2012, 10, 20120788. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Xia, H.; He, W.; Li, Z.; Zhao, J.; Liu, B.; Wang, Y.; Lei, Q.; Kong, Y.; Bai, Y.; et al. Controlled water vapor transmission rate promotes wound-healing via wound re-epithelialization and contraction enhancement. Sci. Rep. 2016, 6, 24596. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; Malajian, D.; Khattri, S.; Correa da Rosa, J.; Dutt, R.; Finney, R.; Dhingra, N.; Xiangyu, P.; Xu, H.; Estrada, Y.D.; et al. Petrolatum: Barrier repair and antimicrobial responses underlying this “inert” moisturizer. J. Allergy Clin. Immunol. 2016, 137, 1091–1102.e7. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; Ku, A.T.; Nishino, Y.; Howard, J.M.; Rao, A.S.; Shaver, T.M.; Garcia, G.E.; Le, D.N.; Karlin, K.L.; Westbrook, T.F.; et al. Tcf3 promotes cell migration and wound repair through regulation of lipocalin 2. Nat. Commun. 2014, 5, 4088. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Ushio, H.; Nakano, N.; Ng, W.; Sayama, K.; Hashimoto, K.; Nagaoka, I.; Okumura, K.; Ogawa, H. Antimicrobial peptides human beta-defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J. Investig. Dermatol. 2007, 127, 594–604. [Google Scholar] [CrossRef]
- Field, C.K.; Kerstein, M.D. Overview of wound healing in a moist environment. Am. J. Surg. 1994, 167, S2–S6. [Google Scholar] [CrossRef]
- Omine, Y.; Hinata, N.; Yamamoto, M.; Kasahara, M.; Matsunaga, S.; Murakami, G.; Abe, S.-I. Regional differences in the density of Langerhans cells, CD8-positive T lymphocytes and CD68-positive macrophages: A preliminary study using elderly donated cadavers. Anat. Cell Biol. 2015, 48, 177–187. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Mills, S.J.; Ashworth, J.J. Ageing and wound healing. Biogerontology 2002, 3, 337–345. [Google Scholar] [CrossRef]
- Haustead, D.J.; Stevenson, A.; Saxena, V.; Marriage, F.; Firth, M.; Silla, R.; Martin, L.; Adcroft, K.F.; Rea, S.; Day, P.J.; et al. Transcriptome analysis of human ageing in male skin shows mid-life period of variability and central role of NF-κB. Sci. Rep. 2016, 6, 26846. [Google Scholar] [CrossRef]
- Skopelja-Gardner, S.; An, J.; Tai, J.; Tanaka, L.; Sun, X.; Hermanson, P.; Baum, R.; Kawasumi, M.; Green, R.; Gale, M., Jr.; et al. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci. Rep. 2020, 10, 7908. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Avci, P.; Dai, T.; Huang, Y.-Y.; Hamblin, M.R. Ultraviolet Radiation in Wound Care: Sterilization and Stimulation. Adv. Wound Care 2013, 2, 422–437. [Google Scholar] [CrossRef]
- Hoyle, N.P.; Seinkmane, E.; Putker, M.; Feeney, K.A.; Krogager, T.P.; Chesham, J.E.; Bray, L.K.; Thomas, J.M.; Dunn, K.; Blaikley, J.; et al. Circadian actin dynamics drive rhythmic fibroblast mobilization during wound healing. Sci. Transl. Med. 2017, 9, eaal2774. [Google Scholar] [CrossRef]
- Pick, R.; He, W.; Chen, C.-S.; Scheiermann, C. Time-of-Day-Dependent Trafficking and Function of Leukocyte Subsets. Trends Immunol. 2019, 40, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, A.L.; Rendon, J.L.; Ramirez, L.; Choudhry, M.A.; Kovacs, E.J. Reduced Neutrophil Chemotaxis and Infiltration Contributes to Delayed Resolution of Cutaneous Wound Infection with Advanced Age. J. Immunol. 2013, 190, 1746. [Google Scholar] [CrossRef]
- Kopcewicz, M.; Walendzik, K.; Bukowska, J.; Kur-Piotrowska, A.; Machcinska, S.; Gimble, J.M.; Gawronska-Kozak, B. Cutaneous wound healing in aged, high fat diet-induced obese female or male C57BL/6 mice. Aging 2020, 12, 7066–7111. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Garcia, B.; Murray, C.K.; Vrahas, M.S.; Hamblin, M.R. UVC Light Prophylaxis for Cutaneous Wound Infections in Mice. Antimicrob. Agents Chemother. 2012, 56, 3841. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, J.; Meller, S.; Conrad, C.; Di Nardo, A.; Homey, B.; Lauerma, A.; Arai, N.; Gallo, R.L.; Digiovanni, J.; Gilliet, M. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J. Exp. Med. 2010, 207, 2921–2930. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yue, J.; Lei, Q.; Gou, X.; Chen, S.-Y.; He, Y.-Y.; Wu, X. Ultraviolet B Inhibits Skin Wound Healing by Affecting Focal Adhesion Dynamics. Photochem. Photobiol. 2015, 91, 909–916. [Google Scholar] [CrossRef]
- Azzouz, D.; Khan, M.A.; Sweezey, N.; Palaniyar, N. Two-in-one: UV radiation simultaneously induces apoptosis and NETosis. Cell Death Discov. 2018, 4, 51. [Google Scholar] [CrossRef]
- Patra, V.; Wagner, K.; Arulampalam, V.; Wolf, P. Skin Microbiome Modulates the Effect of Ultraviolet Radiation on Cellular Response and Immune Function. iScience 2019, 15, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Paatela, E.; Munson, D.; Kikyo, N. Circadian Regulation in Tissue Regeneration. Int. J. Mol. Sci. 2019, 20, 2263. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, N.; Cermakian, N. Circadian Clocks in the Immune System. J. Biol. Rhythm. 2015, 30, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.K.; Liou, Y.-H.; Knudsen, N.H.; Starost, K.A.; Xu, C.; Hyde, A.L.; Liu, S.; Jacobi, D.; Liao, N.-S.; Lee, C.-H. Bmal1 integrates mitochondrial metabolism and macrophage activation. eLife 2020, 9, e54090. [Google Scholar] [CrossRef]
- Greenberg, E.N.; Marshall, M.E.; Jin, S.; Venkatesh, S.; Dragan, M.; Tsoi, L.C.; Gudjonsson, J.E.; Nie, Q.; Takahashi, J.S.; Andersen, B. Circadian control of interferon-sensitive gene expression in murine skin. Proc. Natl. Acad. Sci. USA 2020, 117, 5761–5771. [Google Scholar] [CrossRef]
- Keller, M.; Mazuch, J.; Abraham, U.; Eom, G.D.; Herzog, E.D.; Volk, H.-D.; Kramer, A.; Maier, B. A circadian clock in macrophages controls inflammatory immune responses. Proc. Natl. Acad. Sci. USA 2009, 106, 21407. [Google Scholar] [CrossRef]
- Chopra, K.; Calva, D.; Sosin, M.; Tadisina, K.K.; Banda, A.; De La Cruz, C.; Chaudhry, M.R.; Legesse, T.; Drachenberg, C.B.; Manson, P.N.; et al. A Comprehensive Examination of Topographic Thickness of Skin in the Human Face. Aesthetic Surg. J. 2015, 35, 1007–1013. [Google Scholar] [CrossRef]
- Rittié, L. Cellular mechanisms of skin repair in humans and other mammals. J. Cell Commun. Signal. 2016, 10, 103–120. [Google Scholar] [CrossRef]
- Sinha, M.; Sen, C.K.; Singh, K.; Das, A.; Ghatak, S.; Rhea, B.; Blackstone, B.; Powell, H.M.; Khanna, S.; Roy, S. Direct conversion of injury-site myeloid cells to fibroblast-like cells of granulation tissue. Nat. Commun. 2018, 9, 936. [Google Scholar] [CrossRef]
- Deckers, J.; Hammad, H.; Hoste, E. Langerhans Cells: Sensing the Environment in Health and Disease. Front. Immunol. 2018, 9, 93. [Google Scholar] [CrossRef]
- Tur, E.; Tur, M.; Maibach, H.I.; Guy, R.H. Basal perfusion of the cutaneous microcirculation: Measurements as a function of anatomic position. J. Investig. Dermatol. 1983, 81, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Eaglstein, W.H. Wound healing and aging. Dermatol. Clin. 1986, 4, 481–484. [Google Scholar] [CrossRef]
- Sgonc, R.; Gruber, J. Age-Related Aspects of Cutaneous Wound Healing: A Mini-Review. Gerontology 2013, 59, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Horan, M.A.; Ferguson, M.W. Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab. Investig. 1998, 78, 47–58. [Google Scholar]
- Thornton, M.J. Estrogens and aging skin. Dermatoendocrinology 2013, 5, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Dodsworth, J.; van Boxtel, E.; Tarnuzzer, R.W.; Horan, M.A.; Schultz, G.S.; Ferguson, M.W. Estrogen accelerates cutaneous wound healing associated with an increase in TGF-beta1 levels. Nat. Med. 1997, 3, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Keyes, B.E.; Liu, S.; Asare, A.; Naik, S.; Levorse, J.; Polak, L.; Lu, C.P.; Nikolova, M.; Pasolli, H.A.; Fuchs, E. Impaired Epidermal to Dendritic T Cell Signaling Slows Wound Repair in Aged Skin. Cell 2016, 167, 1323–1338.e14. [Google Scholar] [CrossRef]
- Solé-Boldo, L.; Raddatz, G.; Schütz, S.; Mallm, J.-P.; Rippe, K.; Lonsdorf, A.S.; Rodríguez-Paredes, M.; Lyko, F. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 2020, 3, 188. [Google Scholar] [CrossRef] [PubMed]
- Ray, G.T.; Suaya, J.A.; Baxter, R. Incidence, microbiology, and patient characteristics of skin and soft-tissue infections in a U.S. population: A retrospective population-based study. BMC Infect. Dis. 2013, 13, 252. [Google Scholar] [CrossRef]
- Kim, D.H.; Je, Y.J.; Kim, C.D.; Lee, Y.H.; Seo, Y.J.; Lee, J.H.; Lee, Y. Can Platelet-rich Plasma Be Used for Skin Rejuvenation? Evaluation of Effects of Platelet-rich Plasma on Human Dermal Fibroblast. Ann. Dermatol. 2011, 23, 424–431. [Google Scholar] [CrossRef]
- Balduini, C.L.; Noris, P. Platelet count and aging. Haematologica 2014, 99, 953–955. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirchner, S.; Lei, V.; MacLeod, A.S. The Cutaneous Wound Innate Immunological Microenvironment. Int. J. Mol. Sci. 2020, 21, 8748. https://doi.org/10.3390/ijms21228748
Kirchner S, Lei V, MacLeod AS. The Cutaneous Wound Innate Immunological Microenvironment. International Journal of Molecular Sciences. 2020; 21(22):8748. https://doi.org/10.3390/ijms21228748
Chicago/Turabian StyleKirchner, Stephen, Vivian Lei, and Amanda S. MacLeod. 2020. "The Cutaneous Wound Innate Immunological Microenvironment" International Journal of Molecular Sciences 21, no. 22: 8748. https://doi.org/10.3390/ijms21228748
APA StyleKirchner, S., Lei, V., & MacLeod, A. S. (2020). The Cutaneous Wound Innate Immunological Microenvironment. International Journal of Molecular Sciences, 21(22), 8748. https://doi.org/10.3390/ijms21228748