Abstract
The Thyroid hormone Receptor Interacting Protein 12 (TRIP12) protein belongs to the 28-member Homologous to the E6-AP C-Terminus (HECT) E3 ubiquitin ligase family. First described as an interactor of the thyroid hormone receptor, TRIP12’s biological importance was revealed by the embryonic lethality of a murine model bearing an inactivating mutation in the TRIP12 gene. Further studies showed the participation of TRIP12 in the regulation of major biological processes such as cell cycle progression, DNA damage repair, chromatin remodeling, and cell differentiation by an ubiquitination-mediated degradation of key protein substrates. Moreover, alterations of TRIP12 expression have been reported in cancers that can serve as predictive markers of therapeutic response. The TRIP12 gene is also referenced as a causative gene associated to intellectual disorders such as Clark–Baraitser syndrome and is clearly implicated in Autism Spectrum Disorder. The aim of the review is to provide an exhaustive and integrated overview of the different aspects of TRIP12 ranging from its regulation, molecular functions and physio-pathological implications.
1. Introduction
Thyroid hormone Receptor Interacting Protein 12 cDNA was cloned from the human myeloid KG-1 cell line and named the unidentified KIAA0045 human gene. The deduced coding sequence of KIAA0045 showed partial identity to that of the S. cerevisiae UFD4 protein (Ubiquitin Fusion Degradation 4) [1]. Another study identified Thyroid hormone Receptor Interacting Protein 12 (TRIP12) as a member of the structurally and functionally related E3 ubiquitin ligases based on the identification of a domain homologous to the E6-associated protein carboxyl terminus (HECT domain), which is a protein that induces the ubiquitin-dependent degradation of P53 in the presence of the E6 protein from the papillomavirus [2].
TRIP12 was later characterized as a thyroid hormone receptor-interacting protein by the trap yeast interaction system to isolate proteins that interact with the ligand binding/dimerization/transcriptional activation domain of the rat TR-β1 (Thyroid Receptor-β1) [3]. Fifteen cDNAs encoding Thyroid hormone Receptor Interacting Proteins (TRIPs) were isolated. All the TRIPs interacted with the Thyroid hormone Receptor (TR), but only when thyroid hormone T3 was present (TRIP1 to TRIP11) or only when T3 was absent (TRIP12 to TRIP15). Protein sequences of the fifteen TRIPs are unrelated, but sequences similarities with known proteins and functional motifs were found for nine of them based on blast alignment analyses with no demonstration of functional significance. The TRIP12 sequence was found to show 55% identity with the C-terminus of E6-associated protein, and more than a decade following its identification, TRIP12 was demonstrated to function as an E3 ubiquitin ligase [4]. Evidence of functional roles was later provided for the other TRIPs proteins (Supplemental Table). Among them, TRIP12 is the only TRIP family member bearing an E3 ubiquitin ligase activity.
The biological importance of TRIP12 was revealed by the embryonic lethality of a murine model bearing an inactivating mutation in the TRIP12 gene [5]. Several molecular functions of TRIP12 in important cellular processes and signaling pathways have been demonstrated in recent years. Accumulating evidence indicate that TRIP12 ubiquitinates key proteins for cell homeostasis, regulates gene expression (See Section 5), and plays important roles in cancers and neurological diseases. Up to now, more than 58 publications related to TRIP12 have been referenced in PubMed, half of them in the last 5 years. We have recently contributed to broadening knowledge on the role of TRIP12 by discovering a new substrate essential for pancreatic acinar cells differentiation [6] and by demonstrating that TRIP12 is a new chromatin-associated protein with several implications in the cell cycle progression and in the maintenance of genome integrity [7]. Due to its implication in several important physiological and pathological processes, TRIP12 now emerges as a protein of interest for molecular mechanisms researches and as a potential new therapeutic target.
In this review, we attempt to comprehensively summarize the current knowledge from the literature and databases on TRIP12. This summary includes the gene and protein expression, the protein structure, the protein interactors and functions. We address the physio-pathological roles of TRIP12 in essential biological pathways in relation with some of its substrates. We also present the alterations of the TRIP12 gene found in neurodevelopmental disorders and cancers as well as the alterations of TRIP12 protein expression in cancers, and their consequences. Finally, we discuss the potential role of TRIP12 as a therapeutic target and highlight the need for further research studies on this protein.
2. TRIP12 mRNA and Protein Expression
2.1. TRIP12 Gene Organization
In the human genome, the gene encoding for TRIP12 mRNA is located on the long arm of chromosome 2 at the locus q36.3. It spreads over nearly 168 kb (Genome Browser (see Section Software and databases)). TRIP12 mRNA expression is driven by a bidirectional promoter that also initiates the transcription of FBXO36 (F-box Only protein 36) mRNA in a head-to-head opposite direction. Bidirectional promoters are often used to control genes sharing the same regulation or encoding proteins exhibiting a similar biological process [8]. The FBXO36 gene encodes a protein that belongs to the F-box protein family. F-box is a ≈50 aa-domain that functions as a site for protein–protein interaction. Up to now, the functions of FBXO36 are unknown. In general, F-box proteins are part of SCF (SKP1, Cullin, F-box protein) ubiquitin ligase complexes, in which they bind to substrates for ubiquitin-mediated proteolysis [8,9]. This would be consistent with the fact that TRIP12 and FBXO36 expression are co-regulated. The 555 bp-bidirectional promoter is embedded in a 1753 bp long-CG-rich region also called CpG (Cytosine-phosphate-Guanosine) island, but the regulatory regions and methylation level of TRIP12/FBXO36 promoter remain unstudied.
2.2. TRIP12 mRNA Expression
To date, 26 different TRIP12 splicing mRNA variants have been identified (www.ncbi.nlm.nih.gov) (Table 1). Their length fluctuates from 9,065 (variant 4) to 10,492 nt (variant 20) with 41 to 43 exons with the exception of variants 24, 25, and 26 that contain only the first six (variants 24 and 25) or seven exons (variant 26) with a length of 5,951 (variant 26) to 6,052 nt (variant 25). Little is known regarding the expression level of the different splicing variants in human cells. It is suggested TRIP12 mRNA transcripts are a mixture of all possible isoforms, although the variant NM_001284215.1 seems to be predominant in peripheral leukocytes [10]. For the long variants, while the 5′-Untranslated Region (5′-UTR) is ≈180 nt-long, the 3′-Untranslated Region (3′-UTR) represents approximately one-third of the total mRNA length (≈3700 nt), which likely indicates a regulatory function for this region (Figure 1). In fact, the existence of a short (340 nt) and a long form (3716 nt) of TRIP12 3′-UTR mRNA was reported [11]. The different sites of transcriptional termination are explained by the presence of two consensus polyadenylation signals (Figure 1). These two variants display different translation efficiency in response to an activation of the mTORC (mammalian Target Of Rapamycin Complex 1) signaling pathway where the short form is more efficiently translated [11]. With reference to the Human Protein Atlas (see Section Software and databases), the expression of TRIP12 mRNA is ubiquitous in human tissues even if levels of mRNA vary between tissues. For example, TRIP12 mRNA is highly expressed in testis and in muscles but weakly expressed in pancreatic tissue [1,12,13].

Table 1.
List of the human mRNA variants and protein isoforms of TRIP12.
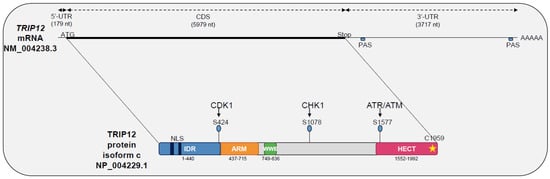
Figure 1.
Schematic representation of Thyroid hormone Receptor Interacting Protein 12 (TRIP12) mRNA (NM_004238.3) and TRIP12 protein (isoform c, NP_004229.1) with functional domains, kinase associated-phosphorylated residues and cysteine catalytic site (yellow star). UTR: Untranslated Region, CDS: Coding Sequence, PAS: Poly-Adenylation Signal, AAA: Poly-adenylated tail, ATG: translation Start codon, Stop: translation Stop codon, NLS: Nuclear Localization Signal, S: serine residue, aa: amino acid, IDR: Intrinsically Disordered Regions, ARM: Armadillo repeats, HECT: Homologous to the E6-AP C-Terminus.
2.3. TRIP12 Protein Expression and Cellular Localisation
TRIP12 protein translation can occur from 26 different mRNA splicing variants (Table 1). This leads to the generation of 14 different TRIP12 protein isoforms (isoforms a to m) with a length comprised between 1722 aa and 2068 aa for the long isoforms a to l (Figure 2). The two short isoforms (m and n) contain only 440 and 411 aa, respectively. Multiple alignment analysis COBALT (Constraint-based Multiple Alignment Tool) (see Section Software and databases) reveals a high similarity between the long isoforms (Figure 2). The two most studied isoforms b and c (2025 and 1992 aa, respectively) only differ by a six aa deletion in the IDR domain and a 27 aa deletion in the WWE domain in the isoform c that potentially modifies the protein–protein interaction capacity of this isoform (see Section 3.2.). For further description of the TRIP12 protein, the 1992 aa isoform c (NP_004229.1) will be used as a reference. Even though several protein isoforms of TRIP12 potentially exist in human cells, it is still unknown whether the different isoforms are cell-specific or if they co-exist and in what proportion in the same type of cells. Unfortunately, commercially available antibodies are directed against epitopes that are common to the long isoforms of TRIP12 (except the isoform d). Therefore, they cannot discriminate the different long isoforms. However, immunostaining analysis using these antibodies reveals a nuclear localization of TRIP12 in several cell lines [7,14] and in human tissues (Human Protein Atlas). It can be explained by the presence of two putative bipartite variants of the classical basically Nuclear Localization Signals (NLS) (aa 73–94 and 136–152) (ELM software (see Section Software and databases)) (Figure 1). The importance of the N-terminal region in the maintenance of TRIP12 nuclear localization was demonstrated in our recent study [7]. Within the nucleus, two studies noted an absence of TRIP12 in the nucleoli [7,15].
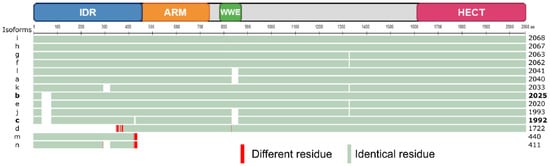
Figure 2.
Schematic representation of a multiple alignment of human TRIP12 isoforms using the COBALT multiple alignment tool (see Section Software and databases). TRIP12 isoform i (2068 aa) was defined as the sequence of reference (master sequence). The two most studied isoforms b and c (and size) are written in bold. Residues in green are identical and in red are different. The different domains of TRIP12 are indicated above the alignment.
3. TRIP12 Protein Structure and Domains
TRIP12 is a large protein with an apparent molecular weight estimated between 189 and 227 kDa for the long forms (Table 1). It comprises several characterized protein domains: a catalytic HECT domain, two protein/protein interaction domains (WWE (Tryptophan-Tryptophan-Glutamate) and ARM (Armadillo repeats)), and a recently identified intrinsically disordered region (IDR) (Figure 1). These domains confer structure and functional roles to the protein. TRIP12 protein domains are conserved during evolution and are subject to post-translational modifications that likely modulate its structure and activity. Several studies reported HECT, ARM repeats, and WWE crystal structure of other proteins [16,17,18]. However, TRIP12 has yet to be recovered in crystal form to establish its complete 3D structure.
3.1. The HECT Domain
HECT domains (≈40 kDa) were first identified in the C-terminal part of the viral E6-AP protein; hence their name, Homologous to the E6-AP C-Terminus [2]. They are generally located in the C-terminal extremity of proteins and are constituted of two lobes: N-lobe domains (N-terminal end) and a C-lobe (C-terminal end) linked by a hinge domain [19,20]. The N-lobe is responsible for the interaction with the ubiquitin-conjugating enzyme E2 during the ubiquitination reaction (detailed in Section 4.1). The C-lobe carries the characteristic catalytic cysteine residue [21,22], which is highly conserved between species and essential to the ubiquitination reaction [2]. The TRIP12 HECT domain is located at residues 1552–1592, is structured in N and C-lobes, and contains a cysteine in position 1959 for which the replacement by an alanine residue abolishes TRIP12 ubiquitin ligase activity (Figure 1) [2,23]. Mechanistically, the 28 known HECT E3 ubiquitin ligases form an intermediate complex with ubiquitin before linking it to a lysine substrate via a thioester bond [20].
3.2. The WWE Domain
A WWE domain is a globular region of ≈80 amino acids characterized by an enrichment with hydrophobic and aromatic residues in its two extremities and the conservation of two tryptophan residues (W) and one glutamate (E) [24]. The C- and N-terminal parts of WWE domains form β-sheets, while the central part is an α-helix. WWE domains are required for protein–protein interaction. They are essential for two families of proteins: E3 ubiquitin ligases (HECT, RING, and RING-H2 types) and in a sub-family of poly (ADP-ribose) polymerases (PARP). PARPs add multiple ADP-ribose moieties to protein targets (parylation) and participate in various cellular functions such as DNA repair and chromatin dynamics. The WWE domains were demonstrated as poly(ADP-ribose) (PAR) binding domains [25]. The WWE domain of TRIP12 is located between the residues 749 and 836 (InterProScan) [26] and is required for the interaction of TRIP12 with APP-BP1 (Amyloid Protein-Binding Protein 1) [4], PTF1a (Pancreas Transcription Factor 1a) [6], and parylated PARP1 (Poly-(ADP ribose) polymerase 1) [27] (Figure 1).
3.3. The Armadillo-Repeats Domain
Armadillo (ARM) repeat-containing proteins are characterized by the presence of imperfect tandem repeats of 42 hydrophobic amino acids. An ARM repeat is composed of three α-helices (H1, H2, and H3). Successive ARM repeats are able to assemble themselves to form a super-helix also named a solenoid. As such, ARM-repeat motifs are strongly implicated in protein/protein interactions. ARM repeat-containing proteins are involved in pleiotropic functions (cell/cell adhesion, cell signaling, cytoskeleton organization, nuclear trafficking, and ubiquitination) [28]. ARM repeats were first identified in the TRIP12 orthologues KAKTUS (Arabidopsis thaliana) and UFD4 (Ubiquitin Fusion Degradation 4) (Saccharomyces cerevisiae) (detailed in Section 4.3) [29,30]. The InterProScan database shows that ARM repeats are also present in the human TRIP12 protein. Slightly different ARM-repeat boundaries are found in this database; we arbitrarily chose to represent the smallest and the most quoted one between residues 437 and 715 (Figure 1). Functional studies demonstrated the role of ARM repeats of UFD4 in the recognition of the ubiquitin degradation signal (detailed in Section 4.3) of the substrates in Saccharomyces cerevisiae [30].
3.4. The IDR Domain
Intrinsically disordered regions (IDR) are common in the human proteome, whereby an estimated 44% of the proteome carries an IDR. These regions can be functional but interestingly lack a defined 3D conformation in physiological conditions [31]. The interaction of an IDR-containing protein with a partner can stabilize and/or fold the disordered region into a stable secondary structure. IDRs are sequences enriched in basic, positively charged, and hydrophilic amino acids such as lysine (K), arginine (R), serine (S), and histidine (H). IDR-containing proteins are mostly involved in protein, DNA, and RNA binding/interaction [32,33,34]. Moreover, the presence of N-terminal IDRs is linked to DNA interaction [33,34,35]. We previously showed that TRIP12 interacts with chromatin through an IDR, which corresponds to the N-terminal quarter of the protein (aa 1–440) (Figure 1), and that the deletion of this domain leads to a loss of TRIP12 interaction with chromatin [7]. E3 ubiquitin ligases containing IDR structures are infrequent. Indeed, the IDEAL database (see Section Software and databases) references only 25 E3 ubiquitin ligases containing IDR among the ≈600 E3 ubiquitin ligases. According to the IDEAL (Intrinsically Disordered proteins with Extensive Annotations and Literature) database, TRIP12 IDR displays identity with well-characterized IDR-containing proteins such as MED1 (Mediator Complex Subunit 1, 25% of identity), AF9 (ALL1-Fused gene from chromosome 9, 23% of identity), and AF4/FMR2 (ALL1-Fused gene from chromosome 4/Fragile X Mental Retardation, 22% of identity). Numerous IDR-containing proteins are involved in transcription regulation [36], and these three proteins promote RNA polymerase II catalytic activity by being a coactivator (i.e., MED1) or by being part of the Super Elongation Complex (i.e., AF9, AF4/FMR2) [37,38]. Taken together with the discovery of TRIP12 chromatin interaction, TRIP12 may act as a new transcriptional regulator. Liu et al. described an APC (Adenomatous polyposis coli) domain, which is known to be positively charged and involved in microtubules interaction, within TRIP12 IDR [39]. However, the function of this APC domain was not further explored. It is also now well documented that IDRs can confer to proteins the particular biophysical property of the liquid-liquid phase separation (LLPS) by which two liquids form two different phases such as oil droplets in water [40,41]. This biophysical process was linked to genome organization and expression [42].
3.5. Conservation of TRIP12 Protein Domains during Evolution
TRIP12 orthologues were differently named depending on the species. For example, the TRIP12 orthologue is CTRIP (Circadian TRIP) in Drosophila melanogaster, KAKTUS (KAK also known as UPL3), in Arabidopsis thaliana (UniProt (see Section Software and databases)) and Ubiquitin Fusion Degradation protein 4 (UFD4) in Saccharomyces cerevisiae (UniProt). The size of TRIP12 orthologues also varies per species. The largest form is found in Drosophila melanogaster (3140 aa), while the smallest sequence is in Saccharomyces cerevisiae (1483 aa) (Figure 3). The TRIP12 amino-acid sequence is highly conserved in vertebrates, demonstrating 82% identity for the zebrafish to 97.7% for the mouse when compared to the human c isoform. This percentage of identity with the human sequence decreases in invertebrates, 38% for Drosophila melanogaster, 31.4% for Arabidopsis thaliana, 28% for Schmidtea mediterranea, and 27.2% for Saccharomyces cerevisiae. HECT and ARM-repeats domains are found in all species, whereas the WWE domain is found among all vertebrates and Schmidtea mediterranea but not in Saccharomyces cerevisiae and Drosophila melanogaster. The HECT domain, which is required for polyubiquitination, is present in less evolved species, underlying the importance of E3 ubiquitin ligase activity of TRIP12 during evolution. IDRs are fast evolving regions. Their evolution rate is higher than of structured domains due to the lack of 3D structure [31]. Highly conserved IDRs are often found in proteins involved in transcription regulation and DNA binding [31]. An N-terminal IDR is present in all the TRIP12 orthologues mentioned above. According to IUPred (see Section Software and databases), the TRIP12 IDR length varies from 1,042 aa in Drosophila melanogaster CTRIP to 117 aa in Schmidtea mediterranea. Vertebrate TRIP12 orthologues showed an IDR average size of 440 residues. Taken together, the high conservation of TRIP12 domains during evolution assumes essential functions for this protein in living organisms.
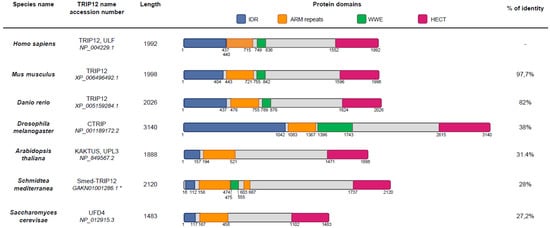
Figure 3.
Schematic representation of protein domains in TRIP12 orthologues. Domain boundaries were defined using InterProScan analysis. Percentage of identity was determined by Emboss matcher (see Section Software and databases) pairwise sequence alignment (using Homo sapiens TRIP12 protein sequence as reference [43]). *: Schmidtea mediterranea cDNA sequence was manually corrected to perform the analysis.
3.6. Post-Translational Modifications of TRIP12
Similar to many other proteins, TRIP12 is post-translationally modified. The PhosphoSitePlus® database (see Section Software and databases) provides an exhaustive list of the post-translationally modified residues identified by mass spectrometry and other approaches (Figure 4). Among the modifications, the most abundant are the phosphorylation and the ubiquitination. TRIP12 can also be acetylated and mono-methylated. TRIP12 IDR is subject to abundant post-translational modifications, since it is a common feature of IDR domains [31], which is in contrast to the ARM repeats-domain that is poorly modified. Only few phosphorylation sites were attributed to specific kinases such as the serine S424, S1078, and S1577 to the kinases CDK1 (Cyclin Dependent Kinase 1), CHK1 (Checkpoint Kinase 1), and ATM/ATR (Ataxia-Telangiectasia-Mutated/Ataxia Telangiectasia and Rad3-related), respectively [44,45,46] (Figure 1). Despite two independent studies that reported the ubiquitination of TRIP12 [47,48] and numerous identified ubiquitinated residues, the ubiquitin ligases responsible for TRIP12 ubiquitination are unknown. Moreover, we cannot exclude that TRIP12 may exhibit functions of self-ubiquitination. It was demonstrated that the Ubiquitin-Specific Peptidase USP7 deubiquitinates and stabilizes the TRIP12 protein [47,48]. Despite numerous studies dedicated to TRIP12, none of the post-translational modifications are shown to function in the regulation of catalytic activity, interacting capacity, nor in the cellular localization of this E3 ubiquitin ligase.
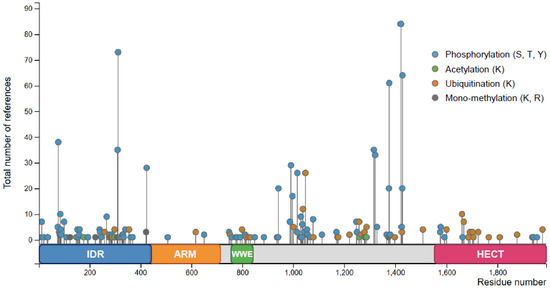
Figure 4.
Post-translational modifications of TRIP12 (isoform c) adapted from PhosphoSitePlus® (see Section Software and databases ). Total number of references represents the number of records in which this modification site was assigned using proteomic discovery mass spectrometry and using other methods. S stands for serine, T stands for threonine, Y stands for tyrosine, and K stands for lysine.
4. Functions of TRIP12 in Ubiquitin-Mediated Proteolysis
A detailed description of ubiquitination process, different ubiquitin ligase families, and functions are excellently reviewed elsewhere [20,49,50,51]. In this section, we will provide an overview of the essential information on TRIP12 actions in ubiquitin-dependent proteolytic pathways.
4.1. Ubiquitination
Ubiquitination is an essential ATP-requiring process leading to protein modification by the covalent conjugation of ubiquitin, which is a highly conserved 76 aa polypeptide (≈8.5 kDa) [49]. Although best known for targeting proteins to degradation by the 26S proteasome, ubiquitination is potentially linked to all cellular processes. Canonical ubiquitin conjugation involves the attachment of the C-terminal glycine 76 (G76) of ubiquitin (Ub) to the ε-amino group of a lysine on the protein substrate. Substrates can be either modified by a single ubiquitin on one (monoubiquitination) or more sites (multi-mono-ubiquitination) or by chains of ubiquitin [51]. In the case of poly (or multi) Ub chains, the C-terminal G76 of one Ub molecule is linked to the ε-NH2 group of one of the seven lysines (K6, K11, K27, K29, K33, K48, or K63) of the preceding ubiquitin. The poly-Ub chains can have different lengths and topologies resulting in different consequences on the fate of the modified protein substrate. For instance, K48-linked polyubiquitination primes proteins for proteolytic destruction by the proteasome, whereas non-K48 linkages target proteins for various cellular functions and responses [50,51].
Ubiquitination is ensured by a three step-concerted action of three enzymes. An Ub-activating enzyme (E1) catalyzes the formation of a thiol-ester linkage between the C-terminal lysine G76 of Ub and the active cysteine (C) residue of E1; the Ub molecule is transferred to the cysteine of the Ub-conjugating enzyme (E2). E3 ligases are classified into three major classes, the RING (Really Interesting New Gene), the RBR (RING-between RING-RING), and the HECT ligases. For HECT and RBR E3 ligases, the transfer of Ub to the substrate lysine-amino group involves a thioester intermediate with a conserved cysteine in the catalytic site of the HECT domain or the Rcat domain, respectively [20]. The RING E3 ligases catalyze the direct transfer of ubiquitin from a thioester-linked E2-ubiquitin conjugate to a substrate [20]. In some cases, the cooperation of an additional E4 ubiquitin ligase, which may encode for independent ligase activity or enhance the processivity of the E3, is required. To act as catalysts, E3s recruit their substrates for degradation by recognizing a specific portion in the protein sequence or degron. Ubiquitin conjugation can be reversed by deubiquitinating enzymes (DUBs) [52].
4.2. Known TRIP12 Substrates
Based on the literature, TRIP12 regulates the stability and promotes the proteolysis of nine protein substrates: ASXL1 (Additional Sex Combs Like 1) [53], USP7 (Ubiquitin-Specific Peptidase 7) [39], PTF1a (Pancreas Transcription Factor 1a) [6], SOX6 (SRY-Box Transcription Factor 6) [12], RNF168 (Ring Finger Protein 168) [14], BAF57 (BRG1-Associated Factor 57) also known as SMARCE1 (SWI/SNF Related, Matrix Associated, Actin-Dependent Regulator Of Chromatin, Subfamily E, Member 1) [54], P14/ARF (P14/Alternate Reading Frame) also known as CDKN2A (Cyclin Dependent Kinase Inhibitor 2A), for which TRIP12 was named ULF (Ubiquitin Ligase for ARF) [15], APP-BP1 (Amyloid Protein-Binding Protein 1) also known as NAE1 (NEDD8-Activating Enzyme E1 Subunit 1) [4] and PARP1 (Poly-(ADP ribose) polymerase 1) [27]. These proteins exert important functions in biological and/or pathological processes that will be outlined in Section 5.
The TRIP12 HECT domain (detailed in Section 3.4) is essential for the degradation of these proteins, but the role of TRIP12 as direct interacting E3 ligase is still questionable for ASXL1 [53] and RNF168 [14] for which experimental approaches that prove ubiquitination by TRIP12 are lacking. TRIP12 polyubiquitinates its substrates with the exception of APP-BP1, which is monoubiquitinated [4]. APP-BP1 polyubiquitination requires an additional E4 activity [4]. TRIP12 mediated-K48-linked polyubiquitination was demonstrated only for USP7 [39] and PTF1a [6]. While the interaction between the E3 ligase and its substrates was validated, the interaction domains of TRIP12 have been mapped only in few cases. Although these studies used different isoforms of TRIP12, they all emphasize the role of consensus protein/protein interaction domains of TRIP12. A region (aa 611–1259 of isoform c) that includes the WWE domain but not ARM repeats is responsible for the interaction with APP-BP1 [4]. A sequence of 577 aa (aa 234–810 of isoform a) preceding the WWE domains interacts with USP7 [39]. In addition, TRIP12 interacts with parylated PARP1 via its WWE (aa 797–911 of isoform a) domain. TRIP12 is freshly considered as an identified PAR-targeted ubiquitin ligase (PTUbL) controlling PARP1 turnover [27]. Finally, we demonstrated that PTF1a binds a region containing ARM repeats and the WWE domain (aa 446–1552 of isoform c) [6].
Interacting domains or ubiquitinated residues were identified for certain proteins regulated by TRIP12. A sequence of 80 aa within and flanking the central coiled-coil region of BAF57 was found to be critical for BAF57 stabilization and to contain lysine residues essential for ubiquitination [55]. The N-terminal part of Tumor necrosis factor-receptor associated factor (TRAF), also known as the Meprin And TRAF-C Homology (MATH) domain of USP7, is responsible for its interaction with TRIP12 [39]. We found that the lysine 312 of PTF1a is essential for its TRIP12-mediated degradation [6]. It was previously reported that the tumor suppressor ARF is a natural lysine-less protein that undergoes N-terminal polyubiquitination [56].
Intriguingly, TRIP12 targets several proteins that are unbound to their physiological binding partners. This is the case for BAF57, which is a subunit of the SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complex (detailed in Section 5.4.) [54]. Most if not all SWI/SNF subunits are assembled into a complex for which maintenance of the stoichiometry is essential and no free subunits exist in the cell; otherwise, they are degraded. The stabilization and protection of BAF57 from proteasome-mediated degradation operates via protein–protein interaction with the BAF155 subunit [57]. TRIP12 was demonstrated to compete with BAF155 for BAF57 and to degrade only an unbound form of BAF57 [55]. The same type of interaction was found for the ubiquitination of the neddylation E1 enzyme subunit APP-BP1 [4]. In fact, TRIP12 does not interact with APP-BP1 in the heterodimeric form bound to Uba3 (Ubiquitin-Like Modifier-Activating Enzyme 3), which is the specific active E1 of the NEDD8 pathway. A similar scheme is described for ARF, which is stabilized and protected from TRIP12 ubiquitination when sequestered by nucleophosmin and TRADD (NFR1-Associated Death Domain Protein) in the nucleolus (detailed in Section 5.2.) [15,58]. Indirect evidence suggests a similar mode of action for the degradation of RNF168 [14]. Indeed, the destabilization of RNF168 induced by a silencing of its binding partner HERC2 (HECT and RLD Domain Containing E3 Ubiquitin Protein Ligase 2) could be mitigated by a depletion of TRIP12. The TRIP12-ubiquitinated lysine 312 of PTF1a is located near the domain interacting with RBP (mammalian suppressor of Hairless) essential for the transcriptional activity of PTF1a. One may hypothesize that free PTF1a could be ubiquitinated, while binding to RBP could prevent TRIP12-mediated PTF1a degradation. Therefore, TRIP12 appears as a regulator of the balance between bound and unbound forms of its protein substrates to maintain them in their complexed forms.
4.3. Role of TRIP12 in Ubiquitin Fusion Degradation and N-Degron Pathways
TRIP12 is sequelogous to the UFD4 HECT E3 ligase, which catalyzes the ubiquitination of Ubiquitin Fusion Degradation (UFD) substrates in yeast [59]. These substrates are artificial fusion proteins consisting of an N-terminal uncleavable ubiquitin moiety that acts as a degron. The UFD pathway consists in the poly-ubiquitination of the uncleavable N-terminal ubiquitin. TRIP12 was shown to mediate the degradation of the aberrant form of ubiquitin named UBB+1 [60]. UBB+1 is a physiological human UFD substrate resulting from a dinucleotide deletion in the mRNA of the ubiquitin B gene. UBB+1 accumulates in the brain in neurodegenerative disorders [61]. The HECT E3 HUWE1 (HECT, UBA, and WWE domain containing E3 ubiquitin protein ligase 1) was reported to cooperate with TRIP12 for the degradation of UBB+1 [62]. Although the conservation of the UFD pathway from yeast to human attests its importance for proteolysis in cells, except for UBB+1, no other native substrates have been identified. Most studies use artificial substrates for specific applications. In particular, enhanced immune response after UFD ubiquitination of antigens opens up interesting perspectives in the development of tumor vaccines [63].
The N-degron pathway (formerly N-end rule pathway) is another ubiquitin-dependent proteolytic pathway that regulates the half-life of proteins according to N-terminal degradation signals or N-degrons. UBR1, UBR2, UBR4, and UBR5 (ubiquitin protein ligase E3 component N-recognin) are the four E3s (or N-recognins) encoded by the mammalian genomes that can recognize Arg/N-degrons [64]. Interestingly, a study demonstrated that UBR1 and UFD4 E3s interact and cooperate to enhance substrate ubiquitination in both pathways in yeast [65]. This mutually cooperative physical interaction between E3s operating in parallel pathways are likely to be relevant to all eukaryotes and might apply to TRIP12 and UBR5 control of RNF168 accumulation [14].
5. The Physio-Pathological Roles of TRIP12
Proteomic approaches have allowed for the identification of several TRIP12 binding partners (Figure 5 and Table 2). However, the molecular function of only a restricted number of these interactions is known. Thus far, TRIP12 was implicated in the regulation of several important molecular pathways such as cell cycle progression, DNA damage repair (DDR), chromatin remodeling, and cell differentiation via a small number of identified protein interactors, which leaves wide open the field of investigation of molecular processes implicating TRIP12.
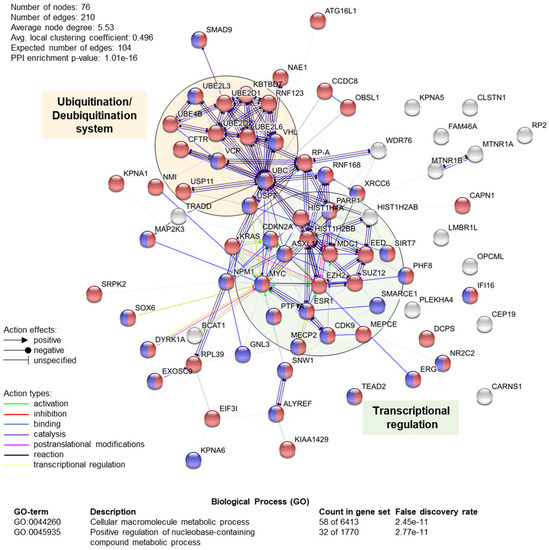
Figure 5.
Schematic representation of human TRIP12-interacting protein–protein network (n = 76) and functional enrichment analysis using STRING software (see Section Software and databases). Network nodes represent proteins. Red nodes represent proteins belonging to cellular macromolecule metabolic process (GO term: 0044260), and purple nodes represent proteins belonging to the positive regulation of the nucleobase-containing compound metabolic process (GO-term: 0045935). Edges represent protein–protein molecular actions (indicated in the figure). Line shape indicates the predicted mode of action (indicated in the figure). Active interaction sources: text mining, experiments, databases, co-expression, neighborhood and co-occurrence. Minimum required interaction score: medium confidence (0.400). Green and orange filled circles indicate proteins implicated in “transcriptional regulation” and “ubiquitination/deubiquitination system”, respectively.
Table 2.
List of the different protein interactors of human TRIP12.
5.1. TRIP12 Protein Interactors
According to BioGrid resources (see Section Software and databases) and the literature, 76 unique proteins interact with human TRIP12 (Table 2). They were identified by different experimental approaches using either these proteins (n = 58), either TRIP12 (n = 7) or both as bait (n = 11). Among them, eight were further classified as TRIP12-ubiquitinated substrates (detailed in Section 4.2). A STRING protein–protein network and functional enrichment analyses (see Section Software and databases) reveals that 76% (58 out of 76) are implicated in cellular macromolecule metabolic process (red circles), which corresponds to proteins involved in the adding of low molecular mass protein units (i.e., ubiquitin) (Figure 5). In parallel, 42% (32 out of 76) are involved in the positive regulation of nucleobase-containing compound metabolism (purple circles), which corresponds to cellular processes that activate chemical reactions/pathways involving nucleic acid-related compounds. Moreover, we can clearly distinguish two protein-protein functional clusters including proteins involved in transcriptional regulation (i.e., PTF1a, EZH2, SUZ12, etc.) and in the ubiquitination/deubiquitination system (i.e., USP7, VHL, USP11, etc.). However, it is important to keep in mind that all the interactions of the TRIP12 with protein partners might depend on the type of cells, the stage of differentiation, or the cell cycle phase. Altogether, these analyses position TRIP12, via its interacting partners, as a potential central regulator of protein stability and genome expression.
5.2. Roles in Cell Cycle Progression
The P53 dependent-activating P14/ARF (P14/Alternative Reading Frame also known as CDKN2A (Cyclin-Dependent Kinase Inhibitor 2A)) pathway plays a major activating role in cell cycle progression. Chen et al. identified TRIP12 as the E3 ubiquitin ligase responsible for the degradation of ARF and renamed it ULF for Ubiquitin ligase for ARF [96]. TRIP12 decreases the stability of ARF after polyubiquitination and degradation by the proteasome. This leads to a proteasome-mediated destabilization of P53 by the E3 ubiquitin ligase MDM2 (Mouse Double Minute 2 homolog) and a cell cycle progression (Figure 6, left panel). The authors further showed the relationship between TRIP12 and two positive regulators of ARF: the nucleophosmin (NPM), a major component of nucleoli, and the transcription factor c-MYC. They reported that NPM sequestrates ARF in the nucleoli, isolating it from the ubiquitinating action of TRIP12 present only in the nucleoplasm (Figure 6, left panel). Then, they demonstrated that c-MYC enhances the ARF half-life by destabilizing the TRIP12/ARF nucleoplasmic complex (Figure 6, right panel). Therefore, the interaction of ARF with MDM2 prevents the ubiquitination-mediated degradation of P53, which in turn, induces a cell cycle arrest and apoptosis [114]. Later, the nucleostemin (also known as Guanine nucleotide-binding protein-like 3) was demonstrated to stabilize the NPM/ARF complex in the nucleoli and to destabilize the TRIP12/ARF complex in the nucleoplasm to inhibit ARF degradation by TRIP12 ubiquitination [75]. The translocation of TRADD (TNF-R Associated Death Domain Protein) from the cytoplasm to the nucleus was also shown to protect ARF from degradation by inhibiting its interaction with TRIP12 [58]. The participation of TRIP12 in cell cycle progression was assessed in non-cancerous cells subject to an oncogenic stress provoked by an overexpression of c-MYC [115]. Paradoxical properties of c-MYC overexpression have been reported in oncogenic processes [116]. Indeed, a mild expression of c-MYC stimulates cell proliferation, whereas a strong overexpression inhibits cell proliferation by activating the ARF/P53 response. In both cases, ARF mRNA levels were induced. However, the authors demonstrate that only a high concentration of c-MYC is able to inhibit the degradation of ARF mediated by TRIP12, which in turn leads to the activation of the ARF/P53 pathway and cell cycle arrest. Moreover, Kajiro et al. generated TRIP12 knock-in mutant mice by homozygous mutation in TRIP12 exon 33 (TRIP12mt/mt) disrupting the ubiquitin ligase activity [5]. This resulted in embryonic lethality at the embryonic stage E11.5. TRIP12mt/mt ES cells exhibited a significant decrease of growth rate caused by the abnormal induction of the cyclin-dependent kinase inhibitor P16 and ARF associated to a P53 protein increased stability. TRIP12mt/mt ES cells also display an abnormal accumulation in G2/M and sub-G1 phases. These data support that TRIP12 functions in the cell cycle control in vivo.
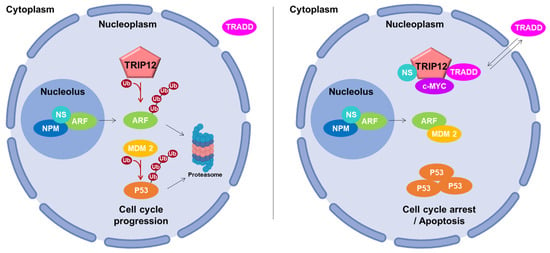
Figure 6.
Schematic representation of TRIP12, ARF, and P53 pathway. On the left, TRIP12 controls ARF and indirectly P53 ubiquitination degradation by proteasome, while TRIP12 participates in cell cycle progression [58,75,96,115]. On the right, ARF is protected from TRIP12 when bound to nucleophosmin (NPM)/nucleostemin (NS) and localized in the nucleolus. Moreover, in the nucleoplasm, TRIP12 is sequestered by different proteins (in the presence of NS or TRADD and in high level c-MYC condition), blocking its interaction with ARF. ARF binds and inhibits MDM2, which leads to P53 accumulation and cell cycle arrest.
TRIP12 is tightly implicated in the control of cell cycle progression. Until very recently, little was known on cell cycle regulation of TRIP12 expression. We showed that the presence of TRIP12 in the nucleus strongly depends on the phase of the cell cycle. The TRIP12 protein appears in the nucleus in the late S phase and endures during the G2 phase [7]. We found that while tightly bound to the chromosomes during mitosis, TRIP12 is rapidly degraded in the early G1 phase and disappears in the nucleus in the late G1 and early S phase. We further demonstrated that TRIP12 plays a role in phase S by controlling DNA replication timing. Via its chromatin-interacting IDR, TRIP12 coordinates mitotic entry and progression [7]. The important function of TRIP12 during mitosis was reinforced by mass spectrometry-based approaches showing that TRIP12 is specifically retained to mitotic chromatin [117]. With regard to variations to TRIP12 expression during the cell cycle, it would be important to reconsider TRIP12 molecular functions according to its presence or absence from the nucleus.
5.3. Roles in DNA Damage Response
5.3.1. Via the ARF/P53 Signaling Pathway
Oncogenic stimuli trigger the activation of checkpoints that delay or prevent tumorigenesis. The DNA damage repair (DDR) pathway and ARF protein are two major checkpoints that both act by the protein P53. Initially studied independently, the interconnection between these two checkpoints in tumorigenesis is gaining notoriety. For example, Velimezi et al. discovered that the key DDR kinase ATM negatively regulates the abundance of ARF with the implication of TRIP12 [118]. In response to DNA damage, the kinase ATM activates the protein phosphatase 1 (PP1), which in turn, dephosphorylates the nucleolar protein NPM (Figure 7). This dephosphorylation liberates ARF from NPM, rendering it susceptible to degradation via the action of TRIP12 in the nucleoplasm. Chen et al. further reported the contribution of TRIP12 in ARF degradation in response to DNA damage induced by doxorubicin [115]. The data showed that after DNA damage, the degradation of ARF is strongly attenuated in TRIP12 knockout mouse embryonic fibroblasts (MEFs). Another study identified the serine S1577 of TRIP12 (Figure 1) as phosphorylated by the kinases ATM/ATR, which suggests that post-translation modifications are possibly critical for a proper implication of TRIP12 in the DDR network [45].
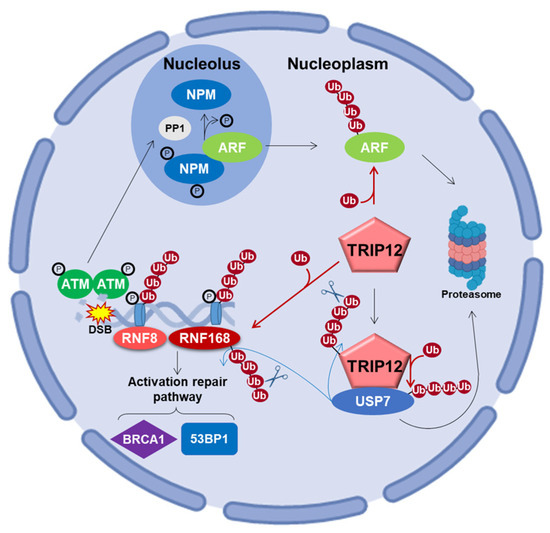
Figure 7.
Schematic representation of TRIP12 in the DNA damage repair (DDR) pathway in response to DNA double-strand break (DSB) [14,39,115,118,119,120,121]. In response to double-strand breaks (DSB), ATM, RNF8, and RNF168 are recruited on the damage and trigger the propagation of the ubiquitination of H2A and H2AX far from the initial DNA lesions. TRIP12 controls the nuclear pool of RNF168 for the ubiquitination of chromatin after DNA disruption. Indeed, TRIP12 ubiquitinates and induces the degradation of RNF168 by the proteasome. TRIP12 controls Ubiquitin-Specific Peptidase 7 (USP7) ubiquitination, which in turn stabilizes it.
5.3.2. Via the Control of Histone Ubiquitination Spreading by RNF168
Histone ubiquitination plays a crucial role in the recruitment of proteins responsible for the detection and the repair of DNA damage. The E3 ubiquitin ligase RNF8 (Ring Finger Protein 8) initiates the ubiquitination of histones H2A and H2AX at the damaged sites (Figure 7) [119]. This priming of ubiquitination reaction is amplified by another E3 ubiquitin ligase, RNF168. The produced polyubiquitination (K63 chain) chain acts as a platform for the interaction of repair signaling factors, such as 53BP1 and the complex RAP80/BRCA1 (Receptor-Associated Protein 80/Breast Cancer 1) [122]. If not properly controlled, RNF168 over-amplifies the ubiquitination signal beyond the DNA breakage on neighboring chromatin, which can have dreadful consequences, notably by sequestrating the genome keepers 53BP1 (P53 Binding Protein 1) and BRCA1. The retention of 53BP1 and BRCA1 at a non-damaged DNA site prevents their recruitment at damaged DNA sites and subsequent repair. It was shown that TRIP12 and UBR5 are two proteins that prevent this over-amplification after the DNA damage induction of double-strand break (DSB) [14]. In physiological conditions, TRIP12 and UBR5 control the quantity of RNF168 required to restrain histone ubiquitination around DNA lesions (Figure 7). A depletion of TRIP12 and UBR5 provokes an accumulation of RNF168 to supra-physiological levels that amplifies histone ubiquitination beyond the DSB sites. Moreover, TRIP12 was demonstrated as the E3 ubiquitin ligase implicated in the stability of RNF168.
5.3.3. Via the Deubiquitinase USP7
It is accepted that the deubiquitinase USP7 plays a major role in the response of DNA damage by stabilizing the expression of key proteins such as P53 and RNF168 [120,121]. TRIP12 interacts with USP7 and polyubiquitinates (K48 chain) it to induce its degradation by the proteasome (Figure 7) [39]. Consequently, the TRIP12-mediated degradation of USP7 affects the stability of 53BP1 and CHK1 proteins, which are two important regulators of DDR regulated by USP7. Importantly, as mentioned in Section 3.6, the TRIP12 protein is also stabilized by USP7, suggesting a relevant interconnected feedback between these proteins. Taken together, these studies show that TRIP12 and USP7 mutually control their expression level. In addition, they both regulate the expression level of RNF168, which is a key factor in DSB repair. This indicates that a fine-tuning of these three enzymes is certainly required to ensure a proper managing of DNA damage repair via RNF168 quantity control.
5.4. The Roles of TRIP12 in Chromatin Remodeling
5.4.1. Via the Control of SWI/SNF Complex Integrity
The level of chromatin compaction participates in highly dynamic processes such as transcription, DNA repair, and recombination. When the chromatin is compacted, genes are globally transcriptionally inactive due, in part, to a low accessibility of the transcriptional machinery to promoters [123]. The modification of chromatin compaction and consequently the modulation of transcriptional activity is partially controlled by the SWI/SNF complex [124]. It is a multi-protein complex constituted by a dozen sub-units. The sub-units BRG1 (hSWI/SNF Brahma-related Gene) and BRM-1 (BRachyury Modifier 1) ensure the ATPase catalytic activity. The function of the SWI/SNF complex is to remodel nucleosome positioning by sliding, inserting, or ejecting histone octamers into DNA in an ATPase-dependent manner. As such, it controls numerous cellular processes [125]. To reach an optimal efficiency, the SWI/SNF complex requires the presence of BAF proteins (BRG-1 associated factors), which interact with BRG-1. Therefore, the regulation of expression of BAF proteins by proteasome-dependent degradation is crucial. It was demonstrated that TRIP12 is the main E3 ubiquitin ligase that polyubiquitinates the free cellular pool of BAF57 (also known as SMARCE1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily E member)) by a mechanism that we described in Section 4.2. [54]. Therefore, TRIP12 appears to be involved in a protein quality control system of the SWI/SNF complex. The TRIP12 mediated-degradation of BAF57 could affect the loading of SWI/SNF on its target genes and consequently disturb gene expression.
5.4.2. Via the Maintenance of Silenced Genes by the Polycomb Complex
Epigenetic modifications of DNA bases play a crucial role in the control of gene transcription. Most of these modifications were first identified in prokaryotes and then in higher eukaryotes. The methylation of the nitrogen atom in position 6 of an adenine (6mA) is the most frequent modification in prokaryotes. This modification was recently described in eukaryotes and particularly in vertebrates [126]. In mammals, the methyltransferase METTL44 (Methyltransferase Like 4) and the dioxygenase ALKBH4 (ALKB Homolog 4, Lysine Demethylase) are responsible for the deposit and the suppression of the 6mA on DNA, respectively [53]. In mammals, the 6mA is associated with epigenetic repressive marks such as the mono-ubiquitination on the K119 of histone H2A (K119 Ub-H2A) and the trimethylation on K27 of histone H3 (K27me3-H3). These repressive marks are used for silencing promoters during the stem cells differentiation and embryonic development. They are mainly added on the chromatin by the Polycomb Repressive Complexes (PRC1 and PRC2) [127,128]. These two marks are reversible. The K119 Ub-H2A is removed by the deubiquitinase complex PR-DUB (Polycomb Repressive-Deubiquitinase complex) formed by the proteins ASXL1 (Additional Sex Combs like 1) and BAP1 (BRAC1 Associated Protein 1) [129]. The BAP1 protein bears the deubiquitinase activity, and it is stabilized by ASXL1 [130]. Mass spectrometry based-proteomic analyzes identified the protein-protein interaction between TRIP12 and major components of PRCs such as SUZ12 (Polycomb protein SUZ12), EZH2 (Histone-lysine N-methyltransferase) [84] and EED (Embryonic Ectoderm Development protein) [80] but with no mechanistic findings. A recent study characterized the role of 6mA deposit and the implication of the E3 ubiquitin ligase TRIP12 in the maintenance of the repressive marks added by the Polycomb complex [53]. The addition of 6mA on PRC1/PRC2-inactivated genes leads to the recruitment of the PR-DUB complex. This recruitment on the chromatin leads to the degradation of the complex by TRIP12-mediated polyubiquitination of ASXL1 (discussed in Section 4.2.) and preserves the level of K119 Ub-H2A. A homozygous deletion of the METTL4 gene in a murine model induces a decrease of 6mA deposit and provokes a craniofacial dysmorphism in a subset of pups. Interestingly, de novo nonsense mutations of the ASXL1 gene were identified in patients with Bohring–Opitz syndrome who display facial dysmorphism [131]. In humans, mutations of the TRIP12 gene are also associated to craniofacial dysmorphism (detailed in Section 6.1). Therefore, we can speculate that an inactivation of TRIP12 could provoke an accumulation of ASXL1 and a stabilization of the PR-DUB complex that would subsequently alter the expression of genes implicated in facial development.
5.5. Roles in Cell Differentiation
Different studies have proven the strong involvement of TRIP12 in the cell differentiation. Differentiation is the process by which dividing cells change their functional or phenotypical type. It involves the coordinated regulation of genes by a multitude of cellular pathways. The first study showing the participation of TRIP12 in stem cells and regeneration mechanisms was performed in the freshwater planarian Schmidtea mediterranea [132]. Schmidtea mediterranea is a model for regeneration, stem cells, and the development of tissues. This worm is able to replace cell loss during normal cellular turnover or tissue loss through the regenerative capacity of adult stem cells called neoblasts. The authors demonstrated that 11 genes of HECT E3 ubiquitin ligases were expressed in neoblasts. Using RNA interference (RNAi), the authors showed that the TRIP12 orthologue also known as “smed-TRIP12” is essential for the establishment and the maintenance of posterior tissues.
TRIP12 displays another important function in the control in muscle fiber homeostasis. An et al. showed that the transcription factor SOX6 (SRY-Box Transcription Factor 6) is a target of TRIP12 in mammals. SOX6 is involved in muscle fiber homeostasis by the transcriptional repression of “slow switch muscle fiber” genes in fetal mice [133]. They showed that a TRIP12 knockdown in myotubes-derived cell lines leads to an increase of SOX6 protein levels and results in a dysregulation of muscle fiber homeostasis. TRIP12 binds to SOX6 protein, inducing its polyubiquitination and its degradation by the proteasome [12].
TRIP12 is also implicated in the homeostasis of pancreatic epithelial cells. We demonstrated that TRIP12 polyubiquitinates and provokes the degradation of the transcription factor PTF1a (Pancreas Transcription Factor 1a) by the proteasome [6]. PTF1a is a transcription factor that controls the expression of pancreatic acinar specific genes such as amylase and chymotrypsin genes. This factor is essential in pancreatic development and pancreatic cell homeostasis by maintaining the acinar phenotype. The expression of PTF1a is lost during acinar to ductal cells transdifferentiation [133,134]. PTF1a is also required for the specification of inhibitory neurons in various regions of the developing central nervous system including the spinal cord, brainstem, cerebellum, and retina [135,136,137]. Thus, it is possible that TRIP12 regulates neuronal specification via PTF1a degradation.
6. TRIP12 in Pathologies
6.1. Alterations of TRIP12 Gene in Autism and Intellectual Disability
TRIP12 (OMIM 604506 (Online Mendelian Inheritance in Man) (see Section Software and databases)) is a neurodevelopmental disorder (NDD)-associated gene. Several publications allowed its identification as a primary gene for Intellectual Disability (ID), which is the most common developmental disorder affecting 1–3% of the world’s population. Most IDs are monogenic with 1,396 genes causing ID [138]. The TRIP12 gene is referenced as a causative gene associated with Clark–Baraitser syndrome (CLABARS; OMIM617752), formerly autosomal-dominant mental retardation-49 (MRD49) in the OMIM database. Although originally thought to be an X-linked disorder [139], Clark-Baraitser syndrome was shown to have an autosomal dominant mode of inheritance [140]. In addition to mental retardation, craniofacial dysmorphism, behavioral psychiatric manifestations, and obesity are the main clinical features of the syndrome. CLABARS is caused by heterozygous mutation in the TRIP12 gene.
The first indication of a putative role of the TRIP12 gene in ID was the report of a large interstitial ≈5.6-Mb deletion on the long arm of chromosome 2 (2q36.2q36.3) in a 17-year-old girl. Twenty-four genes were located in the deleted fragment where TRIP12 was disrupted. The phenotype included severe mental retardation, facial dysmorphism, and multiple renal cysts [141]. The identification of TRIP12 among 10 new ID genes was later accomplished by meta-analysis of large-scale exome sequencing data of a combined cohort including 2104 patient-parent trios [142] where two different de novo heterozygous mutations in the TRIP12 gene were identified in two unrelated patients.
Of importance, several de novo mutations (DNM) of the TRIP12 gene were associated with a broader phenotype than ID in line with the overlap of genes and pathways involved in NDDs. Mutual comorbidities of ID and Autism Spectrum Disorder (ASD) also raised the question of whether TRIP12 can be validated as a gene associated with ASD. Resequencing sixty-four ASD risk genes in two ASD cohorts newly identified mutations in the TRIP12 gene, thus proposing that it could be a novel candidate gene for clinical follow-up [143,144]. Recent detailed clinical information characterizing the phenotype of 11 individuals with mild to moderate ID or learning disability finally demonstrated that TRIP12 is a risk gene for ID with (8/11) or without ASD [10]. However, there is no correlation for the presence or absence of autistic features with the type of mutations in this study. Other typical clinical features of the TRIP12-associated spectrum such as unspecific craniofacial dysmorphism, speech delay, and seizures were reported in another cohort of nine pathogenic variants that further documented that TRIP12 haploinsufficiency causes childhood-onset neurodevelopmental disorders [145]. Recent exome sequencing of Japanese trios and integrative analysis conducted by combining published DNM data identify TRIP12 among 61 significant genes for ASD and suggest that it is a genuine disease-causative gene [146]. The Trip12 gene is a high confidence gene for autism in the SFARI GENE database (see Section Software and databases).
Pathogenic TRIP12 variants are now documented in gene variant databases related to human health LOVD (Leiden Open Variation Database (see Section Software and databases)) and ClinVar (Clinical Variation (see Section Software and databases)) and can be also found in ID/autism websites. According to the LOVD, most pathogenic variants (87%) have DNA changes in coding regions, 8% in spliced regions, 5% in multiple regions. These changes are substitutions (53%), deletions (29%), duplications (13%), and 5% of these variants have still unknown modifications. Consequences at the protein level include stop changes (32% of variants) and frame shifts (26%) generating premature termination, missense changes (18%), no protein production (8%), or unknown (16%). Selected variants are placed on the TRIP12 protein sequence according to the transcript variant 3, NM_004238.3 (Figure 8).
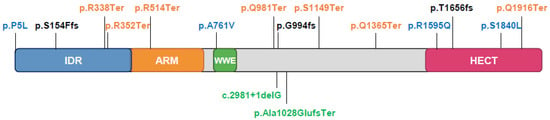
Figure 8.
Schematic illustration of published TRIP12 gene mutations in intellectual disorders. Mutations in blue are missense, mutations in orange are nonsense, mutations in black are frameshift. Mutations in green are located in splice donor sites. From [10,142,143,144,145].
Analysis was performed to identify protein/protein interaction networks and signaling pathways associated with a set of autism and ID genes. These frequently implicate neuronal signaling and development, chromatin remodeling, transcriptional regulation, mRNA splicing, ion transport, cell cycle, and microtubule cytoskeleton. They converge on neuronal and cognitive functions such as the regulation of synaptic transmission, learning, and memory [146,147,148]. To date, only one pilot study in Drosophila melanogaster investigated the role of TRIP12 using inducible RNA interference. This study reported no significant change in behavioral assay after TRIP12 knock-down, suggesting no role in the development of the nervous system [148]. Given the important function for protein degradation in the nervous system, the synaptic plasticity, and the maintenance of neuronal structures, one might speculate that TRIP12 regulates the synaptic proteome. However, physiological roles of TRIP12 suggest its potential contribution to other essential pathways that are deregulated in ID pathologies. Therefore, additional functional analysis using relevant models is necessary to conclude a causal association between TRIP12 gene variants and these diseases.
6.2. Alterations of TRIP12 Gene, mRNA and Protein Expression, and Function in Cancers
6.2.1. Alteration of TRIP12 Gene in Cancers
The cBioPortal database (see Section Software and databases) provides the frequency of CNA (Copy Number Alteration) appearance in TRIP12 gene in cancers [149,150]. It includes mutations, fusions, amplifications, deep deletions, and multiple alterations (Figure 9). On average, the TRIP12 gene is altered in 1.84% of cancer patients (over 27,235 patients). This rate remains relatively weak compared to other genes, for instance, TP53 (39.8%). TRIP12 gene alteration frequency varies from 11.7% in endometrial carcinoma to 1.06% in glioblastoma patients. Up to now, 463 mutations have been located in the human TRIP12 gene (Figure 10). These punctual alterations do not accumulate in hot spots but rather spread all along the protein sequence. Among them, 323 were classified as missense, 120 were classified as truncating mutation, and one arises from nucleotides insertion without an open reading frameshift. Additionally, 20 alterations result from a fusion between the TRIP12 gene and other genes such as PID1 (PTB-containing, cubilin and LRP1-interacting protein) or KIF1A (Kinesin-like protein). However, a nonsense mutation is retrieved 10 times at the residue S1195 generating a truncated protein: six times in colon cancer, two times in uterine cancer, and once in lung and breast cancer. The main issue regarding TRIP12 gene alterations and in particular point mutations is whether these mutations play a causal role in cancer initiation or progression (“driver” mutations) or if they only randomly appear during cancer establishment as a consequence of, for instance, DNA repair failure (“passenger” mutations).
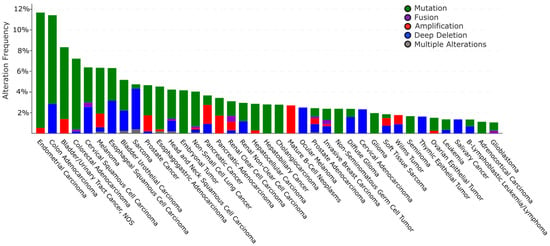
Figure 9.
Copy number alterations (CNA) and mutations of the TRIP12 gene in human cancers in cBioPortal (see Section Software and databases) obtained from 176 non-redundant studies containing more than 100 cases with both mutations and CNA data (27,235 patients and 28,253 samples). Only cancer types with more than 1% of altered cases are represented.
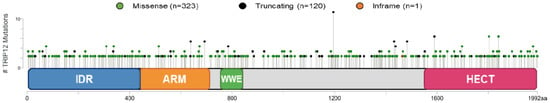
Figure 10.
Schematic representation of mutations on the TRIP12 gene in human cancers in cBioPortal (see Section Software and databases) obtained from 176 non-redundant studies containing more than 100 cases with both mutations and CNA data (27,235 patients and 28,253 samples). Mutation diagram circles are colored with respect to the corresponding mutation types indicated in the legend. In case of different mutation types at a single position, the color of the circle is determined with respect to the most frequent mutation type. Truncating mutations include nonsense, nonstop, frameshift deletion, frameshift insertion, and splice site. Inframe mutations include inframe deletion and inframe insertion.
Only six publications have reported alterations in the TRIP12 gene in cancers. Frameshift mutations of genes containing mononucleotide repeats are features of gastric and colorectal carcinomas with microsatellite instability (MSI). The TRIP12 gene contains repetitions of seven adenines in exon 3 and seven thymidines in exon 24. In a cohort of 27 gastric and 32 colorectal cancer samples, Yoo et al. identified an adenine insertion in the exon 3-adenine repetition in one case of gastric cancer and a thymidine deletion in the exon 24-thymidine repetition in one case of colorectal cancer [151]. These frameshift mutations predict the formation of a truncated TRIP12 protein with no catalytic activity. Unfortunately, the authors did not investigate the functional consequences of this insertion. Another study aiming to identify the genetic loci with an elevated risk of breast cancer related to menopausal hormone therapy (MHT) in post-menopausal women showed a strong linkage between a Single Nucleotide Polymorphism (SNP) located in the exon 1-2 intronic region of the TRIP12 gene and MHT [152]. However, they were not successful in replicating TRIP12 SNP × MHT interaction in an independent cohort. Corresponding analyses of public data of global RNA expression were obtained from 12 lung adenocarcinomas and six controls to identify mutated genes with high-frequency-risk expression [153]. They identified the TRIP12 gene as the most frequently mutated (missense mutation on exon 16) (nine cases out of 12). A recent study reported somatic mutations of the TRIP12 gene in three out 20 cases of HPV-positive anal squamous cell carcinoma (HPV-ASCC) [154]. A missense mutation, a frameshift internal deletion, and a nonsense mutation were identified on exons 3, 37, and 33, respectively. However, the functional consequences of these mutations were not further investigated. A RNA-seq analysis of 139 patients with pediatric Acute Myeloid Leukemia (AML) revealed the existence in one patient of a new gene rearrangement leading to the fusion between TRIP12 and NPM (nucleophosmin) proteins [155]. Although the properties of this new fusion protein were not further studied, the authors suggest that the TRIP12-NPM protein may inhibit the function of ARF. TRIP12 somatic mutations at splice sites were identified in pancreatic cancer recurrence of two patients but not in matched originally resected primary tumor and were qualified as subclonal mutations [156].
Altogether, these studies suggest that although the TRIP12 gene is found mutated in various type of tumors at variable frequencies, the functional consequences of these mutations in cancer development remain largely unidentified. Possible applications to detect these mutations as diagnostic, prognostic, or predictive still remain to be demonstrated.
6.2.2. Alteration of TRIP12 mRNA Expression in Cancers
If we refer to the Gepia2 database (see Section Software and databases), the TRIP12 mRNA level compared to normal corresponding tissues varies depending on the type of cancer. Indeed, among 27 different types of cancer, the down-regulation of TRIP12 mRNA is found in 12 cancer types compared to normal corresponding tissues but reached a statistical importance only in testicular germ cell tumors. In contrast, an overexpression of TRIP12 mRNA is found in 15 types of cancer with a statistical significance for lymphoid neoplasm diffuse large B-cell lymphoma, thymoma and pancreatic adenocarcinoma (Figure 11). Of note, the TRIP12 mRNA level is decreased in tumor types for which TRIP12 mRNA is elevated in normal corresponding tissue, and inversely, it is up-regulated in tumors for which the TRIP12 mRNA level is low in corresponding normal tissues (i.e., pancreatic adenocarcinoma). However, the mechanisms responsible for the alteration of TRIP12 mRNA in tumor samples remain largely unknown. In certain types of cancer, a high level of TRIP12 mRNA is associated with a shorter disease-free survival (i.e., bladder urothelial carcinoma, pancreatic adenocarcinoma), which suggests that the TRIP12 mRNA level could be a useful prognostic marker. Additionally, RNA-seq analysis of PBMC (Peripheral Blood Mononuclear Cells) obtained from a 47-year-old female patient with Acute Myeloid Leukemia (AML) with complete remission before and after induction therapy (fludarabine, cytarabine, and G-CSF (FLAG) protocol) revealed, among others, an exon 3 skipping in the TRIP12 gene [157]. The authors claim that the alternative splicing of TRIP12 mRNA could inhibit the TRIP12-mediated degradation of ARF, leading to an activation of the ARF/P53 signaling pathway and an induction of apoptosis of cell with aberrant growth. This process could contribute to the remission.
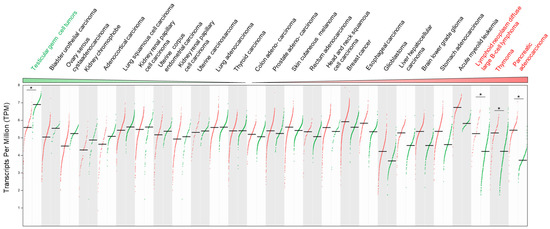
Figure 11.
TRIP12 mRNA expression level in 27 different types of cancer adapted from Gepia2 website (see Section Software and databases). Only tumor/normal tissue matched pairs with more than 25 cases were considered. Each dot represents a normal (green) or a tumor (red) sample. The horizontal black bar represents the mean. The TRIP12 mRNA level was obtained by RNA-seq and expressed as transcripts per million (TPM). Cancer types in which TRIP12 mRNA is statistically differentially expressed (ANOVA test with a q value < 0.01) are marked by an asterisk.
6.2.3. Alterations of TRIP12 Protein Expression in Cancers and Consequences
Available data on TRIP12 protein expression in cancer are much less abundant than those describing TRIP12 mRNA levels. The Human Protein Atlas indicates a heterogeneous expression of TRIP12 depending on cancer type with a high TRIP12 frequency in head-and-neck cancer, testis cancer, and glioma patients. Only a few molecular events leading to alterations of TRIP12 protein expression are known. For instance, Cai et al. demonstrated an overexpression of the deubiquitinase USP7 in hepatocellular carcinoma (HCC) tissues that is correlated with poor prognosis [47]. They showed that USP7 is required for the proliferation of HCC-derived cell lines. Mechanistically, the study reveals that USP7 overexpression stabilizes the TRIP12 protein, which in turn ubiquitinates and degrades ARF to promote HCC-derived cell proliferation. Interestingly, they established that the expressions of TRIP12 and/or USP7 are independent prognosis markers of HCC. Helicobacter pylori (H. pylori) is one of the most common human pathogens. This bacterium is recognized as the strongest identified risk factor for gastric malignancies. Horvat et al. demonstrated that infection by H. pylori of gastric cancer derived-cell lines induces TRIP12 protein expression through the implication of the virulence factor CagA (Cytotoxin-Associated Gene A) by a still unknown mechanism [158]. TRIP12 overexpression was observed in human gastric mucosa infected with H. pylori. The consequence of TRIP12-induced expression is a polyubiquitination-mediated degradation of ARF that leads to an inhibition of autophagy in gastric cells. Patients with human papillomavirus (HPV)-positive head and neck squamous cell carcinoma (HNSCC) have a better response to radiotherapy and higher overall survival rate than those with HPV-negative HNSCC. Indeed, HPV infection leads to P16 overexpression, which in turn provokes a destabilization of the TRIP12 protein by an unknown mechanism [159]. As TRIP12 is a DDR regulator (detailed in Section 5.3.), the down-regulation of TRIP12 sensitizes HPV-positive HNSCC to radiotherapy. Likewise, in 18 HPV-positive HNSCC patients, the overexpression of TRIP12 is associated with a poor survival rate. These results were confirmed recently by the same group [160]. Recently, Gatti et al. identified that the TRIP12 protein level could provide a useful marker for PARP inhibitors (PARPi) sensitivity [27]. Indeed, responses to this therapy are promising, but predictions based on PARP1 gene expression abundance and/or activity are not accurate enough. TRIP12 limits PARP1 presence at the chromatin by polyubiquitinating it and, as a consequence, it decreases the efficiency of PARP1 trapping by PARPi. Of importance, the TRIP12 protein level is negatively correlated with PARP1 protein abundance in two cohorts of breast and ovarian cancer patients.
We described in Section 5.2. the essential role of ARF in the activation of the P53 pathway and its degradation mediated by TRIP12. We also described how the nucleophosmin (NPM) protects ARF from TRIP12-mediated degradation by sequestrating it into the nucleoli [15]. The NPM gene is mutated in one-third of Acute myeloid leukemia (AML), which provokes the translocation of NPM protein into the cytoplasm and the loss of its capacity to maintain ARF localization in the nucleolus [96]. Combined with an overexpression of TRIP12, a frequent loss of NPM nucleolar localization strongly impedes the activation of the ARF/P53 pathway in AML-derived cells and favors their proliferation.
7. Conclusions and Perspectives
In this review, we provide a synthesis of the different knowledge on the E3 ubiquitin ligase TRIP12. First described as a thyroid hormone receptor interacting protein, it appears that the TRIP12 protein displays several important functions in essential cellular processes such as cell cycle progression, DNA damage repair, chromatin remodeling, and cell differentiation. Although the number of studies related to this E3 ubiquitin ligase is constantly growing, numerous aspects of its regulation and functions in physiology and pathologies are not entirely clear.
High-throughput sequencing analyses have determined the level of expression of TRIP12 mRNA in a myriad of healthy and pathological human tissues such as cancers. However, the molecular mechanisms that regulate the production and the stability of TRIP12 mRNA remain largely unknown. The same observation can be made for the post-translational regulation of TRIP12. Indeed, mass spectrometry approaches identified numerous post-translationally modified residues in TRIP12. Unfortunately, none of them have been linked to biological functions, even if three phosphorylation sites are attributed to specific kinases involved in cell cycle (CDK1) and DNA damage response (ATR/ATM, CHK1). We can speculate that these phosphorylated residues modify the protein-interacting properties or the ubiquitin ligase capacity of TRIP12, which in turn participates in cell cycle control and DDR. We could also envisage the possibility that TRIP12 phosphorylated residues result from the activity of kinases implicated in other signaling pathways (i.e., MAP kinases, PI3 kinases). The development of antibodies specific to TRIP12-phosphorylated residues is crucial to decipher the biological consequences of these post-translational modifications. Likewise, the identification of the ubiquitin ligase(s) responsible for the ubiquitinated residues of TRIP12 would certainly help to better perceive the regulation of the TRIP12 protein during the cell cycle. The anaphase-promoting complex/cyclosome (APC/C) system ubiquitinates several cell cycle-regulated proteins early after mitosis (i.e., CYCLIN A and B), thereby mediating degradation by the proteasome. TRIP12 possesses putative KEN box (aa 1496–1570/UniProt source) and Destruction boxes (aa 859–867 and aa 1546–1554/ELM resource) specifically recognized by the APC/C, system making this degradation system a legitimate candidate for the control of TRIP12 stability. To confirm these multiple hypotheses, a complete and exhaustive interactome/substratome of TRIP12 is required, keeping in mind that due to its ubiquitous expression, the interactome of TRIP12 will possibly vary depending on the cell type. We can logically expect other important cellular functions for this E3 ubiquitin ligase depending on the cell type or the cellular context.
This review raises questions, one of which is whether TRIP12 is an appropriate therapeutic target. In the case of cancers in which TRIP12 is overexpressed (i.e., pancreatic cancer) and given its role in the maintenance of a low level of P53 mediated by ARF degradation, it seems logical that the inhibition of TRIP12 could be a promising therapeutic strategy. The inhibition of TRIP12 ubiquitin ligase activity or its interaction with protein partners might counteract the proliferation rate of cancer cells or sensitize them to chemotherapies (by affecting DNA damages repair induced by chemotherapies). To this purpose, the crystallization of the entire TRIP12 protein is a prerequisite to obtain the 3D structure of the protein to facilitate the screening and/or the design of specific inhibitors. PROTACs that are bifunctional molecules made up of an E3 ubiquitin ligase and a target protein connected via a linker could also be used to target TRIP12.
In pathological contexts where the TRIP12 gene is mutated or truncated (numerous types of cancer and intellectual disorders), we do not yet understand whether the TRIP12 protein is indeed expressed or if the mRNA is degraded. If expressed, the functional consequences of mutated and truncated forms of TRIP12 in these pathologies are still unknown. We can only speculate that mutant proteins would act as dominant negative proteins, leading to an alteration of genome expression. The development of murine transgenic models expressing Trip12 mutated forms could address these issues. Moreover, several truncating mutations identified in cancers and IDs theoretically lead to TRIP12 proteins lacking the HECT catalytic domain or protein–protein interacting domains (WWE/ARM). Therefore, therapeutic strategies inhibiting its catalytic activity or protein interaction might be useless or, even worse, deleterious for the patients.
To conclude, the TRIP12 protein is emerging as an important protein in several essential cellular processes as well as in pathological alterations. Uncovering the function of TRIP12 offers a glimpse for potential exciting therapeutic strategies. However, numerous aspects of its regulations and functions need to be better described prior to its application in therapeutic objectives.
8. Software and Data Bases
BioGrid: The Biological General Repository for Interaction Datasets (BioGrid) is an open access database that houses genetic and protein interactions curated from the primary biomedical literature for all major model organism species and humans (https://thebiogrid.org/).
cBioPortal: The cBio Cancer Genomics Portal is an open-access resource for the interactive exploration of multidimensional cancer genomics datasets, currently providing access to data from more than 5000 tumor samples from 20 cancer studies (www.cbioportal.org).
ClinVar: ClinVar is a public database of variant interpretations that has steadily grown to become the largest publicly available genetic variant database and provides an ever-growing resource to study genotype–phenotype correlations (https://www.ncbi.nlm.nih.gov/clinvar/).
COBALT: The constraint-based Multiple Alignment Tool is a multiple sequence alignment tool that finds a collection of pairwise constraints derived from conserved domain database, protein motif database, and sequence similarity, using RPS-BLAST, BLASTP, and PHI-BLAST. Then, pairwise constraints are incorporated into a progressive multiple alignment (www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi).
EMBOSS matcher: EMBOSS Matcher identifies local similarities in two input sequences using a rigorous algorithm based on Bill Pearson’s lalign application (www.ebi.ac.uk).
ELM: The Eukaryotic Linear Motif resource is a computational biology resource for investigating short linear motifs (SLiMs) in eukaryotic proteins. It is currently the largest collection of linear motif classes with annotated and experimentally validated linear motif instances (http://elm.eu.org/).
Genome browser: A genome browser is a graphical interface for the display of information from a biological database for genomic data (www.genome.ucsc.edu).
Gepia2: The Gene Expression Profiling Interactive Analysis 2 web server is a valuable and highly cited resource for gene expression analysis based on tumor and normal samples from the TCGA and the GTEx databases (http://gepia2.cancer-pku.cn).
Human Protein Atlas: The Human Protein Atlas is a program with the aim to map all the human proteins in cells, tissues, and organs using an integration of various omics technologies, including antibody-based imaging, mass spectrometry-based proteomics, transcriptomics, and systems biology (www.proteinatlas.org).
IDEAL: Intrinsically Disordered proteins with Extensive Annotations and Literature is a collection of knowledge on experimentally verified intrinsically disordered proteins. IDEAL contains manual annotations by curators on intrinsically disordered regions, interaction regions to other molecules, post-translational modification sites, references, and structural domain assignments (www.ideal-db.org).
InterProScan: InterProScan is a tool that scans given protein sequences against the protein signatures of the InterPro member databases, currently—PROSITE, PRINTS, Pfam, ProDom, and SMART (https://www.ebi.ac.uk/interpro/).
IUPred: Intrinsically unstructured/disordered proteins have no single well-defined tertiary structure in their native, functional state. This server recognizes such regions from the amino acid sequence based on the estimated pairwise energy content (https://iupred.elte.hu/).
LOVD: The Leiden Variation Database is a free, flexible web-based open source database developed in the Leiden University Medical Center in the Netherlands, which is designed to collect and display variants in the DNA sequence. The focus of an LOVD is usually the combination between a gene and a genetic (heritable) disease (https://www.lovd.nl/).
OMIM: Online Mendelian Inheritance in Man is a comprehensive, authoritative compendium of human genes and genetic phenotypes that is freely available and updated daily (https://www.omim.org/).
PhosphoSitePlus®: PhosphoSitePlus® provides comprehensive information and tools for the study of protein post-translational modifications (PTMs) including phosphorylation, acetylation, and more (https://phosphosite.org/).
SFARI GENE: SFARI GENE is an evolving database for the autism research community that is centered on genes implicated in autism susceptibility (https://gene.sfari.org/).
STRING: Search Tool for the Retrieval of Interacting Genes/Proteins is a biological database and web resource of known and predicted protein–protein interactions. The STRING database contains information from numerous sources, including experimental data, computational prediction methods, and public text collections. It is freely accessible and it is regularly updated (https://string-db.org/).
UniProt: UniProt is the Universal Protein resource, a central repository of protein data created by combining the Swiss-Prot, TrEMBL and PIR-PSD databases. (www.uniprot.org).
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/21/22/8515/s1.
Author Contributions
M.B., C.V., D.L., J.T., M.D. conception, drafting, critical revising of the review (text, figures and tables). All authors have read and agreed to the published version of the manuscript.
Funding
University Paul Sabatier (Toulouse, France). Ligue Nationale contre le Cancer. Fondation Toulouse Cancer Santé (2018CS079). Ligue Régionale contre le Cancer (comité Haute-Garonne, comité Ariège). Institut National de la Santé et de la Recherche Médicale.
Acknowledgments
C. Vargas is funded by the University Paul Sabatier (Toulouse, France). M. Brunet is funded by the University Paul Sabatier (Toulouse, France) and the Ligue Nationale contre le Cancer. D. Larrieu was funded by the Ligue Nationale contre le Cancer. We thank the “Fondation Toulouse Cancer Santé” and the Ligue Nationale contre le Cancer (comité Haute-Garonne, comité Ariège) for financial support. We thank Dina Arvanitis (Inserm U1038, Institute of Cardiovascular and Metabolic Disease, Toulouse, France) for the critical reading of the manuscript. We apologize for all the studies that were not quoted in this manuscript due to a lack of space.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nomura, N.; Nagase, T.; Miyajima, N.; Sazuka, T.; Tanaka, A.; Sato, S.; Seki, N.; Kawarabayasi, Y.; Ishikawa, K.-I.; Tabata, S. Prediction of the Coding Sequences of Unidentified Human Genes. II. The Coding Sequences of 40 New Genes (KIAA0041-KIAA0080) Deduced by Analysis of cDNA Clones from Human Cell Line KG-1. DNA Res. 1994, 1, 223–229. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Beaudenon, S.; Howley, P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 1995, 92, 2563–2567. [Google Scholar] [CrossRef]
- Lee, J.W.; Choi, H.S.; Gyuris, J.; Brent, R.; Moore, D.D. Two classes of proteins dependent on either the presence or absence of thyroid hormone for interaction with the thyroid hormone receptor. Mol. Endocrinol. 1995, 9, 243–254. [Google Scholar]
- Park, Y.; Yoon, S.K.; Yoon, J.-B. TRIP12 functions as an E3 ubiquitin ligase of APP-BP1. Biochem. Biophys. Res. Commun. 2008, 374, 294–298. [Google Scholar] [CrossRef]
- Kajiro, M.; Tsuchiya, M.; Kawabe, Y.-I.; Furumai, R.; Iwasaki, N.; Hayashi, Y.; Katano, M.; Nakajima, Y.; Goto, N.; Watanabe, T.; et al. The E3 Ubiquitin Ligase Activity of Trip12 Is Essential for Mouse Embryogenesis. PLoS ONE 2011, 6, e25871. [Google Scholar] [CrossRef]
- Hanoun, N.; Fritsch, S.; Gayet, O.; Gigoux, V.; Cordelier, P.; Dusetti, N.; Torrisani, J.; Dufresne, M. The E3 Ubiquitin Ligase Thyroid Hormone Receptor-interacting Protein 12 Targets Pancreas Transcription Factor 1a for Proteasomal Degradation. J. Biol. Chem. 2014, 289, 35593–35604. [Google Scholar] [CrossRef]
- Larrieu, D.; Brunet, M.; Vargas, C.; Hanoun, N.; Ligat, L.; Dagnon, L.; Lulka, H.; Pommier, R.M.; Selves, J.; Jády, B.E.; et al. The E3 ubiquitin ligase TRIP12 participates in cell cycle progression and chromosome stability. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Trinklein, N.D.; Aldred, S.F.; Hartman, S.J.; Schroeder, D.I.; Otillar, R.P.; Myers, R.M. An Abundance of Bidirectional Promoters in the Human Genome. Genome Res. 2003, 14, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Tekcham, D.S.; Chen, D.; Liu, Y.; Ling, T.; Zhang, Y.; Chen, H.; Wang, W.; Otkur, W.; Qi, H.; Xia, T.; et al. F-box proteins and cancer: An update from functional and regulatory mechanism to therapeutic clinical prospects. Theranostics 2020, 10, 4150–4167. [Google Scholar] [CrossRef] [PubMed]
- Bramswig, N.C.; Lüdecke, H.-J.; Pettersson, M.; Albrecht, B.; Bernier, R.A.; Cremer, K.; Eichler, E.E.; Falkenstein, D.; Gerdts, J.; Jansen, S.; et al. Identification of new TRIP12 variants and detailed clinical evaluation of individuals with non-syndromic intellectual disability with or without autism. Qual. Life Res. 2017, 136, 179–192. [Google Scholar] [CrossRef]
- Chang, J.-W.; Zhang, W.; Yeh, H.S.; De Jong, E.P.; Jun, S.; Kim, K.H.; Bae, S.S.; Beckman, K.; Hwang, T.H.; Kim, K.S.; et al. mRNA 3’-UTR shortening is a molecular signature of mTORC1 activation. Nat. Commun. 2015, 6, 7218. [Google Scholar] [CrossRef] [PubMed]
- An, C.-I.; Ganio, E.; Hagiwara, N. Trip12, a HECT domain E3 ubiquitin ligase, targets Sox6 for proteasomal degradation and affects fiber type-specific gene expression in muscle cells. Skelet. Muscle 2013, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Sadkowski, T.; Jank, M.; Zwierzchowski, L.; Oprzadek, J.; Motyl, T. Comparison of skeletal muscle transcriptional profiles in dairy and beef breeds bulls. J. Appl. Genet. 2009, 50, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, T.; Altmeyer, M.; Savic, V.; Toledo, L.; Dinant, C.; Grøfte, M.; Bartkova, J.; Poulsen, M.; Oka, Y.; Bekker-Jensen, S.; et al. TRIP12 and UBR5 Suppress Spreading of Chromatin Ubiquitylation at Damaged Chromosomes. Cell 2012, 150, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shan, J.; Zhu, W.-G.; Qin, J.; Gu, W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nat. Cell Biol. 2010, 464, 624–627. [Google Scholar] [CrossRef]
- Huber, A.H.; Nelson, W.J.; Weis, W.I. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 1997, 90, 871–882. [Google Scholar] [CrossRef]
- Ries, L.K.; Liess, A.K.L.; Feiler, C.G.; Spratt, D.E.; Lowe, E.D.; Lorenz, S. Crystal structure of the catalytic C-lobe of the HECT-type ubiquitin ligase E6AP. Protein Sci. 2020, 29, 1550–1554. [Google Scholar] [CrossRef]
- Zweifel, M.E.; Leahy, D.J.; Barrick, D. Structure and Notch Receptor Binding of the Tandem WWE Domain of Deltex. Structure 2005, 13, 1599–1611. [Google Scholar] [CrossRef]
- Sluimer, J.; Distel, B. Regulating the human HECT E3 ligases. Cell. Mol. Life Sci. 2018, 75, 3121–3141. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Huang, L. Structure of an E6AP-UbcH7 Complex: Insights into Ubiquitination by the E2-E3 Enzyme Cascade. Science 1999, 286, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. HECT E3 ubiquitin ligases—Emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020, 133, jcs228072. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L. The WWE domain: A common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem. Sci. 2001, 26, 273–275. [Google Scholar] [CrossRef]
- Wang, Z.; Michaud, G.A.; Cheng, Z.; Zhang, Y.; Hinds, T.R.; Fan, E.; Cong, F.; Xu, W. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012, 26, 235–240. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2018, 47, D351–D360. [Google Scholar] [CrossRef]
- Gatti, M.; Imhof, R.; Huang, Q.; Baudis, M.; Altmeyer, M. The Ubiquitin Ligase TRIP12 Limits PARP1 Trapping and Constrains PARP Inhibitor Efficiency. Cell Rep. 2020, 32, 107985. [Google Scholar] [CrossRef]
- Tewari, R.; Bailes, E.; Bunting, K.A.; Coates, J.C. Armadillo-repeat protein functions: Questions for little creatures. Trends Cell Biol. 2010, 20, 470–481. [Google Scholar] [CrossRef]
- El Refy, A.; Perazza, D.; Zekraoui, L.; Valay, J.-G.; Bechtold, N.; Brown, S.; Hülskamp, M.; Herzog, M.; Bonneville, J.-M. The Arabidopsis KAKTUS gene encodes a HECT protein and controls the number of endoreduplication cycles. Mol. Genet. Genom. 2003, 270, 403–414. [Google Scholar] [CrossRef]
- Ju, D.; Wang, X.; Xu, H.; Xie, Y. The armadillo repeats of the Ufd4 ubiquitin ligase recognize ubiquitin-fusion proteins. FEBS Lett. 2006, 581, 265–270. [Google Scholar] [CrossRef]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.W.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Toretsky, J.A.; Wright, P.E. Assemblages: Functional units formed by cellular phase separation. J. Cell Biol. 2014, 206, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y. Intrinsically Disordered Regions as Affinity Tuners in Protein-DNA Interactions. Biophys. J. 2012, 102, 632a. [Google Scholar] [CrossRef]
- Watson, M.; Stott, K. Disordered domains in chromatin-binding proteins. Essays Biochem. 2019, 63, 147–156. [Google Scholar] [CrossRef]
- Vuzman, D.; Levy, Y. DNA search efficiency is modulated by charge composition and distribution in the intrinsically disordered tail. Proc. Natl. Acad. Sci. USA 2010, 107, 21004–21009. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef]
- Leach, B.I.; Kuntimaddi, A.; Schmidt, C.R.; Cierpicki, T.; Johnson, S.A.; Bushweller, J.H. Leukemia Fusion Target AF9 Is an Intrinsically Disordered Transcriptional Regulator that Recruits Multiple Partners via Coupled Folding and Binding. Structure 2013, 21, 176–183. [Google Scholar] [CrossRef]
- Chen, Y.; Cramer, P. Structure of the super-elongation complex subunit AFF4 C-terminal homology domain reveals requirements for AFF homo- and heterodimerization. J. Biol. Chem. 2019, 294, 10663–10673. [Google Scholar] [CrossRef]
- Liu, X.; Yang, X.; Li, Y.; Zhao, S.; Li, C.; Ma, P.; Mao, B. Trip12 is an E3 ubiquitin ligase for USP7/HAUSP involved in the DNA damage response. FEBS Lett. 2016, 590, 4213–4222. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, H.O.L.A.A.; Rosen, S.F.B.M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Posey, A.E.; Holehouse, A.S.; Pappu, R.V. Phase Separation of Intrinsically Disordered Proteins. Methods Enzymol. 2018, 611, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Gallego, L.D.; Schneider, M.; Mittal, C.; Romanauska, A.; Carrillo, R.M.G.; Schubert, T.; Pugh, B.F.; Köhler, A. Phase separation directs ubiquitination of gene-body nucleosomes. Nat. Cell Biol. 2020, 579, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed]
- Blasius, M.; Forment, J.V.; Thakkar, N.; Wagner, S.A.; Choudhary, C.; Jackson, S.P. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 2011, 12, R78. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Petrone, A.; Adamo, M.E.; Cheng, C.; Kettenbach, A.N. Identification of Candidate Cyclin-dependent kinase 1 (Cdk1) Substrates in Mitosis by Quantitative Phosphoproteomics. Mol. Cell. Proteom. 2016, 15, 2448–2461. [Google Scholar] [CrossRef]
- Cai, J.-B.; Shi, G.-M.; Dong, Z.-R.; Ke, A.-W.; Ma, H.-H.; Gao, Q.; Shen, Z.-Z.; Huang, X.-Y.; Chen, H.; Yu, D.-D.; et al. Ubiquitin-specific protease 7 accelerates p14 ARF degradation by deubiquitinating thyroid hormone receptor-interacting protein 12 and promotes hepatocellular carcinoma progression. Hepatology 2015, 61, 1603–1614. [Google Scholar] [CrossRef]
- Georges, A.; Marcon, E.; Greenblatt, J.; Frappier, L. Identification and Characterization of USP7 Targets in Cancer Cells. Sci. Rep. 2018, 8, 15833. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.D. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997, 11, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Kweon, S.-M.; Chen, Y.; Moon, E.; Kvederaviciutė, K.; Klimasauskas, S.; Feldman, D.E. An Adversarial DNA N6-Methyladenine-Sensor Network Preserves Polycomb Silencing. Mol. Cell 2019, 74, 1138–1147.e6. [Google Scholar] [CrossRef] [PubMed]
- Keppler, B.R.; Archer, T.K. Ubiquitin-dependent and Ubiquitin-independent Control of Subunit Stoichiometry in the SWI/SNF Complex. J. Biol. Chem. 2010, 285, 35665–35674. [Google Scholar] [CrossRef]
- Hah, N.; Kolkman, A.; Ruhl, D.D.; Pijnappel, W.W.M.P.; Heck, A.J.R.; Timmers, H.T.M.; Kraus, W.L. A Role for BAF57 in Cell Cycle–Dependent Transcriptional Regulation by the SWI/SNF Chromatin Remodeling Complex. Cancer Res. 2010, 70, 4402–4411. [Google Scholar] [CrossRef]
- Kuo, M.-L.; Besten, W.D.; Sherr, C.J. N-Terminal Polyubiquitination of the ARF Tumor Suppressor, A Natural Lysine-Less Protein. Cell Cycle 2004, 3, 1367–1369. [Google Scholar] [CrossRef]
- Chen, J.; Archer, T.K. Regulating SWI/SNF Subunit Levels via Protein-Protein Interactions and Proteasomal Degradation: BAF155 and BAF170 Limit Expression of BAF57. Mol. Cell. Biol. 2005, 25, 9016–9027. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Sasaki, M.; Ghazarian, D.; Moreno, J.; Done, S.J.; Ueda, T.; Inoue, S.; Chang, Y.-L.; Chen, N.-J.; Mak, T.W. TRADD contributes to tumour suppression by regulating ULF-dependent p19Arf ubiquitylation. Nat. Cell Biol. 2012, 14, 625–633. [Google Scholar] [CrossRef]
- Johnson, E.S.; Ma, P.C.M.; Ota, I.M.; Varshavsky, A. A Proteolytic Pathway That Recognizes Ubiquitin as a Degradation Signal. J. Biol. Chem. 1995, 270, 17442–17456. [Google Scholar] [CrossRef]
- Park, Y.; Yoon, S.K.; Yoon, J.-B. The HECT Domain of TRIP12 Ubiquitinates Substrates of the Ubiquitin Fusion Degradation Pathway. J. Biol. Chem. 2008, 284, 1540–1549. [Google Scholar] [CrossRef]
- Van Leeuwen, F.; De Kleijn, D.P.; Hurk, H.H.V.D.; Neubauer, A.; Sonnemans, M.A.; Sluijs, J.A.; Köycü, S.; Ramdjielal, R.D.; Salehi, A.; Martens, G.J.; et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science 1998, 279, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, E.G.; Steinhauer, C.; Lees, M.; Lauridsen, A.-M.; Ellgaard, L.; Hartmann-Petersen, R. HUWE1 and TRIP12 Collaborate in Degradation of Ubiquitin-Fusion Proteins and Misframed Ubiquitin. PLoS ONE 2012, 7, e50548. [Google Scholar] [CrossRef] [PubMed]
- Setz, C.; Friedrich, M.; Hahn, S.; Dörrie, J.; Schaft, N.; Schuler, G.; Schubert, U. Just One Position-Independent Lysine Residue Can Direct MelanA into Proteasomal Degradation following N-Terminal Fusion of Ubiquitin. PLoS ONE 2013, 8, e55567. [Google Scholar] [CrossRef]
- Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.-S.; Shemorry, A.; Auerbach, D.; Varshavsky, A. The N-end rule pathway is mediated by a complex of the RING-type Ubr1 and HECT-type Ufd4 ubiquitin ligases. Nat. Cell Biol. 2010, 12, 1177–1185. [Google Scholar] [CrossRef]
- Havugimana, P.C.; Hart, G.T.; Nepusz, T.; Yang, H.; Turinsky, A.L.; Li, Z.; Wang, P.I.; Boutz, D.R.; Fong, V.; Phanse, S.; et al. A Census of Human Soluble Protein Complexes. Cell 2012, 150, 1068–1081. [Google Scholar] [CrossRef]
- Arbogast, F.; Arnold, J.; Hammann, P.; Kuhn, L.; Chicher, J.; Murera, D.; Weishaar, J.; Muller, S.; Fauny, J.-D.; Gros, F. ATG5 is required for B cell polarization and presentation of particulate antigens. Autophagy 2018, 15, 280–294. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nat. Cell Biol. 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Nakayama, M.; Kikuno, R.; Ohara, O. Protein-Protein Interactions between Large Proteins: Two-Hybrid Screening Using a Functionally Classified Library Composed of Long cDNAs. Genome Res. 2002, 12, 1773–1784. [Google Scholar] [CrossRef]
- Hanson, D.; Stevens, A.; Murray, P.; Black, G.C.M.; Clayton, P.E. Identifying biological pathways that underlie primordial short stature using network analysis. J. Mol. Endocrinol. 2014, 52, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, C.; Forget, D.; Bouchard, A.; Li, Q.; Chua, G.; Poitras, C.; Thérien, C.; Bergeron, D.; Bourassa, S.; Greenblatt, J.; et al. Systematic Analysis of the Protein Interaction Network for the Human Transcription Machinery Reveals the Identity of the 7SK Capping Enzyme. Mol. Cell 2007, 27, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Cho, Y.-E.; Kim, S.-H.; Kim, Y.-J.; Park, J.-H. GLTSCR2 promotes the nucleoplasmic translocation and subsequent degradation of nucleolar ARF. Oncotarget 2016, 8, 16293–16302. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Z.; Hou, J.; Sun, L.; Wen, T.; Wang, L.; Zhao, X.; Xie, Q.; Zhang, S.Q. NMI mediates transcription-independent ARF regulation in response to cellular stresses. Mol. Biol. Cell 2012, 23, 4635–4646. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.; Zhang, Y.; Dai, M.-S.; Sun, X.-X.; Zeng, S.X.; Lu, H. Nucleostemin stabilizes ARF by inhibiting the ubiquitin ligase ULF. Oncogene 2014, 34, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.D.; Coyaud, É.; Gonçalves, J.; Mojarad, B.A.; Liu, Y.; Wu, Q.; Gheiratmand, L.; Comartin, D.; Tkach, J.M.; Cheung, S.W.; et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 2015, 163, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venable, J.; Lapointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 Cochaperone Aha1 Downregulation Rescues Misfolding of CFTR in Cystic Fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef]
- Yamauchi, T.; Masuda, T.; Canver, M.C.; Seiler, M.; Semba, Y.; Shboul, M.; Al-Raqad, M.; Maeda, M.; Schoonenberg, V.A.; Cole, M.A.; et al. Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018, 33, 386–400.e5. [Google Scholar] [CrossRef]
- Guard, S.E.; Poss, Z.C.; Ebmeier, C.C.; Pagratis, M.; Simpson, H.; Taatjes, D.J.; Old, W.M. The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, X.; Zhao, M.; Yang, R.; Malik, R.; Qiao, Y.; Poliakov, A.; Yocum, A.K.; Li, Y.; Chen, W.; et al. The central role of EED in the orchestration of polycomb group complexes. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef]
- Wang, S.; Kollipara, R.K.; Humphries, C.G.; Ma, S.-H.; Hutchinson, R.; Li, R.; Siddiqui, J.; Tomlins, S.A.; Raj, G.V.; Kittler, R. The ubiquitin ligase TRIM25 targets ERG for degradation in prostate cancer. Oncotarget 2016, 7, 64921–64931. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, R.; Bamundo, A.; Nassa, G.; Nola, E.; Paris, O.; Ambrosino, C.; Facchiano, A.; Baumann, M.; Nyman, T.A.; Weisz, A. Identification of proteins associated with ligand-activated estrogen receptor α in human breast cancer cell nuclei by tandem affinity purification and nano LC-MS/MS. Proteom. Clin. Appl. 2011, 5, 470. [Google Scholar] [CrossRef]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Oliviero, G.; Cagney, G.; Waston, A.; Streubel, G.; Jerman, E.; Andrews, D.; Doyle, B.; Munawar, N.; Wynne, K.; Crean, J.; et al. Dynamic Protein Interactions of the Polycomb Repressive Complex 2 during Differentiation of Pluripotent Cells. Mol. Cell. Proteom. 2016, 15, 3450–3460. [Google Scholar] [CrossRef]
- Gilmore, J.M.; Sardiu, M.E.; Groppe, B.D.; Thornton, J.L.; Liu, X.; Dayebgadoh, G.; Banks, C.A.; Slaughter, B.D.; Unruh, J.R.; Workman, J.L.; et al. WDR76 Co-Localizes with Heterochromatin Related Proteins and Rapidly Responds to DNA Damage. PLoS ONE 2016, 11, e0155492. [Google Scholar] [CrossRef]
- Lambert, J.-P.; Tucholska, M.; Go, C.; Knight, J.D.; Gingras, A.-C. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteom. 2015, 118, 81–94. [Google Scholar] [CrossRef]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Cesa, L.C.; Faull, P.A.; Lukauskas, S.; Frimurer, T.; et al. H4K20me0 recognition by BRCA1–BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef]
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol. Cell. Proteom. 2015, 14, 2341–2356. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 1–17. [Google Scholar] [CrossRef]
- Hauri, S.; Comoglio, F.; Seimiya, M.; Gerstung, M.; Glatter, T.; Hansen, K.; Aebersold, R.; Paro, R.; Gstaiger, M.; Beisel, C. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 2016, 17, 583–595. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Mercer, J.; Shrestha, Y.; Oliver, R.; Tamayo, P.; Doench, J.G.; Tirosh, I.; Piccioni, F.; Hartenian, E.; Horn, H.; et al. Genetic and Proteomic Interrogation of Lower Confidence Candidate Genes Reveals Signaling Networks in β-Catenin-Active Cancers. Cell Syst. 2016, 3, 302–316.e4. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Zhong, X.; McAlpine, W.; Liao, T.-C.; Zhang, D.-W.; Fang, B.; Russell, J.; Ludwig, S.; Nair-Gill, E.; Zhang, Z.; et al. LMBR1L regulates lymphopoiesis through Wnt/β-catenin signaling. Science 2019, 364, eaau0812. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Ma, S.; Shan, L.; Wang, Y.; Wang, Y.; Cao, C.; Liu, B.; Yang, C.; Wang, L.; Tian, S.; et al. Ubiquitin-specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J. Clin. Investig. 2018, 128, 4280–4296. [Google Scholar] [CrossRef] [PubMed]
- Benleulmi-Chaachoua, A.; Chen, L.; Sokolina, K.; Wong, V.; Jurisica, I.; Emerit, M.B.; Darmon, M.; Espin, A.; Stagljar, I.; Tafelmeyer, P.; et al. Protein interactome mining defines melatonin MT1receptors as integral component of presynaptic protein complexes of neurons. J. Pineal Res. 2015, 60, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Heidelberger, J.B.; Voigt, A.; Borisova, M.E.; Petrosino, G.; Ruf, S.; Wagner, S.A.; Beli, P. Proteomic profiling of VCP substrates links VCP to K6-linked ubiquitylation and c-Myc function. EMBO Rep. 2018, 19, e44754. [Google Scholar] [CrossRef]
- Chen, D.; Yoon, J.-B.; Gu, W. Reactivating the ARF-p53 axis in AML cells by targeting ULF. Cell Cycle 2010, 9, 2946–2951. [Google Scholar] [CrossRef]
- Yu, L.; Jearawiriyapaisarn, N.; Lee, M.P.; Hosoya, T.; Wu, Q.; Myers, G.; Lim, K.-C.; Kurita, R.; Nakamura, Y.; Vojtek, A.B.; et al. BAP1 regulation of the key adaptor protein NCoR1 is critical for γ-globin gene repression. Genes Dev. 2018, 32, 1537–1549. [Google Scholar] [CrossRef]
- Shami Shah, A.; Batrouni, A.G.; Kim, D.; Punyala, A.; Cao, W.; Han, C.; Goldberg, M.L.; Smolka, M.B.; Baskin, J.M. PLEKHA4/kramer Attenuates Dishevelled Ubiquitination to Modulate Wnt and Planar Cell Polarity Signaling. Cell Rep. 2019, 27, 2157–2170.e8. [Google Scholar] [CrossRef]
- Khanna, R.; Krishnamoorthy, V.; Parnaik, V.K. E3 ubiquitin ligase RNF 123 targets lamin B1 and lamin-binding proteins. FEBS J. 2018, 285, 2243–2262. [Google Scholar] [CrossRef]
- Maréchal, A.; Li, J.-M.; Ji, X.Y.; Wu, C.-S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jiménez, A.E.; Jin, J.; Zou, L. PRP19 Transforms into a Sensor of RPA-ssDNA after DNA Damage and Drives ATR Activation via a Ubiquitin-Mediated Circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Greco, T.M.; Boonmee, A.; Miteva, Y.; Cristea, I.M. Functional Proteomics Establishes the Interaction of SIRT7 with Chromatin Remodeling Complexes and Expands Its Role in Regulation of RNA Polymerase I Transcription. Mol. Cell. Proteom. 2012, 11, 60–76. [Google Scholar] [CrossRef] [PubMed]
- Colland, F.; Jacq, X.; Trouplin, V.; Mougin, C.; Groizeleau, C.; Hamburger, A.; Meil, A.; Wojcik, J.; Legrain, P.; Gauthier, J.-M. Functional Proteomics Mapping of a Human Signaling Pathway. Genome Res. 2004, 14, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Llères, D.; Denegri, M.; Biggiogera, M.; Ajuh, P.; Lamond, A.I. Direct interaction between hnRNP-M and CDC5L/PLRG1 proteins affects alternative splice site choice. EMBO Rep. 2010, 11, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Lipp, J.J.; Marvin, M.C.; Shokat, K.M.; Guthrie, C. SR protein kinases promote splicing of nonconsensus introns. Nat. Struct. Mol. Biol. 2015, 22, 611–617. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.; Wang, J.; Malovannaya, A.; Xi, Y.; Li, W.; Guerra, R.; Hawke, D.H.; Qin, J.; Chen, J. Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol. 2015, 11, 775. [Google Scholar] [CrossRef]
- Baranes-Bachar, K.; Levy-Barda, A.; Oehler, J.; Reid, D.A.; Soria-Bretones, I.; Voss, T.C.; Chung, D.; Park, Y.; Liu, C.; Yoon, J.B.; et al. The Ubiquitin E3/E4 Ligase UBE4A Adjusts Protein Ubiquitylation and Accumulation at Sites of DNA Damage, Facilitating Double-Strand Break Repair. Mol. Cell 2018, 69, 866–878.e7. [Google Scholar] [CrossRef]
- Mulder, M.P.; Witting, K.; Berlin, I.; Pruneda, J.N.; Wu, K.-P.; Chang, J.-G.; Merkx, R.; Bialas, J.; Groettrup, J.B.M.; Vertegaal, A.C.; et al. A cascading activity-based probe sequentially targets E1–E2–E3 ubiquitin enzymes. Nat. Chem. Biol. 2016, 12, 523–530. [Google Scholar] [CrossRef]
- Schwarz, S.E.; Rosa, J.L.; Scheffner, M. Characterization of Human hect Domain Family Members and Their Interaction with UbcH5 and UbcH7. J. Biol. Chem. 1998, 273, 12148–12154. [Google Scholar] [CrossRef]
- Pao, K.-C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; Van Aalten, D.M.F.; Virdee, S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nat. Cell Biol. 2018, 556, 381–385. [Google Scholar] [CrossRef]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the Human Deubiquitinating Enzyme Interaction Landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef]
- Georges, A.; Coyaud, E.; Marcon, E.; Greenblatt, J.; Raught, B.; Frappier, L. USP7 Regulates Cytokinesis through FBXO38 and KIF20B. Sci. Rep. 2019, 9, 2724. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-C.; Yang, J.-C.; Chang, Y.-C.; Chuang, J.-G.; Lin, C.-W.; Wu, M.-S.; Chow, L.-P. VCP Phosphorylation-Dependent Interaction Partners Prevent Apoptosis in Helicobacter pylori-Infected Gastric Epithelial Cells. PLoS ONE 2013, 8, e55724. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.; Schild-Poulter, C. Mapping the Ku Interactome Using Proximity-Dependent Biotin Identification in Human Cells. J. Proteome Res. 2018, 18, 1064–1077. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Evan, G.I. p53—A Jack of all trades but master of none. Nat. Rev. Cancer 2009, 9, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Kon, N.; Zhong, J.; Zhang, P.; Yu, L.; Gu, W. Differential Effects on ARF Stability by Normal versus Oncogenic Levels of c-Myc Expression. Mol. Cell 2013, 51, 46–56. [Google Scholar] [CrossRef]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct Thresholds Govern Myc’s Biological Output In Vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef]
- Ginno, P.A.; Burger, L.; Seebacher, J.; Iesmantavicius, V.; Schübeler, D. Cell cycle-resolved chromatin proteomics reveals the extent of mitotic preservation of the genomic regulatory landscape. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A Dynamic Role of HAUSP in the p53-Mdm2 Pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Zhu, Q.; Sharma, N.; Qianzheng, Z.; Wani, G.; A Wani, A. USP7 deubiquitinase promotes ubiquitin-dependent DNA damage signaling by stabilizing RNF168. Cell Cycle 2015, 14, 1413–1425. [Google Scholar] [CrossRef]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, G.; Groudine, M. Controlling the double helix. Nat. Cell Biol. 2003, 421, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.W.M.; Orkin, S.H. The SWI/SNF complex—Chromatin and cancer. Nat. Rev. Cancer 2004, 4, 133–142. [Google Scholar] [CrossRef]
- Vignali, M.; Hassan, A.H.; Neely, K.E.; Workman, J.L. ATP-Dependent Chromatin-Remodeling Complexes. Mol. Cell. Biol. 2000, 20, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Beaulaurier, J.; Deikus, G.; Wu, T.P.; Strahl, M.; Hao, Z.; Luo, G.; Gregory, J.A.; Chess, A.; He, C.; et al. Mapping and characterizing N6-methyladenine in eukaryotic genomes using single-molecule real-time sequencing. Genome Res. 2018, 28, 1067–1078. [Google Scholar] [CrossRef]
- Kirmizis, A. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004, 18, 1592–1605. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nat. Cell Biol. 2004, 431, 873–878. [Google Scholar] [CrossRef]
- Scheuermann, J.C.; Alonso, A.G.D.A.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Müller, J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nat. Cell Biol. 2010, 465, 243–247. [Google Scholar] [CrossRef]
- Kloet, S.L.; Makowski, M.M.; Baymaz, H.I.; Van Voorthuijsen, L.; Karemaker, I.D.; Santanach, A.; Jansen, P.W.; Di Croce, A.S.L.; Vermeulen, M. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol. 2016, 23, 682–690. [Google Scholar] [CrossRef]
- Hoischen, A.; Van Bon, B.W.; Rodríguez-Santiago, B.; Gilissen, C.; Vissers, L.E.L.M.; De Vries, P.; Janssen, I.; Van Lier, B.; Hastings, R.; Smithson, S.F.; et al. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat. Genet. 2011, 43, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.M.; Nisperos, S.V.; Weeks, J.; Ghulam, M.; Marín, I.; Zayas, R.M. Identification of HECT E3 ubiquitin ligase family genes involved in stem cell regulation and regeneration in planarians. Dev. Biol. 2015, 404, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, N.; Yeh, M.; Liu, A. Sox6 is required for normal fiber type differentiation of fetal skeletal muscle in mice. Dev. Dyn. 2007, 236, 2062–2076. [Google Scholar] [CrossRef] [PubMed]
- Krah, N.M.; De La O, J.-P.; Swift, G.H.; Hoang, C.Q.; Willet, S.G.; Pan, F.C.; Cash, G.M.; Bronner, M.P.; Wright, C.V.; Macdonald, R.J.; et al. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015, 4, e07125. [Google Scholar] [CrossRef]
- Borromeo, M.D.; Meredith, D.M.; Castro, D.S.; Chang, J.C.; Tung, K.-C.; Guillemot, F.; Johnson, J.E. A transcription factor network specifying inhibitory versus excitatory neurons in the dorsal spinal cord. Development 2014, 141, 2803–2812. [Google Scholar] [CrossRef]
- Lelièvre, E.; Lek, M.; Boije, H.; Houille-Vernes, L.; Brajeul, V.; Slembrouck, A.; Roger, J.; Sahel, J.-A.; Matter, J.-M.; Sennlaub, F.; et al. Ptf1a/Rbpj complex inhibits ganglion cell fate and drives the specification of all horizontal cell subtypes in the chick retina. Dev. Biol. 2011, 358, 296–308. [Google Scholar] [CrossRef]
- Millen, K.J.; Steshina, E.Y.; Iskusnykh, I.Y.; Chizhikov, V.V. Transformation of the cerebellum into more ventral brainstem fates causes cerebellar agenesis in the absence of Ptf1a function. Proc. Natl. Acad. Sci. USA 2014, 111, E1777–E1786. [Google Scholar] [CrossRef]
- Ilyas, M.; Mir, A.; Efthymiou, S.; Houlden, H. The genetics of intellectual disability: Advancing technology and gene editing. F1000Research 2020, 9, 22. [Google Scholar] [CrossRef]
- Mendicino, A.; Sabbadini, G.; Pergola, M.S. Clark—Baraitser syndrome: Report of a new case and review of the literature. Clin. Dysmorphol. 2005, 14, 133–135. [Google Scholar] [CrossRef]
- Louie, R.J.; Friez, M.J.; Skinner, C.; Baraitser, M.; Clark, R.D.; Schwartz, C.E.; Stevenson, R.E. Clark-Baraitser syndrome is associated with a nonsense alteration in the autosomal gene TRIP12. Am. J. Med Genet. Part A 2019, 182, 595–596. [Google Scholar] [CrossRef]
- Doco-Fenzy, M.; Landais, E.; Andrieux, J.; Schneider, A.; Delemer, B.; Sulmont, V.; Melin, J.-P.; Ploton, M.; Thevenard, J.; Monboisse, J.-C.; et al. Deletion 2q36.2q36.3 with multiple renal cysts and severe mental retardation. Eur. J. Med. Genet. 2008, 51, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.-J.; De Vries, P.; De Vries, B.B.A.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-analysis of 2104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nat. Cell Biol. 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Stessman, H.A.; Boyle, E.A.; Witherspoon, K.T.; Martin, B.; Lee, C.; Vives, L.; Baker, C.; Hiatt, J.B.; Nickerson, D.A.; et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun. 2014, 5, 1–6. [Google Scholar] [CrossRef]
- Zhang, J.; Gambin, T.; Yuan, B.; Szafranski, P.; Rosenfeld, J.A.; Al Balwi, M.; Alswaid, A.; Al-Gazali, L.; Al Shamsi, A.M.; Komara, M.; et al. Haploinsufficiency of the E3 ubiquitin-protein ligase gene TRIP12 causes intellectual disability with or without autism spectrum disorders, speech delay, and dysmorphic features. Qual. Life Res. 2017, 136, 377–386. [Google Scholar] [CrossRef]
- Takata, A.; Miyake, N.; Tsurusaki, Y.; Fukai, R.; Miyatake, S.; Koshimizu, E.; Kushima, I.; Okada, T.; Morikawa, M.; Uno, Y.; et al. Integrative Analyses of De Novo Mutations Provide Deeper Biological Insights into Autism Spectrum Disorder. Cell Rep. 2018, 22, 734–747. [Google Scholar] [CrossRef]
- Ruzzo, E.K.; Pérez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef]
- Stessman, H.A.F.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Yoo, N.J.; Park, S.W.; Lee, S.H. Frameshift mutations of ubiquitination-related genes HERC2, HERC3, TRIP12, UBE2Q1 and UBE4B in gastric and colorectal carcinomas with microsatellite instability. Pathology 2011, 43, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Hein, R.; Network, T.G.; Flesch-Janys, D.; Dahmen, N.; Beckmann, L.; Lindström, S.; Schoof, N.; Czene, K.; Mittelstraß, K.; Illig, T.; et al. A genome-wide association study to identify genetic susceptibility loci that modify ductal and lobular postmenopausal breast cancer risk associated with menopausal hormone therapy use: A two-stage design with replication. Breast Cancer Res. Treat. 2013, 138, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yi, S.; Yang, F.; Zhou, Y.; Ji, Q.; Cai, J.; Mei, Y. Identification of mutant genes with high-frequency, high-risk, and high-expression in lung adenocarcinoma. Thorac. Cancer 2014, 5, 211–218. [Google Scholar] [CrossRef]
- Cacheux, W.; Dangles-Marie, V.; Rouleau, E.; Lazartigues, J.; Girard, E.; Briaux, A.; Mariani, P.; Richon, S.; Vacher, S.; Buecher, B.; et al. Exome sequencing reveals aberrant signalling pathways as hallmark of treatment-naive anal squamous cell carcinoma. Oncotarget 2017, 9, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Shiba, N.; Yoshida, K.; Hara, Y.; Yamato, G.; Shiraishi, Y.; Matsuo, H.; Okuno, Y.; Chiba, K.; Tanaka, H.; Kaburagi, T.; et al. Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia. Blood Adv. 2019, 3, 3157–3169. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Attiye, M.; Gerold, J.M.; Makohon-Moore, A.P.; Hayashi, A.; Hong, J.; Kappagantula, R.; Zhang, L.; Melchor, J.P.; Reiter, J.G.; et al. The Evolutionary Origins of Recurrent Pancreatic Cancer. Cancer Discov. 2020, 10, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Jin, Z.; Cheng, Y.; Cao, X. RNA-Seq analysis identifies aberrant RNA splicing of TRIP12 in acute myeloid leukemia patients at remission. Tumor Biol. 2014, 35, 9585–9590. [Google Scholar] [CrossRef]
- Horvat, A.; Noto, J.M.; Ramatchandirin, B.; Zaika, E.; Palrasu, M.; Wei, J.; Schneider, B.G.; El-Rifai, W.; Peek, R.M.; Zaika, A.I. Helicobacter pylori pathogen regulates p14ARF tumor suppressor and autophagy in gastric epithelial cells. Oncogene 2018, 37, 5054–5065. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, P.-J.; Molkentine, D.P.; Chen, C.; Molkentine, J.; Piao, H.; Raju, U.; Zhang, J.; Valdecanas, D.R.; Tailor, R.C.; et al. TRIP12 as a mediator of human papillomavirus/p16-related radiation enhancement effects. Oncogene 2016, 36, 820–828. [Google Scholar] [CrossRef]
- Molkentine, J.M.; Molkentine, D.P.; Bridges, K.A.; Xie, T.; Yang, L.; Sheth, A.; Heffernan, T.P.; Clump, D.A.; Faust, A.Z.; Ferris, R.L.; et al. Targeting DNA damage response in head and neck cancers through abrogation of cell cycle checkpoints. Int. J. Radiat. Biol. 2020, 1–8. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).