Cell-Type-Specific Complement Profiling in the ABCA4−/− Mouse Model of Stargardt Disease

 and
and
Abstract
1. Introduction
2. Results
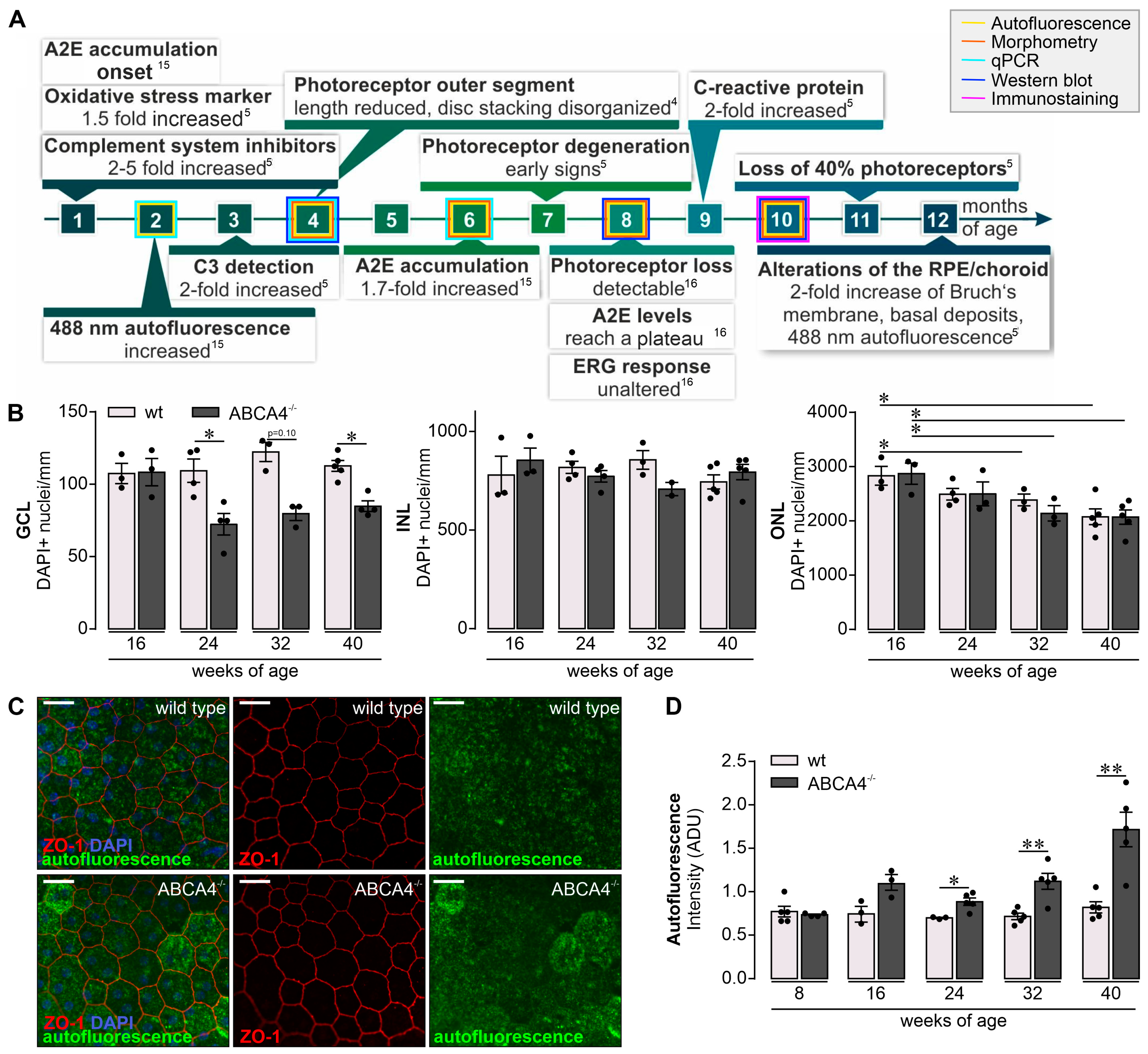
2.1. Retinal Phenotype is Changed in Albino ABCA4−/− Compared to Wild-Type Mice
2.2. Complement Expression Changed in the Different Retinal Cell Types Over Time
2.3. Increase of C3 Cleavage Products in ABCA4−/− Mice Compared to Wild-Type Mice
2.4. Decrease of CFI Levels in the Retina of ABCA4−/− Mice
2.5. Signs of Mild Microglial Activation in Aging ABCA4−/− Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Immunohistochemistry of Retina and RPE/Choroid Flat Mounts
4.3. Isolation of Retinal Cell Populations by Immunomagnetic Enrichment
4.4. qRT-PCR
4.5. Western Blot
4.6. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Molday, R.S. Insights into the Molecular Properties of ABCA4 and Its Role in the Visual Cycle and Stargardt Disease. Prog. Mol. Biol. Transl. Sci. 2015, 134, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Genead, M.A.; Fishman, G.A.; Stone, E.M.; Allikmets, R. The Natural History of Stargardt Disease with Specific Sequence Mutation in the ABCA4 Gene. Investig. Opthalmol. Vis. Sci. 2009, 50, 5867–5871. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Starqardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Taubitz, T.; Tschulakow, A.V.; Tikhonovich, M.; Illing, B.; Fang, Y.; Biesemeier, A.; Julien-Schraermeyer, S.; Schraermeyer, U. Ultrastructural alterations in the retinal pigment epithelium and photoreceptors of a Stargardt patient and three Stargardt mouse models: Indication for the central role of RPE melanin in oxidative stress. PeerJ 2018, 6, e5215. [Google Scholar] [CrossRef] [PubMed]
- Radu, R.A.; Hu, J.; Yuan, Q.; Welch, D.L.; Makshanoff, J.; Lloyd, M.; McMullen, S.; Travis, G.H.; Bok, D. Complement System Dysregulation and Inflammation in the Retinal Pigment Epithelium of a Mouse Model for Stargardt Macular Degeneration. J. Biol. Chem. 2011, 286, 18593–18601. [Google Scholar] [CrossRef]
- Lenis, T.L.; Sarfare, S.; Jiang, Z.; Lloyd, M.B.; Bok, D.; Radu, R.A. Complement modulation in the retinal pigment epithelium rescues photoreceptor degeneration in a mouse model of Stargardt disease. Proc. Natl. Acad. Sci. USA 2017, 114, 3987–3992. [Google Scholar] [CrossRef]
- Zhou, J.; Jang, Y.P.; Kim, S.R.; Sparrow, J.R. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2006, 103, 16182–16187. [Google Scholar] [CrossRef]
- Zhou, J.; Kim, S.R.; Westlund, B.S.; Sparrow, J.R. Complement Activation by Bisretinoid Constituents of RPE Lipofuscin. Investig. Opthalmology Vis. Sci. 2009, 50, 1392–1399. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Clark, S.J.; Bishop, P.N. The eye as a complement dysregulation hotspot. Semin. Immunopathol. 2018, 40, 65–74. [Google Scholar] [CrossRef]
- Shahulhameed, S.; Vishwakarma, S.; Chhablani, J.; Tyagi, M.; Pappuru, R.R.; Jakati, S.; Chakrabarti, S.; Kaur, I. A Systematic Investigation on Complement Pathway Activation in Diabetic Retinopathy. Front. Immunol. 2020, 11, 154. [Google Scholar] [CrossRef]
- Bradley, D.T.; Zipfel, P.F.; E Hughes, A. Complement in age-related macular degeneration: A focus on function. Eye 2011, 25, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Heesterbeek, T.J.; Lechanteur, Y.T.E.; Lorés-Motta, L.; Schick, T.; Daha, M.R.; Altay, L.; Liakopoulos, S.; Smailhodzic, D.; Hollander, A.I.D.; Hoyng, C.B.; et al. Complement Activation Levels Are Related to Disease Stage in AMD. Investig. Opthalmol. Vis. Sci. 2020, 61, 18. [Google Scholar] [CrossRef]
- MacLaren, R.E.; Barnard, A.R.; Singh, M.S.; Carter, E.; Jiang, Z.; Radu, R.A.; Schraermeyer, U.; MacLaren, R.E. Fundus Autofluorescence in theAbca4−/−Mouse Model of Stargardt Disease—Correlation With Accumulation of A2E, Retinal Function, and Histology. Investig. Opthalmol. Vis. Sci. 2013, 54, 5602–5612. [Google Scholar] [CrossRef]
- Wu, L.; Nagasaki, T.; Sparrow, J.R. Photoreceptor Cell Degeneration in Abcr –/– Mice. Retin. Degener. Dis. 2010, 533–539. [Google Scholar] [CrossRef]
- Issa, P.C.; Barnard, A.R.; Herrmann, P.; Washington, I.; MacLaren, R.E. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc. Natl. Acad. Sci. USA 2015, 112, 8415–8420. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Blonska, A.; Flynn, E.; Duncker, T.; Greenberg, J.P.; Secondi, R.; Ueda, K.; Delori, F.C. Quantitative Fundus Autofluorescence in Mice: Correlation with HPLC Quantitation of RPE Lipofuscin and Measurement of Retina Outer Nuclear Layer Thickness. Investig. Opthalmol. Vis. Sci. 2013, 54, 2812–2820. [Google Scholar] [CrossRef]
- Pauly, D.; Agarwal, D.; Dana, N.; Schäfer, N.; Biber, J.; Wunderlich, K.A.; Jabri, Y.; Straub, T.; Zhang, N.R.; Gautam, A.K.; et al. Cell-Type-Specific Complement Expression in the Healthy and Diseased Retina. Cell Rep. 2019, 29, 2835–2848.e4. [Google Scholar] [CrossRef]
- Grosche, A. GFAP expression across purified retinal cell populations after immunomagnetic separation. Unpublished work. 2020. [Google Scholar]
- Rose, K.L.; Paixao-Cavalcante, D.; Fish, J.; Manderson, A.P.; Malik, T.H.; Bygrave, A.E.; Lin, T.; Sacks, S.; Walport, M.J.; Cook, H.T.; et al. Factor I is required for the development of membranoproliferative glomerulonephritis in factor H–deficient mice. J. Clin. Investig. 2008, 118, 608–618. [Google Scholar] [CrossRef]
- Collier, R.J.; Wang, Y.; Smith, S.S.; Martin, E.; Ornberg, R.; Rhoades, K.; Romano, C. Complement Deposition and Microglial Activation in the Outer Retina in Light-Induced Retinopathy: Inhibition by a 5-HT1AAgonist. Investig. Opthalmol. Vis. Sci. 2011, 52, 8108–8116. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, S.; Reinehr, S.; Stute, G.; Rodust, C.; Grotegut, P.; Hensel, A.-T.; Dick, H.B.; Joachim, S.C. Interaction of complement system and microglia activation in retina and optic nerve in a NMDA damage model. Mol. Cell. Neurosci. 2018, 89, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, M.J.; De Jong, E.K.; Hollander, A.I.D. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol. Immunol. 2017, 84, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Park, D.H.; Connor, K.M.; Lambris, J.D. The Challenges and Promise of Complement Therapeutics for Ocular Diseases. Front. Immunol. 2019, 10, 1007. [Google Scholar] [CrossRef] [PubMed]
- McHarg, S.; Clark, S.J.; Day, A.J.; Bishop, P.N. Age-related macular degeneration and the role of the complement system. Mol. Immunol. 2015, 67, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Kassa, E.; Ciulla, T.A.; Hussain, R.M.; Dugel, P.U. Complement inhibition as a therapeutic strategy in retinal disorders. Expert Opin. Biol. Ther. 2019, 19, 335–342. [Google Scholar] [CrossRef]
- Newman, N.M.; A Stevens, R.; Heckenlively, J.R. Nerve fibre layer loss in diseases of the outer retinal layer. Br. J. Ophthalmol. 1987, 71, 21–26. [Google Scholar] [CrossRef]
- Ratra, D.; Jaishankar, D.; Sachidanandam, R.; Yusufali, H.; Ratra, V. Swept-source optical coherence tomography study of choroidal morphology in Stargardt disease. Oman J. Ophthalmol. 2018, 11, 150–157. [Google Scholar]
- Lim, J.I.; Tan, O.; Fawzi, A.A.; Hopkins, J.J.; Gil-Flamer, J.H.; Huang, D. A Pilot Study of Fourier-Domain Optical Coherence Tomography of Retinal Dystrophy Patients. Am. J. Ophthalmol. 2008, 146, 417–426.e2. [Google Scholar] [CrossRef]
- Racz, B.; Varadi, A.; Kong, J.; Allikmets, R.; Pearson, P.G.; Johnson, G.; Cioffi, C.L.; Petrukhin, K. A non-retinoid antagonist of retinol-binding protein 4 rescues phenotype in a model of Stargardt disease without inhibiting the visual cycle. J. Biol. Chem. 2018, 293, 11574–11588. [Google Scholar] [CrossRef]
- Li, Q.; Li, Y.X.; Stahl, G.L.; Thurman, J.M.; He, Y.; Tong, H.H. Essential role of factor B of the alternative complement pathway in complement activation and opsonophagocytosis during acute pneumococcal otitis media in mice. Infect Immun. 2011, 79, 2578–2585. [Google Scholar] [CrossRef] [PubMed]
- Kremlitzka, M.; Nowacka, A.A.; Mohlin, F.C.; Bompada, P.; De Marinis, Y.; Blom, A.M. Interaction of Serum-Derived and Internalized C3 With DNA in Human B Cells—A Potential Involvement in Regulation of Gene Transcription. Front. Immunol. 2019, 10, 493. [Google Scholar] [CrossRef] [PubMed]
- Satyam, A.; Kannan, L.; Matsumoto, N.; Geha, M.; Lapchak, P.H.; Bosse, R.; Shi, G.-P.; Lucca, J.J.D.; Tsokos, M.G.; Tsokos, G.C. Intracellular Activation of Complement 3 Is Responsible for Intestinal Tissue Damage during Mesenteric Ischemia. J. Immunol. 2016, 198, 788–797. [Google Scholar] [CrossRef] [PubMed]
- King, B.C.; Kulak, K.; Krus, U.; Rosberg, R.; Golec, E.; Wozniak, K.; Gomez, M.F.; Zhang, E.; O’Connell, D.J.; Renström, E.; et al. Complement Component C3 Is Highly Expressed in Human Pancreatic Islets and Prevents β Cell Death via ATG16L1 Interaction and Autophagy Regulation. Cell Metab. 2019, 29, 202–210.e6. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.C.; Sim, R.B.; Lea, S.M.; Fremeaux-Bacchi, V.; Blom, A.M. Complement factor I in health and disease. Mol. Immunol. 2011, 48, 1611–1620. [Google Scholar] [CrossRef]
- Roversi, P.; Johnson, S.; Caesar, J.J.E.; McLean, F.; Leath, K.J.; Tsiftsoglou, S.A.; Morgan, B.P.; Harris, C.L.; Sim, R.B.; Lea, S.M. Structural basis for complement factor I control and its disease-associated sequence polymorphisms. Proc. Natl. Acad. Sci. USA 2011, 108, 12839–12844. [Google Scholar] [CrossRef]
- Gallego, J.I.S.; Nita, I.M.; Nilsson, S.C.; Groeneveld, T.W.; Villoutreix, B.O.; Blom, A.M. Analysis of binding sites on complement Factor I that are required for its activity. J Biol Chem. 2010, 285, 6235–6245. [Google Scholar] [CrossRef]
- Van De Ven, J.; Nilsson, S.; Tan, P.; Buitendijk, G.; Ristau, T.; Mohlin, F.; Nabuurs, S.; Schoenmaker-Koller, F.; Smailhodzic, D.; Campochiarro, P.; et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Mol. Immunol. 2013, 56, 247–248. [Google Scholar] [CrossRef]
- Maggs, D.J. Cornea and Sclera. In Slatter’s Fundamentals of Veterinary Ophthalmology; Elsevier Inc.: New York, NY, USA, 2008; pp. 175–202. [Google Scholar]
- Krausgruber, T.; Fortelny, N.; Fife-Gernedl, V.; Senekowitsch, M.; Schuster, L.C.; Lercher, A.; Nemc, A.; Schmidl, C.; Rendeiro, A.F.; Bergthaler, A.; et al. Structural cells are key regulators of organ-specific immune responses. Nat. Cell Biol. 2020, 583, 296–302. [Google Scholar] [CrossRef]
- Voigt, A.P.; Mulfaul, K.; Mullin, N.K.; Flamme-Wiese, M.J.; Giacalone, J.C.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 24100–24107. [Google Scholar] [CrossRef]
- Radu, R.A.; Yuan, Q.; Hu, J.; Peng, J.H.; Lloyd, M.; Nusinowitz, S.; Bok, D.; Travis, G.H. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Investig. Opthalmol. Vis. Sci. 2008, 49, 3821–3829. [Google Scholar] [CrossRef] [PubMed]
- Kohno, H.; Chen, Y.; Kevany, B.M.; Pearlman, E.; Miyagi, M.; Maeda, T.; Palczewski, K.; Maeda, A. Photoreceptor Proteins Initiate Microglial Activation via Toll-like Receptor 4 in Retinal Degeneration Mediated by All-trans-retinal. J. Biol. Chem. 2013, 288, 15326–15341. [Google Scholar] [CrossRef] [PubMed]
- Mukai, R.; Okunuki, Y.; Husain, D.; Kim, C.B.; Lambris, J.D.; Connor, K.M. The Complement System Is Critical in Maintaining Retinal Integrity during Aging. Front. Aging Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Mages, K.; Grassmann, F.; Jägle, H.; Rupprecht, R.; Weber, B.H.F.; Hauck, S.M.; Grosche, A. The agonistic TSPO ligand XBD173 attenuates the glial response thereby protecting inner retinal neurons in a murine model of retinal ischemia. J. Neuroinflamm. 2019, 16, 43. [Google Scholar] [CrossRef]
- Schäfer, N.; Grosche, A.; Schmitt, S.I.; Braunger, B.M.; Pauly, D. Complement Components Showed a Time-Dependent Local Expression Pattern in Constant and Acute White Light-Induced Photoreceptor Damage. Front. Mol. Neurosci. 2017, 10, 197. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Primer Sequences: Forward | Primer Sequences: Reverse | Accession Number |
|---|---|---|---|
| abca4 | 5′ aggagaagcaatcaaatcagga 3′ | 5′ gaagatgctctggatatctctgc 3′ 5′ tgagtaggtgtcattctattctgg 3′ | NM_007378.1 |
| rd1 | 5′ tgacaattactccttttccctcagtctg 3′ 5′ tacccacccttcctaattttctcacgc 3′ | 5′ gtaaacagcaagaggctttattgggaac 3′ | AH002075.2 |
| rd8 | 5′ gtgaagacagctacagttctgatc 3′ 5′gcccctgtttgcatggaggaaacttggaagacagctacagttcttctg 3′ | 5′ gccccatttgcacactgatgac 3′ | NM_133239.2 |
| rd10 | 5′ acaaggaacaagggctctga 3′ | 5′ ccttccactcattgctaggac 3′ | NM_008806.2 |
| rd12 | 5′ tgacacctagttttaatattttgatcc 3′ | 5′cagagcttgaaccccatt 3′ | NM_029987.2 |
| Gene ID | Primer Sequences: Forward | Primer Sequences: Reverse | TaqMan® Probe from Roche | Accession Number |
|---|---|---|---|---|
| idh3b | 5′ gctgcggcatctcaatct 3′ | 5′ ccatgtctcgagtccgtacc 3′ | # 67 | NM_130884.4 |
| c1s | 5′ ggtggatacttctgctcctgtc 3′ | 5′ agggcagtgaacacatctcc 3′ | # 69 | NM_144938.2 |
| c3 | 5′ accttacctcggcaagtttct 3′ | 5′ ttgtagagctgctggtcagg 3′ | # 76 | NM_009778.3 |
| cfb | 5′ ctcgaacctgcagatccac 3′ | 5′ tcaaagtcctgcggtcgt 3′ | # 112 | NM_008198.2 |
| cfp | 5′ tcttgagtggcagctacagg 3′ | 5′ cagaccagccacccatct 3′ | # 56 | NM_008823.4 |
| cfh | 5′ aaaaaccaaagtgccgagac 3′ | 5′ ggaggtgatgtctccattgtc 3′ | # 25 | NM_009888.3 |
| cfi | 5′ tttctcttggctctccacttg 3′ | 5′ tgcagtaagcatttctgatcg 3′ | # 63 | NM_007686.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabri, Y.; Biber, J.; Diaz-Lezama, N.; Grosche, A.; Pauly, D. Cell-Type-Specific Complement Profiling in the ABCA4−/− Mouse Model of Stargardt Disease. Int. J. Mol. Sci. 2020, 21, 8468. https://doi.org/10.3390/ijms21228468
Jabri Y, Biber J, Diaz-Lezama N, Grosche A, Pauly D. Cell-Type-Specific Complement Profiling in the ABCA4−/− Mouse Model of Stargardt Disease. International Journal of Molecular Sciences. 2020; 21(22):8468. https://doi.org/10.3390/ijms21228468
Chicago/Turabian StyleJabri, Yassin, Josef Biber, Nundehui Diaz-Lezama, Antje Grosche, and Diana Pauly. 2020. "Cell-Type-Specific Complement Profiling in the ABCA4−/− Mouse Model of Stargardt Disease" International Journal of Molecular Sciences 21, no. 22: 8468. https://doi.org/10.3390/ijms21228468
APA StyleJabri, Y., Biber, J., Diaz-Lezama, N., Grosche, A., & Pauly, D. (2020). Cell-Type-Specific Complement Profiling in the ABCA4−/− Mouse Model of Stargardt Disease. International Journal of Molecular Sciences, 21(22), 8468. https://doi.org/10.3390/ijms21228468