Molecular Mechanisms of Treatment Resistance in Glioblastoma

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. General Mechanisms of Treatment Resistance
2.1. The Blood–Brain and Blood–Brain–Tumor Barriers
2.2. Intra- and Intertumoral Heterogeneity
2.2.1. Cellular Heterogeneity
2.2.2. Spatial Heterogeneity
2.2.3. Heterogeneity between Primary and Recurrent Tumors
2.3. Heterogeneity of the Tumoral Microenvironment
3. Molecular Mechanisms of Resistance to Conventional Therapy
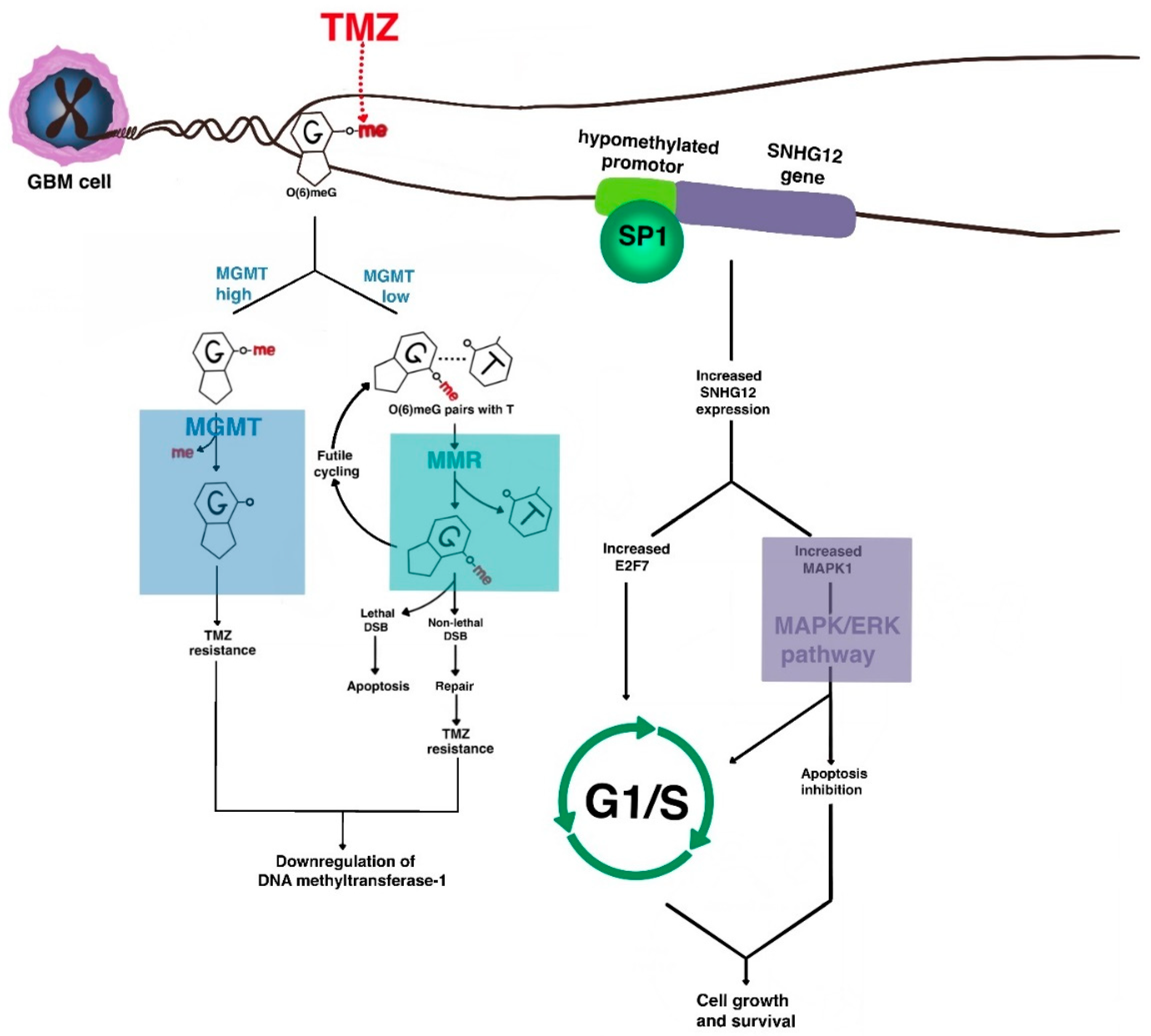
3.1. Resistance to Cytotoxic Chemotherapy
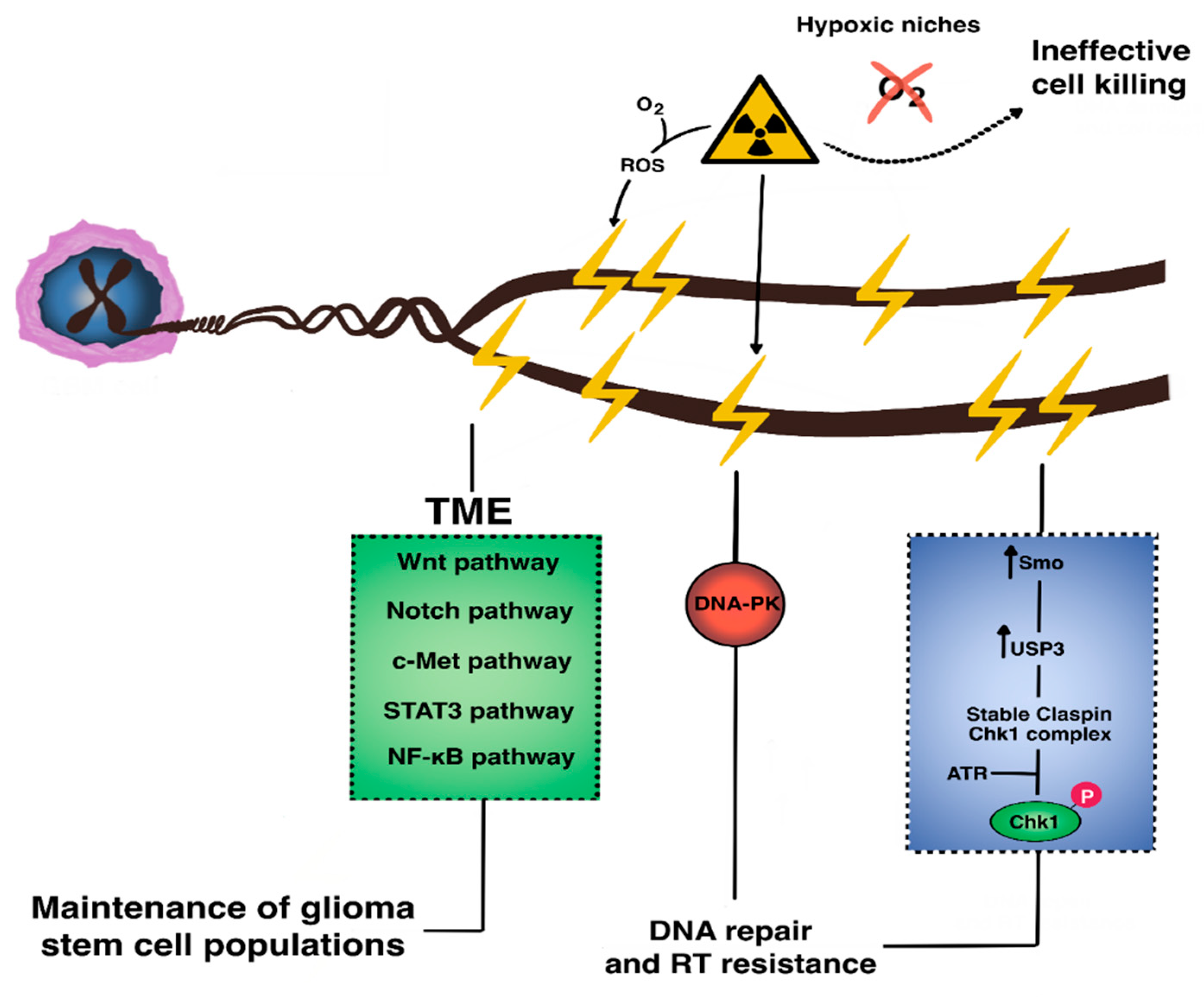
3.2. Resistance to Radiotherapy
4. Molecular Mechanisms of Resistance to Targeted Therapy
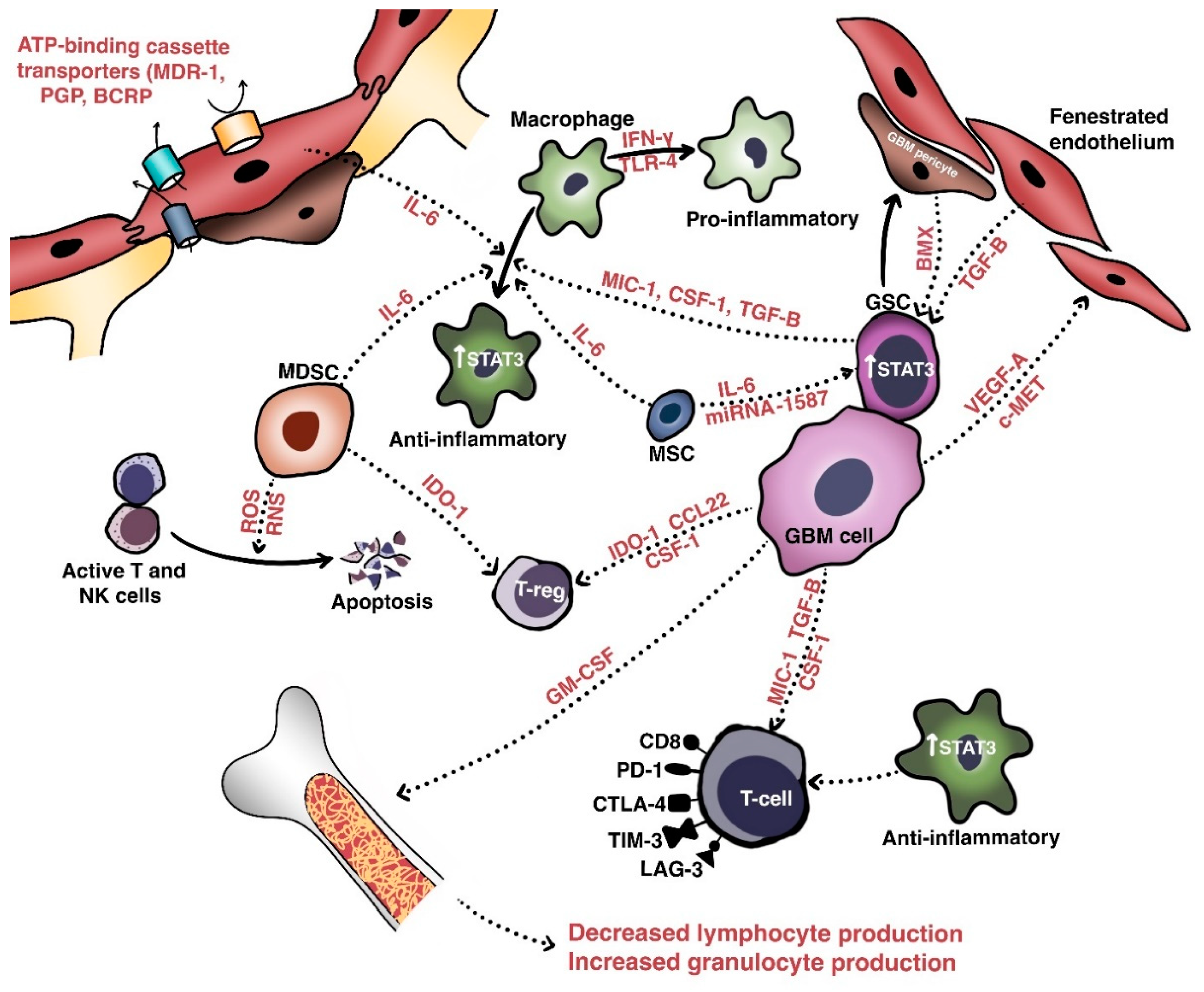
5. Molecular Mechanisms of Resistance to Immunotherapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003, 5, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II Trial of Single-Agent Bevacizumab Followed by Bevacizumab Plus Irinotecan at Tumor Progression in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. New Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy with Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastomaa: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood–brain barrier: An overview: Structure, regulation, and clinical implica-tions. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Pappenheimer, J.R.; Renkin, E.M.; Borrero, L.M. Filtration, Diffusion and Molecular Sieving Through Peripheral Capillary Membranes; a con-tribution to the pore theory of capillary permeability. Am. J. Physiol. Content 1951, 167, 13–46. [Google Scholar] [CrossRef]
- Ueda, K.; Cornwell, M.M.; Gottesman, M.M.; Pastan, I.; Roninson, I.B.; Ling, V.; Riordan, J.R. The mdrl gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem. Biophys. Res. Commun. 1986, 141, 956–962. [Google Scholar] [CrossRef]
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; E Mackie, J.; E Grant, C.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line [published correction appears in Science]. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Tsuji, A.; Terasaki, T.; Takabatake, Y.; Tenda, Y.; Tamai, I.; Yamashima, T.; Moritani, S.; Tsuruo, T.; Yamashita, J. P-glycoprotein as the drug efflux pump in primary cultured bovine brain capillary endothelial cells. Life Sci. 1992, 51, 1427–1437. [Google Scholar] [CrossRef]
- Becker, C.M.; Oberoi, R.K.; McFarren, S.J.; Muldoon, D.M.; Pafundi, D.H.; Pokorny, J.L.; Brinkmann, D.H.; Ohlfest, J.R.; Sarkaria, J.N.; Largaespada, D.A.; et al. Decreased affinity for efflux transporters increases brain penetrance and molecular targeting of a PI3K/mTOR inhibitor in a mouse model of glioblastoma. Neuro Oncol. 2015, 17, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Kizilbash, S.H.; Gupta, S.K.; Chang, K.; Kawashima, R.; Parrish, K.E.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A.; Decker, P.A.; et al. Restricted Delivery of Talazoparib Across the Blood–Brain Barrier Limits the Sensitizing Effects of PARP Inhibition on Temozolomide Therapy in Glioblastoma. Mol. Cancer 2017, 16, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Ratnam, N.M.; Gilbert, M.R.; Giles, A.J. Immunotherapy in CNS cancers: The role of immune cell trafficking. Neuro Oncol. 2018, 21, 37–46. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, H.; Xiong, C.; Liu, Y. Hypoxic glioblastoma release exosomal VEGF-A induce the permeability of blood-brain barrier. Biochem. Biophys. Res. Commun. 2018, 502, 324–331. [Google Scholar] [CrossRef]
- Wang, W.; Dentler, W.L.; Borchardt, R.T. VEGF increases BMEC monolayer permeability by affecting occludin expression and tight junction assembly. Am. J. Physiol. Circ. Physiol. 2001, 280, H434–H440. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nat. Cell Biol. 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Mettang, M.; Meyer-Pannwitt, V.; Karpel-Massler, G.; Zhou, S.; Carragher, N.O.; Föhr, K.J.; Baumann, B.; Nonnenmacher, L.; Enzenmüller, S.; Dahlhaus, M.; et al. Blocking distinct interactions between Glioblastoma cells and their tissue microenvironment: A novel multi-targeted therapeutic approach. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor Tyrosine Kinase BMX Maintains Self-Renewal and Tumorigenic Potential of Glioblastoma Stem Cells by Activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, C.; Shideng, B.; Bian, X.; Gimple, R.C.; Fang, X.; Huang, Z.; Zhai, K.; Ke, S.Q.; Ping, Y.-F.; et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell 2017, 21, 591–603.e4. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Guryanova, O.A.; Zhou, W.; Liu, C.; Huang, Z.; Fang, X.; Wang, X.; Chen, C.; Wu, Q.; He, Z.; et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 2018, 10, eaah6816. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef]
- Findley, M.K.; Koval, M. Regulation and roles for claudin-family tight junction proteins. IUBMB Life 2009, 61, 431–437. [Google Scholar] [CrossRef]
- Roudnicky, F.; Kim, B.K.; Lan, Y.; Schmucki, R.; Küppers, V.; Christensen, K.; Graf, M.; Patsch, C.; Burcin, M.; Meyer, C.A.; et al. Identification of a combination of transcription factors that synergistically increases endothelial cell barrier resistance. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Chang, J.; Mancuso, M.R.; Maier, C.; Liang, X.; Yuki, K.; Yang, L.; Kwong, J.W.; Wang, J.; Rao, V.; Vallon, M.; et al. Gpr124 is essential for blood–brain barrier integrity in central nervous system disease. Nat. Med. 2017, 23, 450–460. [Google Scholar] [CrossRef]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.-C.; et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 2018, 33, 874–889.e7. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Ii, J.E.H.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. New Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the TGF- 2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro Oncol. 2010, 13, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Chang, S.M.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010, 12, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-T.; Lin, Y.-J.; Chai, W.-Y.; Lin, C.-J.; Chen, P.-Y.; Huang, C.-Y.; Kuo, J.S.; Liu, H.-L.; Wei, K.-C. Neuronavigation-guided focused ultrasound (NaviFUS) for transcranial blood-brain barrier opening in recurrent glioblastoma patients: Clinical trial protocol. Ann. Transl. Med. 2020, 8, 673. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Colombeau, L.; Gries, M.; Peterlini, T.; Mathieu, C.; Thomas, N.; Boura, C.; Frochot, C.; Vanderesse, R.; Lux, F.; et al. Ultrasmall AGuIX theranostic nanoparticles for vascular-targeted interstitial photodynamic therapy of glioblastoma. Int. J. Nanomed. 2017, 12, 7075–7088. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Haider, T.; Chandak, A.R.; Chakraborty, A.; Banerjee, S.; Soni, V. Surface modified silk fibroin nanoparticles for improved delivery of doxorubicin: Development, characterization, in-vitro studies [published online ahead of print, 2020 August 3]. Int. J. Biol. Macromol. 2020, 164, 2018–2027. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways [published correction appears in Nature]. Nat. Cell Biol. 2008, 455, 1061–1068. [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Kim, Y.-W.; Koul, D.; Kim, S.H.; Lucio-Eterovic, A.K.; Freire, P.R.; Yao, J.; Wang, J.; Almeida, J.S.; Aldape, K.; Yung, W.A. Identification of prognostic gene signatures of glioblastoma: A study based on TCGA data analysis. Neuro Oncol. 2013, 15, 829–839. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, X.; Hu, B.; Muller, F.; Kim, H.; Squatrito, M.; Millelsen, T.; Scarpace, L.; Barthel, F.; Lin, Y.-H.; et al. Tumor evolution of glioma intrinsic gene expression subtype associates with immunological changes in the microenvironment. bioRxiv 2016, 052076. [Google Scholar] [CrossRef]
- Gill, B.J.; Pisapia, D.J.; Malone, H.R.; Goldstein, H.; Lei, L.; Sonabend, A.; Yun, J.; Samanamud, J.; Sims, J.S.; Banu, M.; et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 12550–12555. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Rennert, R.; Achrol, A.S.; Januszyk, M.; Kahn, S.A.; Liu, T.T.; Liu, Y.; Sahoo, D.; Rodrigues, M.; Maan, Z.N.; Wong, V.W.; et al. Multiple Subsets of Brain Tumor Initiating Cells Coexist in Glioblastoma a: Multiplici-ty Exists Within GBM Initiating Cells. Stem Cells 2016, 34, 1702–1707. [Google Scholar] [CrossRef]
- Pesenti, C.; Navone, S.E.; Guarnaccia, L.; Terrasi, A.; Costanza, J.; Silipigni, R.; Guarneri, S.; Fusco, N.; Fontana, L.; Locatelli, M.; et al. The Genetic Landscape of Human Glioblastoma and Matched Primary Cancer Stem Cells Reveals Intratumour Similarity and Intertumour Heterogeneity. Stem Cells Int. 2019, 2019, 2617030. [Google Scholar] [CrossRef]
- Piccirillo, S.; Colman, S.; Potter, N.E.; Van Delft, F.W.; Lillis, S.; Carnicer, M.-J.; Kearney, L.; Watts, C.; Greaves, M. Genetic and functional diversity of propagating cells in glioblastoma. Stem Cell Rep. 2014, 4, 7–15. [Google Scholar] [CrossRef]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.J.; Potdar, S.; Kyttälä, A.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M.; et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer 2019, 19, 1–14. [Google Scholar] [CrossRef]
- Herting, C.J.; Chen, Z.; Pitter, K.L.; Szulzewsky, F.; Kaffes, I.; Kaluzova, M.; Park, J.C.; Cimino, P.J.; Brennan, C.; Wang, B.; et al. Genetic driver mutations define the expression signature and microenvironmental composition of high-grade gliomas. Glia 2017, 65, 1914–1926. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, N.; Gielen, G.H.; Rauschenbach, L.; Kebir, S.; Till, A.; Reinartz, R.; Simon, M.; Niehusmann, P.; Kleinschnitz, C.; Herrlinger, U.; et al. Longitudinal heterogeneity in glioblastoma: Moving targets in recurrent versus primary tumors. J. Transl. Med. 2019, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, I.-H.; Cho, H.J.; Park, C.-K.; Jung, Y.-S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.-S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Körber, V.; Yang, J.; Barah, P.; Wu, Y.; Stichel, D.; Gu, Z.; Fletcher, M.N.C.; Jones, D.; Hentschel, B.; Lamszus, K.; et al. Evolutionary Trajectories of IDHWT Glioblastomas Reveal a Common Path of Early Tumorigenesis Instigated Years ahead of Initial Diagnosis. Cancer Cell 2019, 35, 692–704.e12. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef]
- Akgül, S.; Patch, A.-M.; D’Souza, R.C.J.; Mukhopadhyay, P.; Nones, K.; Kempe, S.; Kazakoff, S.; Jeffree, R.L.; Stringer, B.W.; Pearson, J.V.; et al. Intratumoural Heterogeneity Underlies Distinct Therapy Responses and Treatment Resistance in Glioblastoma. Cancers 2019, 11, 190. [Google Scholar] [CrossRef]
- Barthel, F.P.; The GLASS Consortium; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef]
- Kim, E.L.; Sorokin, M.; Kantelhardt, S.R.; Kalasauskas, D.; Sprang, B.; Fauss, J.; Ringel, F.; Garazha, A.; Albert, E.; Gaifullin, N.M.; et al. Intratumoral Heterogeneity and Longitudinal Changes in Gene Expression Predict Differential Drug Sensitivity in Newly Diagnosed and Recurrent Glioblastoma. Cancers 2020, 12, 520. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef] [PubMed]
- Heiland, D.H.; Gaebelein, A.; Boerries, M.; Woerner, J.; Pompe, N.; Franco, P.; Heynckes, S.; Bartholomä, M.D.; Hailín, D.Ó.; Carro, M.S.; et al. Microenvironment-Derived Regulation of HIF Signaling Drives Transcriptional Heterogeneity in Glioblastoma Multiforme. Mol. Cancer Res. 2018, 16, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Luoto, S.; Hermelo, I.; Vuorinen, E.M.; Hannus, P.; Kesseli, J.; Nykter, M.; Granberg, K.J. Computational Characterization of Suppressive Immune Microenvironments in Glioblastoma. Cancer Res. 2018, 78, 5574–5585. [Google Scholar] [CrossRef] [PubMed]
- Fianco, G.; Mongiardi, M.P.; Levi, A.; De Luca, T.; Desideri, M.; Trisciuoglio, D.; Del Bufalo, D.; Cinà, I.; Di Benedetto, A.; Mottolese, M.; et al. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. eLife 2017, 6. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Bueno, C.; Ródenas, M.; Moraleda, J.M.; Macian, F.; Martínez, S. Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget 2017, 8, 68614–68626. [Google Scholar] [CrossRef]
- Huang, M.; Liu, T.; Ma, P.; Mitteer, R.A.; Zhang, Z.; Kim, H.J.; Yeo, E.; Zhang, D.; Cai, P.; Li, C.; et al. c-Met–mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J. Clin. Investig. 2016, 126, 1801–1814. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, D.; Wu, J.Y.; Xing, K.; Yeo, E.; Li, C.; Zhang, L.; Holland, E.C.; Yao, L.; Qin, L.; et al. Wnt-mediated endothelial transformation into mesenchymal stem cell–like cells induces chemoresistance in glioblastoma. Sci. Transl. Med. 2020, 12, eaay7522. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Riquelme, D.; Martínez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martínez, S. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef]
- Kast, R.E.; A Hill, Q.; Wion, D.; Mellstedt, H.; Focosi, D.; Karpel-Massler, G.; Heiland, T.; Halatsch, M.-E. Glioblastoma-synthesized G-CSF and GM-CSF contribute to growth and immunosuppression: Potential therapeutic benefit from dapsone, fenofibrate, and ribavirin. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Győrffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Ahn, B.J.; Han, S.J.; Parsa, A.T. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: Implications for immunotherapy. Neuro Oncol. 2012, 14, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.-A.; Zhou, S.; Nonnenmacher, L.; Karpel-Massler, G.; Jennewein, C.; Schneider, M.; Halatsch, M.-E.; Carragher, N.O.; Baumann, B.; Krause, A.; et al. Inhibition of NF- B Signaling Ablates the Invasive Phenotype of Glioblastoma. Mol. Cancer Res. 2013, 11, 1611–1623. [Google Scholar] [CrossRef]
- Xia, S.; Lal, B.; Tung, B.; Wang, S.; Goodwin, C.R.; Laterra, J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. 2016, 18, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Nandhu, M.S.; Kwiatkowska, A.; Bhaskaran, V.; Hayes, J.; Hu, B.; Viapiano, M.S. Tumor-derived fibulin-3 activates pro-invasive NF-κB signaling in glioblastoma cells and their microenvironment. Oncogene 2017, 36, 4875–4886. [Google Scholar] [CrossRef]
- Hu, B.; Nandhu, M.S.; Sim, H.; Agudelo-Garcia, P.A.; Saldivar, J.C.; Dolan, C.E.; Mora, M.E.; Nuovo, G.J.; Cole, S.E.; Viapiano, M.S. Fibulin-3 Promotes Glioma Growth and Resistance through a Novel Paracrine Regulation of Notch Signaling. Cancer Res. 2012, 72, 3873–3885. [Google Scholar] [CrossRef]
- Hiddingh, L.; Tannous, B.A.; Teng, J.; Tops, B.B.; Jeuken, J.; Hulleman, E.; Boots-Sprenger, S.H.; Vandertop, W.P.; Noske, D.P.; Kaspers, G.J.; et al. EFEMP1 induces γ-secretase/Notch-mediated temozolomide resistance in glioblastoma. Oncotarget 2013, 5, 363–374. [Google Scholar] [CrossRef]
- Zeng, A.-L.; Yan, W.; Liu, Y.-W.; Wang, Z.; Hu, Q.; Nie, E.; Zhou, X.; Li, R.; Wang, X.-F.; Jiang, T.; et al. Tumour exosomes from cells harbouring PTPRZ1–MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene 2017, 36, 5369–5381. [Google Scholar] [CrossRef]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.-G.; Joyce, C.; et al. Mesenchymal Stem Cells Isolated from Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; De Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Mizumoto, K.; Nakamura, M. Radiation-induced centrosome overduplication and multiple mitotic spindles in human tumor cells. Exp. Cell. Res. 2000, 255, 321–326. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2006, 26, 186–197. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Shinsato, Y.; Furukawa, T.; Yunoue, S. Reduction of mlh1 and pms2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget 2013, 4, 2261–2270. [Google Scholar] [CrossRef]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 1, 2038–2045. [Google Scholar] [CrossRef]
- Stark, A.M.; Doukas, A.; Hugo, H.-H.; Hedderich, J.; Hattermann, K.; Mehdorn, H.M.; Held-Feindt, J. Expression of DNA mismatch repair proteins MLH1, MSH2, and MSH6 in recurrent glioblastoma. Neurol. Res. 2014, 37, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, K.; Nakao, K.; Ohba, S.; Hirose, Y. Human Glioma Cells Acquire Temozolomide Resistance After Repeated Drug Exposure Via DNA Mismatch Repair Dysfunction. Anticancer. Res. 2020, 40, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Lee, D.H.; Yeon, S.-K.; Jeon, Y.H.; Yoo, J.; Lee, S.W.; Kwon, S.H. Temozolomide-resistant Glioblastoma Depends on HDAC6 Activity Through Regulation of DNA Mismatch Repair. Anticancer. Res. 2019, 39, 6731–6741. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nat. Cell Biol. 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, H.; Liu, T.; Huang, M.; Butter, P.-P.; Li, C.; Zhang, L.; Kao, G.D.; Gong, Y.; Maity, A.; et al. Temporal DNA-PK activation drives genomic instability and therapy resistance in glioma stem cells. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nat. Cell Biol. 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Chang, K.-Y.; Hsu, T.-I.; Hsu, C.-C.; Tsai, S.-Y.; Liu, J.-J.; Chou, S.-W.; Liu, M.-S.; Liou, J.-P.; Ko, C.-Y.; Chen, K.-Y.; et al. Specificity protein 1-modulated superoxide dismutase 2 enhances temozolomide resistance in glioblastoma, which is independent of O 6 -methylguanine-DNA methyltransferase. Redox Biol. 2017, 13, 655–664. [Google Scholar] [CrossRef]
- Lu, C.; Wei, Y.; Wang, X.; Zhang, Z.; Yin, J.; Li, W.; Chen, L.; Lyu, X.; Shi, Z.; Yan, W.; et al. DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma. Mol. Cancer 2020, 19, 1–19. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, G.; Hou, S.-X.; Sha, L.-G. MAPK8 mediates resistance to temozolomide and apoptosis of glioblastoma cells through MAPK signaling pathway. Biomed. Pharm. 2018, 106, 1419–1427. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, K.H.; Lee, J.; Lee, Y.-A.; Kim, M.; Lee, S.J.; Park, K.; Yang, H.; Jin, J.; Joo, K.M.; et al. Wnt activation is implicated in glioblastoma radioresistance. Lab. Investig. 2011, 92, 466–473. [Google Scholar] [CrossRef]
- Huang, K.; Liu, X.; Li, Y.; Wang, Q.; Zhou, J.; Wang, Y.; Dong, F.; Yang, C.; Sun, Z.; Fang, C.; et al. Genome-Wide CRISPR-Cas9 Screening Identifies NF-κB/E2F6 Responsible for EGFRvIII-Associated Temozolomide Resistance in Glioblastoma. Adv. Sci. 2019, 6, 1900782. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.-F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch Promotes Radioresistance of Glioma Stem Cells. Stem Cells 2009, 28, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Harding, A.; Kozlov, S.; Kijas, A.W.; Ensbey, K.S.; Walker, D.G.; Lavin, M.F. A Role for Homologous Recombination and Abnormal Cell-Cycle Progression in Radioresistance of Glioma-Initiating Cells. Mol. Cancer 2012, 11, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Ilkhanizadeh, S.; Wang, S.; Miroshnikova, Y.A.; Salvatierra, N.A.; Wong, R.A.; Schmidt, C.; Weaver, V.M.; Weiss, W.A.; Persson, A.I. STAT3 Blockade Inhibits Radiation-Induced Malignant Progression in Glioma. Cancer Res. 2015, 75, 4302–4311. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cázares, H.; Eberhart, C.G.; et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 9951–9956. [Google Scholar] [CrossRef] [PubMed]
- Lal, B.; Xia, S.; Abounader, R.; Laterra, J. Targeting the c-Met Pathway Potentiates Glioblastoma Responses to -Radiation. Clin. Cancer Res. 2005, 11, 4479–4486. [Google Scholar] [CrossRef]
- Kim, S.-H.; Joshi, K.; Ezhilarasan, R.; Myers, T.R.; Siu, J.; Gu, C.; Nakano-Okuno, M.; Taylor, D.; Minata, M.; Sulman, E.P.; et al. EZH2 Protects Glioma Stem Cells from Radiation-Induced Cell Death in a MELK/FOXM1-Dependent Manner. Stem Cell Rep. 2015, 4, 226–238. [Google Scholar] [CrossRef]
- Liu, H.; Sun, Y.; Qi, X.; Gordon, R.E.; O’Brien, J.A.; Yuan, H.; Zhang, J.; Wang, Z.; Zhang, M.; Song, Y.; et al. EZH2 Phosphorylation Promotes Self-Renewal of Glioma Stem-Like Cells Through NF-κB Methylation. Front. Oncol. 2019, 9, 641. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, P.; Pavlyukov, M.S.; Hongyu, L.; Zhang, Z.; Kim, S.-H.; Minata, M.; Mohyeldin, A.; Xie, W.; Chen, D.; et al. Targeting NEK2 attenuates glioblastoma growth and radioresistance by destabilizing histone methyltransferase EZH2. J. Clin. Investig. 2017, 127, 3075–3089. [Google Scholar] [CrossRef]
- Jeon, H.-Y.; Ham, S.W.; Kim, J.-K.; Jin, X.; Lee, S.Y.; Shin, Y.J.; Choi, C.-Y.; Sa, J.K.; Kim, S.H.; Chun, T.; et al. Ly6G+ inflammatory cells enable the conversion of cancer cells to cancer stem cells in an irradiated glioblastoma model. Cell Death Differ. 2019, 26, 2139–2156. [Google Scholar] [CrossRef]
- Tukachinsky, H.; Lopez, L.V. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell. Biol. 2010, 18, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-C.; Shieh, S.-Y. Deubiquitinating enzyme USP3 controls CHK1 chromatin association and activation. Proc. Natl. Acad. Sci. USA 2018, 115, 5546–5551. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Chen, Z.; Zhao, P.; Sun, G.; Bao, Z.; Chao, H.; Fan, L.; Li, C.; You, Y.; Qu, Y.; et al. Smoothened Promotes Glioblastoma Radiation Resistance Via Activating USP3-Mediated Claspin Deubiquitination. Clin. Cancer Res. 2020, 26, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, J. Replication protein A and more: Single-stranded DNA-binding proteins in eukaryotic cells. Acta Biochim. Et Biophys. Sin. 2016, 48, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.; Obara, E.A.A.; Elbæk, K.J.; Vitting-Serup, K.; Hamerlik, P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. Int. J. Mol. Sci. 2020, 21, 1588. [Google Scholar] [CrossRef]
- Bartkova, B., Jr.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nat. Cell Biol. 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Gaudet, S.; Branton, D.; Lue, R.A. Characterization of PDZ-binding kinase, a mitotic kinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5167–5172. [Google Scholar] [CrossRef]
- Mao, P.; Bao, G.; Wang, Y.-C.; Du, C.-W.; Yu, X.; Guo, X.-Y.; Li, R.-C.; Wang, M.-D. PDZ-Binding Kinase-Dependent Transcriptional Regulation of CCNB2 Promotes Tumorigenesis and Radio-Resistance in Glioblastoma. Transl. Oncol. 2020, 13, 287–294. [Google Scholar] [CrossRef]
- Ichimura, K.; Pearson, D.M.; Kocialkowski, S.; Bäcklund, L.M.; Chan, R.; Jones, D.T.; Collins, V.P. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009, 11, 341–347. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nat. Cell Biol. 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Ellingson, B.M.; Touat, M.; Maher, E.; De La Fuente, M.I.; Holdhoff, M.; Cote, G.M.; Burris, H.; Janku, F.; Young, R.J.; et al. Ivosidenib in Isocitrate Dehydrogenase 1–Mutated Advanced Glioma. J. Clin. Oncol. 2020, 38, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; A Goldlust, S.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, 399. [Google Scholar] [CrossRef]
- Uhm, J.H.; Ballman, K.V.; Wu, W.; Giannini, C.; Krauss, J.; Buckner, J.C.; James, C.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J.; et al. Phase II Evaluation of Gefitinib in Patients with Newly Diagnosed Grade 4 Astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. 2011, 80, 347–353. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. 2013, 15, 490–496. [Google Scholar] [CrossRef]
- AbbVie Provides Update on Depatuxizumab Mafodotin (Depatux-M), An Investigational Medicine for Newly Diagnosed Glioblastoma, an AGGRESSIVE Form of Brain Cancer. Available online: https://news.abbvie.com/news/press-releases/abbvie-provides-update-on-depatuxizumab-mafodotin-depatux-m-an-investigational-medicine-for-newly-diagnosed-glioblastoma-an-aggressive-form-brain-cancer.htm (accessed on 24 December 2020).
- Hasselbalch, B.; Lassen, U.; Hansen, S.; Holmberg, M.; Sørensen, M.; Kosteljanetz, M.; Broholm, H.; Stockhausen, M.-T.; Poulsen, H.S. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: A phase II trial. Neuro Oncol. 2010, 12, 508–516. [Google Scholar] [CrossRef]
- Klingler, S.; Guo, B.; Yao, J.; Yan, H.; Zhang, L.; Vaseva, A.V.; Chen, S.; Canoll, P.; Horner, J.W.; Wang, Y.A.; et al. Development of Resistance to EGFR-Targeted Therapy in Malignant Glioma Can Occur through EGFR-Dependent and -Independent Mechanisms. Cancer Res. 2015, 75, 2109–2119. [Google Scholar] [CrossRef]
- Lassman, A.B.; Pugh, S.L.; Gilbert, M.R.; Aldape, K.D.; Geinoz, S.; Beumer, J.H.; Christner, S.M.; Komaki, R.; DeAngelis, L.M.; Gaur, R.; et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro Oncol. 2015, 17, 992–998. [Google Scholar] [CrossRef] [PubMed]
- di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Savatovsky, M.G.J.; et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin. Cancer Res. 2015, 21, 3307–3317. [Google Scholar] [CrossRef] [PubMed]
- Chi, A.S.; Batchelor, T.T.; Kwak, E.L.; Clark, J.W.; Wang, D.L.; Wilner, K.D.; Louis, D.N.; Iafrate, A.J. Rapid Radiographic and Clinical Improvement After Treatment of a MET-Amplified Recurrent Glioblastoma with a Mesenchymal-Epithelial Transition Inhibitor. J. Clin. Oncol. 2012, 30, e30–e33. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol. 2015, 17, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Lassen, U.; Mau-Sørensen, M.; Gaziel, T.B.; Hasselbalch, B.; Poulsen, H.S. Phase II study of bevacizumab and temsirolimus combination therapy for recurrent glioblastoma multiforme. Anticancer. Res. 2013, 33, 1657–1660. [Google Scholar] [PubMed]
- Schreck, K.C.; A Grossman, S.; Pratilas, C. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262. [Google Scholar] [CrossRef] [PubMed]
- Hottinger, A.F.; Ben Aissa, A.; Espeli, V.; Squiban, D.; Dunkel, N.; I Vargas, M.; Hundsberger, T.; Mach, N.; Schaller, K.; Weber, D.C.; et al. Phase I study of sorafenib combined with radiation therapy and temozolomide as first-line treatment of high-grade glioma. Br. J. Cancer 2014, 110, 2655–2661. [Google Scholar] [CrossRef]
- Costa, B.; Bendinelli, S.; Gabelloni, P.; Da Pozzo, E.; Daniele, S.; Scatena, F.; Vanacore, R.; Campiglia, P.; Bertamino, A.; Gomez-Monterrey, I.; et al. Human Glioblastoma Multiforme: p53 Reactivation by a Novel MDM2 Inhibitor. PLoS ONE 2013, 8, e72281. [Google Scholar] [CrossRef]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.S.; Labussière, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef]
- Tien, A.-C.; Li, J.; Bao, X.; Derogatis, A.; Kim, S.; Mehta, S.; Sanai, N. A Phase 0 Trial of Ribociclib in Recurrent Glioblastoma Patients Incorporating a Tumor Pharmacodynamic- and Pharmacokinetic-Guided Expansion Cohort. Clin. Cancer Res. 2019, 25, 5777–5786. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.V.; Searle, K.; Jerzak, K.J. Central nervous system-specific efficacy of CDK4/6 inhibitors in randomized controlled trials for metastatic breast cancer. Oncotarget 2019, 10, 6317–6322. [Google Scholar] [CrossRef] [PubMed]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.-S.; Zhong, W.-Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic Inhibition of Cyclin-Dependent Kinases 4 and 6 Arrests the Growth of Glioblastoma Multiforme Intracranial Xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 1. [Google Scholar]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; De Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: CheckMate 143. Neuro Oncol. 2017, 19, iii21. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors [published correction appears in Nat Med]. Nat. Med. 2019, 24, 1459–1468. [Google Scholar] [CrossRef]
- Dieckmann, D.; Plottner, H.; Berchtold, S.; Berger, T.; Schuler, G. Ex Vivo Isolation and Characterization of Cd4+Cd25+ T Cells with Regulatory Properties from Human Blood. J. Exp. Med. 2001, 193, 1303–1310. [Google Scholar] [CrossRef]
- Fecci, P.E.; Mithcell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E., II; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, A.E.; Lesniak, M.S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol. 2006, 8, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.; Hofmann, W.J.; Hug, H. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells—a mechanism of immune evasion? Nat. Med. 1996, 2, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; DeChant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 Expression Defines a Subset of CD4+CD25highFoxp3+ Regulatory T Cells That Are Expanded at Tumor Sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef]
- Baeza, N.; Weller, M.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. PTEN methylation and expression in glioblastomas. Acta Neuropathol. 2003, 106, 479–485. [Google Scholar] [CrossRef]
- Waldron, J.S.; Yang, I.; Han, S.; Tihan, T.; Sughrue, M.E.; Mills, S.A.; Pieper, R.O.; Parsa, A.T. Implications for immunotherapy of tumor-mediated T-cell apoptosis associated with loss of the tumor suppressor PTEN in glioblastoma. J. Clin. Neurosci. 2010, 17, 1543–1547. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2015, 6, 202–216. [Google Scholar] [CrossRef]
- Chen, Q.; Han, B.; Meng, X.; Duan, C.; Yang, C.; Wu, Z.; Magafurov, D.; Zhao, S.; Safin, S.; Jiang, C.; et al. Immunogenomic analysis reveals LGALS1 contributes to the immune heterogeneity and immunosuppression in glioma. Int. J. Cancer 2019, 145, 517–530. [Google Scholar] [CrossRef]
- Hahn, S.A.; Stahl, H.F.; Becker, C.; Correll, A.; Schneider, F.-J.; Tuettenberg, A.; Jonuleit, H. Soluble GARP has potent antiinflammatory and immunomodulatory impact on human CD4+ T cells. Blood 2013, 122, 1182–1191. [Google Scholar] [CrossRef]
- Zimmer, N.; Kim, E.L.; Sprang, B.; Leukel, P.; Khafaji, F.; Ringel, F.; Sommer, C.J.; Tuettenberg, A. GARP as an Immune Regulatory Molecule in the Tumor Microenvironment of Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 3676. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses1. Neuro Oncol. 2006, 8, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wei, S.; Zou, L.; Altuwaijri, S.; Szeliga, W.; Kolls, J.; Chang, A.; Zou, W. Cutting edge: Th17 and regulatory T cell dynamics and the regulation of IL-2 in the tumor microenvironment. J. Immunol. 2007, 178, 6730–6733. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Kong, L.-Y.; Jordan, J.; Conrad, C.; Madden, T.; Fokt, I.; Priebe, W.; Heimberger, A.B. A Novel Small Molecule Inhibitor of Signal Transducers and Activators of Transcription 3 Reverses Immune Tolerance in Malignant Glioma Patients. Cancer Res. 2007, 67, 9630–9636. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mule, J.J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef]
- Akiyama, Y.; Nonomura, C.; Ashizawa, T.; Iizuka, A.; Kondou, R.; Miyata, H.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; et al. The anti-tumor activity of the STAT3 inhibitor STX-0119 occurs via promotion of tumor-infiltrating lymphocyte accumulation in temozolomide-resistant glioblastoma cell line. Immunol. Lett. 2017, 190, 20–25. [Google Scholar] [CrossRef]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017, 20, 450–461.e4. [Google Scholar] [CrossRef]
- Almand, B.; Clark, J.I.; Nikitina, E.; Van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased Production of Immature Myeloid Cells in Cancer Patients: A Mechanism of Immunosuppression in Cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; A Walentynowicz, K.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef]
- Marigo, I.; Bosio, E.; Solito, S.; Mesa, C.; Fernandez, A.; Dolcetti, L. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010, 6, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Bah, I.; Kumbhare, A.; Nguyen, L.; McCall, C.E.; El Gazzar, M. IL-10 induces an immune repressor pathway in sepsis by promoting S100A9 nuclear localization and MDSC development. Cell. Immunol. 2018, 332, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.G.; Megiel, C.; Russell, S.M.; Bingham, B.; Arger, N.; Woo, T.; Epstein, A.L. Functional characterization of human Cd33+ And Cd11b+ myeloid-derived suppressor cell subsets induced from peripheral blood mononuclear cells co-cultured with a diverse set of human tumor cell lines. J. Transl. Med. 2011, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Schrum, A.G.; Cho, H.-I.; Celis, E.; Gabrilovich, D.I. Mechanism of T Cell Tolerance Induced by Myeloid-Derived Suppressor Cells. J. Immunol. 2010, 184, 3106–3116. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-J.; Lee, S.-H.; Kim, E.-K.; Lee, E.-J.; Baek, J.-A.; Park, S.-H.; Kwok, S.-K.; Cho, M.-L. Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef]
- Comba, A.; Dunn, P.J.; E Argento, A.; Kadiyala, P.; Ventosa, M.; Patel, P.; Zamler, D.B.; Núñez, F.J.; Zhao, L.; Castro, M.G.; et al. Fyn tyrosine kinase, a downstream target of receptor tyrosine kinases, modulates antiglioma immune responses. Neuro Oncol. 2020, 22, 806–818. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; De Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol. 2017, 19, 1047–1057. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.B.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; De Andrea, C.; De Cerio, A.L.-D.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Giles, A.J.; Hutchinson, M.-K.N.D.; Sonnemann, H.M.; Jung, J.; E Fecci, P.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D.; et al. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, C.-K.; Jackson, C.M.; Garzon-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M.; et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [Google Scholar] [CrossRef] [PubMed]
- Dutoit, V.; Philippin, G.; Widmer, V.; Marinari, E.; Vuilleumier, A.; Migliorini, D.; Schaller, K.; Dietrich, P.-Y. Impact of Radiochemotherapy on Immune Cell Subtypes in High-Grade Glioma Patients. Front. Oncol. 2020, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ishikawa, E.; Matsuda, M.; Sugii, N.; Kohzuki, H.; Akutsu, H.; Sakamoto, N.; Takano, S.; Matsumura, A. Infiltration of CD163-positive macrophages in glioma tissues after treatment with anti-PD-L1 antibody and role of PI3Kγ inhibitor as a combination therapy with anti-PD-L1 antibody in in vivo model using temozolomide-resistant murine glioma-initiating cells. Brain Tumor Pathol. 2020, 37, 41–49. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. New Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Mints, M.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2018, 565, 234–239. [Google Scholar] [CrossRef]
- Rampling, R.; Peoples, S.; Mulholland, P.J.; James, A.; Al-Salihi, O.; Twelves, C.J.; McBain, C.; Jefferies, S.; Jackson, A.; Wa, R.J.; et al. A Cancer Research UK First Time in Human Phase I Trial of IMA950 (Novel Multipeptide Therapeutic Vaccine) in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2016, 22, 4776–4785. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.; Glantz, M.; Peereboom, D.M.; et al. A randomized, double-blind, placebo-controlled phase 2 trial of dendritic cell (DC) vaccination with ICT-107 in newly diagnosed glioblastoma (GBM) patients. J. Clin. Oncol. 2005, 2014, 32. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, W.B.; Kleijn, A.; Teunissen, C.E.; Voerman, J.S.; Teodosio, C.; Noske, D.P.; van Dongen, J.J.M.; Dirven, C.M.F.; Lamfers, M.L.M. Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro Oncol. 2018, 20, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen–specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, J.; Mills, L.; Malo, C.S.; Jin, F.; Kurokawa, C.; Geekiyanage, H.; Schroeder, M.; Sarkaria, J.; Johnson, A.J.; Galanis, E. Immunovirotherapy with measles virus strains in combination with anti–PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. 2016, 19, 493–502. [Google Scholar] [CrossRef]
- Wu, A.; Maxwell, R.; Xia, Y.; Cardarelli, P.; Oyasu, M.; Belcaid, Z.; Kim, E.; Hung, A.; Luksik, A.S.; Garzon-Muvdi, T.; et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J. Neuro Oncol. 2019, 143, 241–249. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ou, A.; Yung, W.K.A.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 351. https://doi.org/10.3390/ijms22010351
Ou A, Yung WKA, Majd N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. International Journal of Molecular Sciences. 2021; 22(1):351. https://doi.org/10.3390/ijms22010351
Chicago/Turabian StyleOu, Alexander, W. K. Alfred Yung, and Nazanin Majd. 2021. "Molecular Mechanisms of Treatment Resistance in Glioblastoma" International Journal of Molecular Sciences 22, no. 1: 351. https://doi.org/10.3390/ijms22010351
APA StyleOu, A., Yung, W. K. A., & Majd, N. (2021). Molecular Mechanisms of Treatment Resistance in Glioblastoma. International Journal of Molecular Sciences, 22(1), 351. https://doi.org/10.3390/ijms22010351