Mechanisms of EGFR Resistance in Glioblastoma


Abstract
1. Introduction
2. Results
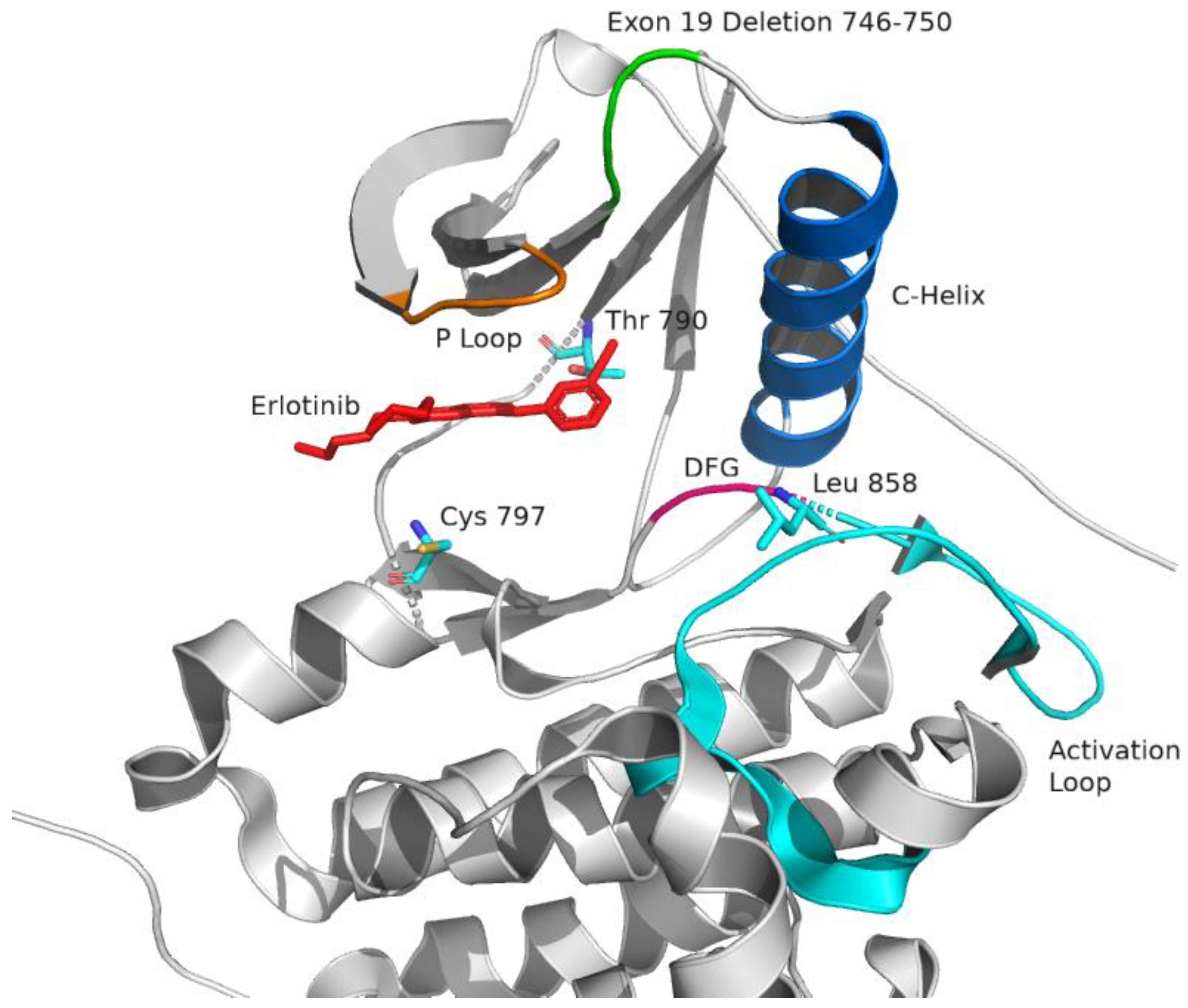
2.1. Epidermal Growth Factor Receptor (EGFR) Structure
2.2. Targeting EGFR
2.3. Specific EGFR Targeted Agents
2.3.1. Gefitinib
2.3.2. Erlotinib
2.3.3. Predicting Response of Erlotinib and Gefitinib in Glioblastoma
2.3.4. Afatinib
2.3.5. Dacomitinib
2.3.6. Osimertinib
2.3.7. Rociletinib
2.3.8. Lapatinib
2.3.9. Neratinib
2.3.10. Cetuximab
2.3.11. Rindopepimut
2.3.12. Depatuxizumab Mafodotin (ABT-414)
3. Discussion
Funding
Conflicts of Interest
Abbreviations
| ADC | Antibody drug conjugate |
| ATP | Adenosine triphosphate |
| BBB | Blood brain barrier |
| BiTE | Bispecific T-cell engager |
| CDK | Cyclin-dependent kinase |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CYP | Cytochrome P450 |
| ecDNA | Extrachromosomal DNA |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EIAED | Enzyme inducing antiepileptic drug |
| EORTC | European Organization for Research and Treatment of Cancer |
| ErbB | Erb-b2 receptor tyrosine kinase |
| FISH | Fluorescence in situ hybridization |
| GBM | Glioblastoma |
| Grb2 | Growth factor receptor-bound protein 2 |
| HB-EGF | Heparin-binding EGF-like growth factor |
| HER | Human epidermal growth factor receptor |
| IC50 | Half maximal inhibitory concentration |
| IGFR1 | Insulin growth factor receptor 1 |
| IHC | Immunohistochemistry |
| INSIGhT | Individualized Screening Trial of Innovative Glioblastoma Therapy clinical trial |
| JAK | Janus kinase |
| KLH | Keyhole limpet hemocyanin |
| KRAS | Kirsten rat sarcoma |
| MAPK | Mitogen-activated protein kinase |
| MET | MET receptor tyrosine kinase; also known as hepatocyte growth factor receptor |
| MGMT | Enzyme O6-methylguanine-DNA methyltransferase |
| MMAF | Monomethyl auristatin F |
| MRI | Magnetic resonance imaging |
| NABTC | North American Brain Tumor Consortium |
| NCCTG | North Central Cancer Treatment Group |
| NSCLC | Non-small cell lung cancer |
| OS | Overall survival |
| PDB | Protein Database |
| PI3K | Phosphoinositide-3 kinase |
| PFS | Progression free survival |
| PKB | Protein kinase B; also known as Akt |
| PKC | Protein kinase C |
| PLCγ | Phospholipase C γ |
| RET | Rearranged during transfection |
| RT | Radiotherapy |
| RTOG | Radiation Therapy Oncology Group |
| SPECT | Single-photon emission computed tomography |
| STAT | Signal transducer and activator of transcription |
| TGF-α | Transforming growth factor-α |
| TKI | Tyrosine kinase inhibitor |
| TMZ | Temozolomide |
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011. Neuro-Oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef]
- Stupp, R.; Weller, M.; Belanger, K.; Bogdahn, U.; Ludwin, S.K.; Lacombe, D.; Mirimanoff, R.O. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.F.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. A phase III randomized controlled trial of short-course radiotherapy with or without concomitant and adjuvant temozolomide in elderly patients with glioblastoma (CCTG CE.6, EORTC 26062-22061, TROG 08.02, NCT00482677). J. Clin. Oncol. 2016, 34, LBA2. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. RTOG 0525: A randomized phase III trial comparing standard adjuvant temozolomide (TMZ) with a dose-dense (dd) schedule in newly diagnosed glioblastoma (GBM). J. Clin. Oncol. 2011, 29, 2006. [Google Scholar] [CrossRef]
- Wick, W.; Platten, M.; Meisner, C.; Felsberg, J.; Tabatabai, G.; Simon, M.; Nikkhah, G.; Papsdorf, K.; Steinbach, J.P.; Sabel, M.; et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: The NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012, 13, 707–715. [Google Scholar] [CrossRef]
- Malmström, A.; Grønberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Jorissen, R.N. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Burgess, A.W. EGFR family: Structure physiology signalling and therapeutic targets. Growth Factors 2008, 26, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal Structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Ward, C.W.; Hoyne, P.A.; Flegg, R.H. Insulin and epidermal growth factor receptors contain the cysteine repeat motif found in the tumor necrosis factor receptor. Proteins Struct. Funct. Bioinform. 1995, 22, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Garrett, T.P.J.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Zhu, H.-J.; Walker, F.; Frenkel, M.J.; Hoyne, P.A.; et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor α. Cell 2002, 110, 763–773. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 1.2r3pre; Schrödinger, LLC.: New York, NY, USA; Available online: http://pymol.sourceforge.net/faq.html (accessed on 11 November 2020).
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Wiley, H.S.; Herbst, J.J.; Walsh, B.J.; Lauffenburger, D.A.; Rosenfeld, M.G.; Gill, G.N. The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 1991, 266, 12. [Google Scholar]
- Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2018, 47, D590–D595. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. STAT-mediated EGFR signaling in cancer. J. Cell. Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-J.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.-D.; Huang, C.-M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.; Cohen, S.; Bishayee, S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: A study with an aberrant epidermal growth factor receptor (EGFRVIII/ EGFR) expressed in cancer cells. J. Biol. Chem. 2000, 276, 5375–5383. [Google Scholar] [CrossRef] [PubMed]
- Orellana, L.; Thorne, A.H.; Lema, R.; Gustavsson, J.; Parisian, A.D.; Hospital, A.; Cordeiro, T.N.; Bernadó, P.; Scott, A.M.; Brun-Heath, I.; et al. Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proc. Natl. Acad. Sci. USA 2019, 116, 10009–10018. [Google Scholar] [CrossRef]
- Arkhipov, A.; Shan, Y.; Das, R.; Endres, N.F.; Eastwood, M.P.; Wemmer, D.E.; Kuriyan, J.; Shaw, D.E. Architecture and membrane interactions of the EGF receptor. Cell 2013, 152, 557–569. [Google Scholar] [CrossRef]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Sutto, L.; Gervasio, F.L. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10616–10621. [Google Scholar] [CrossRef]
- Carey, K.D.; Garton, A.J.; Romero, M.S.; Kahler, J.; Thomson, S.; Ross, S.; Park, F.; Haley, J.D.; Gibson, N.; Sliwkowski, M.X. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006, 66, 8163–8171. [Google Scholar] [CrossRef]
- Tartarone, A.; Lerose, R. Clinical approaches to treat patients with non-small cell lung cancer and epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance. Ther. Adv. Respir. Dis. 2015, 9, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Juchum, M.; Günther, M.; Laufer, S. Fighting cancer drug resistance: Opportunities and challenges for mutation-specific EGFR inhibitors. Drug Resist. Updat. 2015, 20, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Moran, T.; Majem, M.; Reguart, N.; Dalmau, E.; Márquez-Medina, D.; Lianes, P. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: A new era begins. Cancer Treat. Rev. 2014, 40, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Yoshida, K.; Hida, T.; Tsuboi, M.; Tada, H.; Kuwano, H.; Mitsudomi, T. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin. Cancer Res. 2006, 12, 5764–5769. [Google Scholar] [CrossRef] [PubMed]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [Google Scholar] [CrossRef]
- Kobayashi, S.; Jänne, P.A.; Meyerson, M.; Eck, M.J. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.H.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef]
- Hossam, M.; Lasheen, D.S.; Abouzid, K.A. Covalent EGFR inhibitors: Binding mechanisms, synthetic approaches, and clinical profiles. Arch. Pharm. 2016, 349, 573–593. [Google Scholar] [CrossRef]
- Arrieta, O.; Vega-González, M.T.; López-Macías, D.; Martínez-Hernández, J.N.; Bacon-Fonseca, L.; Macedo-Pérez, E.O.; Ramírez-Tirado, L.A.; Flores-Estrada, D.; De La Garza-Salazar, J. Randomized, open-label trial evaluating the preventive effect of tetracycline on afatinib induced-skin toxicities in non-small cell lung cancer patients. Lung Cancer 2015, 88, 282–288. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR Inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Van Der Steen, N.; Caparello, C.; Rolfo, C.; Pauwels, P.; Peters, G.J.; Giovannetti, E. New developments in the management of non-small-cell lung cancer, focus on rociletinib: What went wrong? OncoTargets Ther. 2016, 9, 6065–6074. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, S.; Wang, K.; Sun, S.-Y. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J. Hematol. Oncol. 2019, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Han, J.; Sequist, L.; Cho, B.; Lee, J.; Kim, S.; Su, W.; Tsai, C.; Yang, J.; Yu, H.; et al. OA 09.03 TATTON Ph Ib expansion cohort: Osimertinib plus savolitinib for Pts with EGFR-mutant MET-amplified NSCLC after progression on prior EGFR-TKI. J. Thorac. Oncol. 2017, 12, S1768. [Google Scholar] [CrossRef]
- Deng, L.; Kiedrowski, L.A.; Ravera, E.; Cheng, H.; Halmos, B. Response to dual crizotinib and osimertinib treatment in a lung cancer patient with MET amplification detected by liquid biopsy who acquired secondary resistance to EGFR tyrosine kinase inhibition. J. Thorac. Oncol. 2018, 13, e169–e172. [Google Scholar] [CrossRef]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.; Digumarthy, S.; Gainor, J.; Marcoux, N.; Banwait, M.; Dias-Santagata, D.; Iafrate, A.; Mino-Kenudson, M.; et al. MA26.03 Activity of osimertinib and the selective RET inhibitor BLU-667 in an EGFR-mutant patient with acquired RET rearrangement. J. Thorac. Oncol. 2018, 13, S451. [Google Scholar] [CrossRef]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28, S24–S31. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Shepard, D.R.; Ahluwalia, M.S.; Brewer, C.J.; Agarwal, N.; Stevens, G.H.J.; Suh, J.H.; Toms, S.A.; Vogelbaum, M.A.; Weil, R.J.; et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J. Neuro-Oncol. 2009, 98, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nat. Cell Biol. 2017, 543, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Tsai, C.; Yang, J.; Shepherd, F.; Satouchi, M.; Kim, D.; Bazhenova, L.; Hirashima, T.; Rukazenkov, Y.; Cantarini, M.; et al. 3083 AZD9291 activity in patients with EGFR-mutant advanced non-small cell lung cancer (NSCLC) and brain metastases: Data from phase II studies. Eur. J. Cancer 2015, 51, S625–S626. [Google Scholar] [CrossRef]
- Ballard, P.; Yates, J.W.; Yang, Z.; Kim, D.-W.; Yang, J.C.-H.; Cantarini, M.; Pickup, K.; Jordan, A.; Hickey, M.; Grist, M.; et al. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin. Cancer Res. 2016, 22, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Belda-Iniesta, C.; Carpeño, J.D.C.; Saenz, E.C.; Gutiérrez, M.; Perona, R.; Barón, M.G. Long term responses with cetuximab therapy in glioblastoma multiforme. Cancer Biol. Ther. 2006, 5, 912–914. [Google Scholar] [CrossRef]
- Scott, A.M.; Lee, F.-T.; Tebbutt, N.; Herbertson, R.; Gill, S.S.; Liu, Z.; Skrinos, E.; Murone, C.; Saunder, T.H.; Chappell, B.; et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 4071–4076. [Google Scholar] [CrossRef]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef]
- Bent, M.J.V.D.; Gao, Y.; Kerkhof, M.; Kros, J.M.; Gorlia, T.; Van Zwieten, K.; Prince, J.; Van Duinen, S.; Smitt, P.A.S.; Taphoorn, M.; et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro-Oncol. 2015, 17, 935–941. [Google Scholar] [CrossRef]
- Uhm, J.H.; Ballman, K.V.; Wu, W.; Giannini, C.; Krauss, J.; Buckner, J.C.; James, C.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J.; et al. Phase II Evaluation of Gefitinib in patients with newly diagnosed grade 4 astrocytoma: Mayo/north central cancer treatment group study N0074. Int. J. Radiat. Oncol. 2011, 80, 347–353. [Google Scholar] [CrossRef]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. RTOG 0211: A phase 1/2 Study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int. J. Radiat. Oncol. 2013, 85, 1206–1211. [Google Scholar] [CrossRef]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II Trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Prados, M.D.; Chang, S.M.; Butowski, N.; DeBoer, R.; Parvataneni, R.; Carliner, H.; Kabuubi, P.; Ayers-Ringler, J.; Rabbitt, J.; Page, M.; et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J. Clin. Oncol. 2009, 27, 579–584. [Google Scholar] [CrossRef]
- Brown, P.D.; Krishnan, S.; Sarkaria, J.N.; Wu, W.; Jaeckle, K.A.; Uhm, J.H.; Geoffroy, F.J.; Arusell, R.; Kitange, G.; Jenkins, R.B.; et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North central cancer treatment group study N0177. J. Clin. Oncol. 2008, 26, 5603–5609. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.A.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2009, 12, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O.; Cuatrecasas, M.; Benavides, M.; Segura, P.P.; Berrocal, A.; Erill, N.; Colomer, A.; Quintana, M.J.; Balaña, C.; Gil, M.; et al. Efficacy of erlotinib in patients with relapsed gliobastoma multiforme who expressed EGFRVIII and PTEN determined by immunohistochemistry. J. Neuro-Oncol. 2014, 116, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Norden, A.D.; Cloughesy, T.F.; Robins, H.I.; Lieberman, F.S.; Gilbert, M.R.; Mehta, M.P.; et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol. 2014, 16, 567–578. [Google Scholar] [CrossRef]
- Humphrey, P.A.; Wong, A.J.; Vogelstein, B.; Zalutsky, M.R.; Fuller, G.N.; Archer, G.E.; Friedman, H.S.; Kwatra, M.M.; Bigner, S.H.; Bigner, D.D. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc. Natl. Acad. Sci. USA 1990, 87, 4207–4211. [Google Scholar] [CrossRef]
- Aldape, K.D.; Ballman, K.; Furth, A.; Buckner, J.C.; Giannini, C.; Burger, P.C.; Scheithauer, B.W.; Jenkins, R.B.; James, C.D. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 2004, 63, 700–707. [Google Scholar] [CrossRef]
- Francis, J.M.; Zhang, C.-Z.; Maire, C.L.; Jung, J.; Manzo, V.E.; Adalsteinsson, V.A.; Homer, H.; Haidar, S.; Blumenstiel, B.; Pedamallu, C.S.; et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014, 4, 956–971. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted Therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2013, 343, 72–76. [Google Scholar] [CrossRef]
- Lassman, A.B.; Rossi, M.R.; Razier, J.R.; Abrey, L.E.; Lieberman, F.S.; Grefe, C.N.; Lamborn, K.; Pao, W.; Shih, A.H.; Kuhn, J.G.; et al. Molecular Study of malignant gliomas treated with epidermal growth factor receptor inhibitors: Tissue analysis from north American brain tumor consortium trials 01-03 and 00-01. Clin. Cancer Res. 2005, 11, 7841–7850. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Schulte, A.; Liffers, K.; Kathagen, A.; Riethdorf, S.; Zapf, S.; Merlo, A.; Kolbe, K.; Westphal, M.; Lamszus, K. Erlotinib resistance in EGFR-amplified glioblastoma cells is associated with upregulation of EGFRvIII and PI3Kp110δ. Neuro Oncol. 2013, 15, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.J.; Acquaviva, J.; Chi, D.; Lessard, J.; Zhu, H.; Woolfenden, S.; Bronson, R.T.; Pfannl, R.; White, F.M.; Housman, D.E.; et al. Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene 2011, 31, 3039–3050. [Google Scholar] [CrossRef]
- Velpula, K.K.; Dasari, V.R.; Asuthkar, S.; Gorantla, B.; Tsung, A.J. EGFR and c-Met Cross Talk in Glioblastoma and Its Regulation by Human Cord Blood Stem Cells. Transl. Oncol. 2012, 5, 379–392. [Google Scholar] [CrossRef]
- Ma, Y.; Tang, N.; Thompson, R.C.; Mobley, B.C.; Clark, S.W.; Sarkaria, J.N.; Wang, J. InsR/IGF1R pathway mediates resistance to EGFR inhibitors in glioblastoma. Clin. Cancer Res. 2016, 22, 1767–1776. [Google Scholar] [CrossRef]
- Chakravarti, A.; Loeffler, J.S.; Dyson, N.J. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002, 62, 200–207. [Google Scholar]
- Burtness, B.; Anadkat, M.; Basti, S.; Hughes, M.; Lacouture, M.E.; McClure, J.S.; Myskowski, P.L.; Paul, J.; Perlis, C.S.; Saltz, L.; et al. NCCN task force report: Management of dermatologic and other toxicities associated with EGFR inhibition in patients with cancer. J. Natl. Compr. Cancer Netw. 2009, 7, S-5–S-21. [Google Scholar] [CrossRef]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y.; et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro Oncol. 2014, 17, 430–439. [Google Scholar] [CrossRef]
- Eisenstat, D.D.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.R.; Kavan, P.; Phuphanich, S.; Fu, Y.; Cong, X.J.; Shahidi, M.; et al. A phase II study of daily afatinib (BIBW 2992) with or without temozolomide (21/28 days) in the treatment of patients with recurrent glioblastoma. J. Clin. Oncol. 2011, 29, 2010. [Google Scholar] [CrossRef]
- Zahonero, C.; Aguilera, P.; Ramírez-Castillejo, C.; Pajares, M.; Bolós, M.V.; Cantero, D.; Perez-Nuñez, A.; Hernández-Laín, A.; Sanchez-Gomez, P.; Sepulveda-Sanchez, J. Preclinical test of dacomitinib, an irreversible EGFR inhibitor, confirms its effectiveness for glioblastoma. Mol. Cancer Ther. 2015, 14, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda-Sánchez, J.M.; Ángeles, M.V.; Balana, C.; Gil-Gil, M.; Reynés, G.; Gallego, Ó.; Martínez-García, M.; Vicente, E.; Quindós, M.; Luque, R.; et al. Phase II trial of dacomitinib, a pan–human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol. 2017, 19, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Goss, G.D.; Tsai, C.-M.; Shepherd, F.; Ahn, M.-J.; Bazhenova, L.; Crinò, L.; De Marinis, F.; Felip, E.; Morabito, A.; Hodge, R.; et al. CNS response to osimertinib in patients with T790M-positive advanced NSCLC: Pooled data from two phase II trials. Ann. Oncol. 2018, 29, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Kwatra, M.; Nanni, C.; Roberts, C.; Kwatra, S.; Gilbert, M.R.; Lesser, G.J. Exth-a precision medicine approach to target EGFRVIII in GBM: Osimertinib (azd9291) inhibits the growth of EGFRVIII-positive glioblastoma stem cells and increases survival of mice bearing intracranial EGFRVIII-positive GBM. Neuro Oncol. 2017, 19, vi82. [Google Scholar] [CrossRef]
- Liu, X.; Chen, X.; Shi, L.; Shan, Q.; Cao, Q.; Yue, C.; Li, H.; Li, S.; Wang, J.; Gao, S.; et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 1–14. [Google Scholar] [CrossRef]
- Makhlin, I.; Salinas, R.D.; Zhang, D.; Jacob, F.; Ming, G.-L.; Song, H.; Saxena, D.; Dorsey, J.F.; Nasrallah, M.P.; Morrissette, J.J.; et al. Clinical activity of the EGFR tyrosine kinase inhibitor osimertinib in EGFR-mutant glioblastoma. CNS Oncol. 2019, 8, CNS43. [Google Scholar] [CrossRef]
- Walter, A.O.; Sjin, R.T.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 11. [Google Scholar]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef]
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L.; et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2009, 65, 353–361. [Google Scholar] [CrossRef]
- Karavasilis, V.; Kotoula, V.; Pentheroudakis, G.; Televantou, D.; Lambaki, S.; Chrisafi, S.; Bobos, M.; Fountzilas, G. A phase I study of temozolomide and lapatinib combination in patients with recurrent high-grade gliomas. J. Neurol. 2013, 260, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Faiq, N.; Green, S.; Lai, A.; Green, R.; Hu, J.; Cloughesy, T.F.; Mellinghoff, I.; Nghiemphu, P.L. Report of safety of pulse dosing of lapatinib with temozolomide and radiation therapy for newly-diagnosed glioblastoma in a pilot phase II study. J. Neuro Oncol. 2017, 134, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.M.; Trippa, L.; Gaffey, S.C.; Arrillaga, I.; Lee, E.Q.; Tanguturi, S.K.; Ahluwalia, M.S.; Colman, H.; Galanis, E.; De Groot, J.F.; et al. Individualized screening trial of innovative glioblastoma therapy (INSIGhT). J. Clin. Oncol. 2017, 35, TPS2079. [Google Scholar] [CrossRef]
- Hasselbalch, B.; Lassen, U.; Poulsen, H.S.; Stockhausen, M.-T. Cetuximab insufficiently inhibits glioma cell growth due to persistent EGFR downstream signaling. Cancer Investig. 2010, 28, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, B.; Lassen, U.; Hansen, S.; Holmberg, M.; Mau-Sørensen, M.; Kosteljanetz, M.; Broholm, H.; Stockhausen, M.-T.; Poulsen, H.S. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: A phase II trial. Neuro Oncol. 2010, 12, 508–516. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Mitten, M.J.; Norvell, S.; Falls, H.D.; Devries, P.J.; Cheng, D.; Meulbroek, J.A.; Buchanan, F.G.; et al. ABT-414, an antibody-drug conjugate targeting a tumor-selective EGFR epitope. Mol. Cancer Ther. 2016, 15, 661–669. [Google Scholar] [CrossRef]
- Reilly, E.B.; Phillips, A.C.; Buchanan, F.G.; Kingsbury, G.; Zhang, Y.; Meulbroek, J.A.; Cole, T.B.; Devries, P.J.; Falls, H.D.; Beam, C.; et al. Characterization of ABT-806, a humanized tumor-specific anti-EGFR monoclonal antibody. Mol. Cancer Ther. 2015, 14, 1141–1151. [Google Scholar] [CrossRef]
- Garrett, T.P.J.; Burgess, A.W.; Gan, H.K.; Luwor, R.B.; Cartwright, G.; Walker, F.; Orchard, S.G.; Clayton, A.H.A.; Nice, E.C.; Rothacker, J.; et al. Antibodies specifically targeting a locally misfolded region of tumor associated EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 5082–5087. [Google Scholar] [CrossRef]
- Jungbluth, A.A.; Stockert, E.; Huang, H.J.S.; Collins, V.P.; Coplan, K.; Iversen, K.; Kolb, D.; Johns, T.G.; Scott, A.M.; Gullick, W.J.; et al. A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 639–644. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Dimino, C.; Mansukhani, M.M.; Murty, V.V.; Canoll, P.; Narita, Y.; Muragaki, Y.; Gan, H.K.; Merrell, R.T.; Bent, M.J.V.D.; et al. Effect of therapeutic pressure on stability of EGFR amplification in glioblastoma. J. Clin. Oncol. 2018, 36, 2033. [Google Scholar] [CrossRef]
- Gan, H.K.; Reardon, D.A.; Lassman, A.B.; Merrell, R.; Bent, M.V.D.; Butowski, N.; Lwin, Z.; Wheeler, H.; Fichtel, L.; Scott, A.M.; et al. Safety, pharmacokinetics, and antitumor response of depatuxizumab mafodotin as monotherapy or in combination with temozolomide in patients with glioblastoma. Neuro Oncol. 2017, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Narita, Y.; Muragaki, Y.; Maruyama, T.; Kagawa, N.; Asai, K.; Kuroda, J.; Kurozumi, K.; Nagane, M.; Matsuda, M.; Ueki, K.; et al. Phase I/II study of depatuxizumab mafodotin (ABT-414) monotherapy or combination with temozolomide in Japanese patients with/without EGFR-amplified recurrent glioblastoma. J. Clin. Oncol. 2019, 37, 2065. [Google Scholar] [CrossRef]
- Bent, M.J.V.D.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol. 2019, 22, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Lobbous, M.; Nabors, L.B. A troublesome burden, the amplification of EGFR in glioblastoma! Neuro Oncol. 2020, 22, 594–595. [Google Scholar] [CrossRef]
- AbbVie News Center. AbbVie Provides Update on Depatuxizumab Mafodotin (Depatux-M), an Investigational Medicine for Newly Diagnosed Glioblastoma, an Aggressive Form of Brain Cancer. Available online: https://news.abbvie.com/news/press-releases/abbvie-provides-update-on-depatuxizumab-mafodotin-depatux-m-an-investigational-medicine-for-newly-diagnosed-glioblastoma-an-aggressive-form-brain-cancer.htm (accessed on 10 July 2020).
- Lassman, A.B.; Aldape, K.D.; Ansell, P.J.; Bain, E.; Curran, W.J.; Eoli, M.; French, P.J.; Kinoshita, M.; Looman, J.; Mehta, M.; et al. Epidermal growth factor receptor (EGFR) amplification rates observed in screening patients for randomized trials in glioblastoma. J. Neuro Oncol. 2019, 144, 205–210. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Suki, D.; Yang, D.; Shi, W.; Aldape, K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 2005, 3, 38. [Google Scholar] [CrossRef]
- Inda, M.-D.-M.; Bonavia, R.; Mukasa, A.; Narita, Y.; Sah, D.W.; Vandenberg, S.; Brennan, C.; Johns, T.G.; Bachoo, R.; Hadwiger, P.; et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010, 24, 1731–1745. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 1–14. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.M.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.-F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—A phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Amit, I.; Wides, R.; Yarden, Y. Evolvable signaling networks of receptor tyrosine kinases: Relevance of robustness to malignancy and to cancer therapy. Mol. Syst. Biol. 2007, 3, 151. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Yarden, Y. EGF–ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Pourzia, A.L.; Nourian, A.A.; Williams, K.J.; Nathanson, D.; Babic, I.; Villa, G.R.; Tanaka, K.; Nael, A.; Yang, H.; et al. De-repression of PDGFRβ transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discov. 2013, 3, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Franklin, M.C.; Carey, K.D.; Vajdos, F.F.; Leahy, D.J.; De Vos, A.M.; Sliwkowski, M.X. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. [Google Scholar] [CrossRef]
- Needham, S.R.; Roberts, S.K.; Arkhipov, A.; Mysore, V.P.; Tynan, C.J.; Zanetti-Domingues, L.C.; Kim, E.T.; LoSasso, V.; Korovesis, D.; Hirsch, M.; et al. EGFR oligomerization organizes kinase-active dimers into competent signalling platforms. Nat. Commun. 2016, 7, 13307. [Google Scholar] [CrossRef]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). August 2020. Available online: https://clinicaltrials.gov/ct2/results?term=egfr&cond=glioblastoma&Search=Apply&recrs=b&recrs=a&recrs=f&recrs=d&age_v=&gndr=&type=&rslt= (accessed on 6 August 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Mechanism of Action | |
|---|---|---|
| Gefitinib | Type I TKI | Targets EGFR. Reversible ATP-site competitive inhibitor of EGFR kinase domain. |
| Erlotinib | Type 1 and Type I ½B TKI | Targets EGFR. Reversible ATP-site competitive inhibitor of EGFR kinase domain. |
| Afatinib | Type VI (irreversible) TKI | Targets EGFR. Irreversible ATP-site EGFR kinase inhibitor. Covalent bond with Cys 797 on EGFR kinase domain. |
| Rociletinib | Type VI (irreversible) TKI | Targets EGFR. Irreversible ATP-site EGFR kinase inhibitor. Covalent bond with Cys 797 on EGFR kinase domain. |
| Osimertinib | Type VI (irreversible) TKI | Targets EGFR. Designed for low affinity for wild-type EGFR compared to L858R/T790M, with improved therapeutic index. Irreversible ATP-site EGFR kinase inhibitor. Covalent bond with Cys 797 on EGFR kinase domain. |
| Lapatinib | Type I ½ TKI | Targets EGFR/HER2/HER4. Reversible ATP-site competitive inhibitor, targeting the inactive kinase conformation (αC-helix out). Dual EGFR/HER2 inhibitor. |
| Dacomitinib | Type VI (irreversible) TKI | Pan-ErbB TKI. Irreversible pan-ErbB tyrosine kinase ATP-site kinase inhibitor. Covalent bond with Cys 797 on EGFR kinase domain. |
| Neratinib | Type VI (irreversible) TKI | Targets EGFR/HER2/HER4. Irreversible pan-ErbB tyrosine kinase ATP-site kinase inhibitor. Covalent bond with Cys 797 on EGFR kinase domain. |
| Cetuximab | Monoclonal Antibody | Targets extracellular domain III, interfering with ligand binding. |
| Rindopepimut (CDX-110) | Peptide Vaccine | Peptide vaccine of EGFRvIII-specific peptide conjugated to KLH |
| Depatuxizumab mafodotin (ABT-414) | Antibody Drug Conjugate | Targets cryptic region on extracellular domain II exposed by EGFRvIII mutation, extracellular domain I mutations, and domain II mutations. Conjugated to MMAF (monomethyl auristatin F) payload. |
| Trial | Phase | Therapeutic Approach | Response | Status |
|---|---|---|---|---|
| Mayo/NCCTG N0074 [52] | II | Gefitinib 500–1000 mg daily in new GBM | PFS12 16.7% OS12 54.2% | Closed |
| N0177 [53] | I/II | Erlotinib 150 mg daily in new GBM | OS12 61% | Closed |
| Reardon et al., 2014 [54] and Eisenstat et al., 2011 [55] | II | Afatinib 20–50 mg daily alone, in combination with TMZ, or TMZ alone in recurrent GBM | PFS6 3%, 10%, 23% respectively | Closed |
| Karavasilis et al., 2013 [56] | I | Lapatinib 1000–1500 mg twice daily in recurrent GBM | mOS 5.9 months mPFS 2.4 months | Closed |
| ACT IV [57] | III | Rindopepimut in new GBM | No improvement compared to control | Closed |
| INTELLANCE-2 [58] | II | Depatux-M alone, in combination with TMZ, or control (TMZ or lomustine), in recurrent GBM | OS24 19.8% with combination Depatux-M and TMZ compared to control OS24 5.2%. No benefit in monotherapy Depatux-M arm. | Closed |
| INTELLANCE-1 [59] | III | Depatux-M in combination with RT/TMZ vs. RT/TMZ alone, in new GBM | Stopped for futility; no survival benefit at prespecified endpoint | Closed |
| INSIGhT | II | Neratinib in new GBM | Pending | Ongoing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, P.C.; Magge, R.S. Mechanisms of EGFR Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 8471. https://doi.org/10.3390/ijms21228471
Pan PC, Magge RS. Mechanisms of EGFR Resistance in Glioblastoma. International Journal of Molecular Sciences. 2020; 21(22):8471. https://doi.org/10.3390/ijms21228471
Chicago/Turabian StylePan, Peter C., and Rajiv S. Magge. 2020. "Mechanisms of EGFR Resistance in Glioblastoma" International Journal of Molecular Sciences 21, no. 22: 8471. https://doi.org/10.3390/ijms21228471
APA StylePan, P. C., & Magge, R. S. (2020). Mechanisms of EGFR Resistance in Glioblastoma. International Journal of Molecular Sciences, 21(22), 8471. https://doi.org/10.3390/ijms21228471